INTRODUCTION
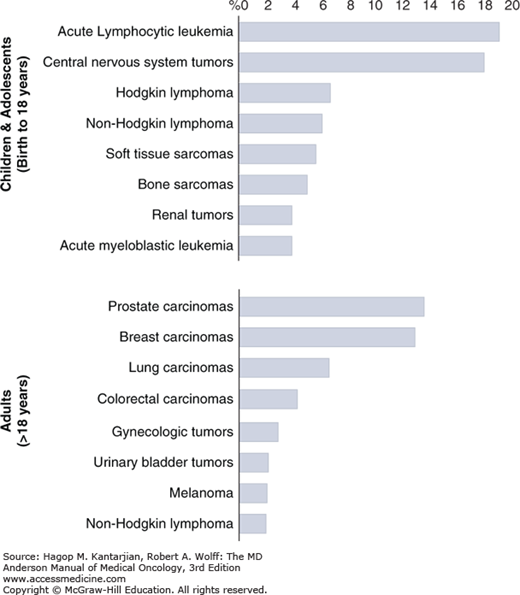
Fortunately, pediatric cancers are rare, with only 1 in 300 children being diagnosed before 18 years of age. And, with overall survival (OS) approaching 80%, there is hope for most of these children and for their families. However, just as in adults, if their cancer is metastatic at diagnosis, if the cancer does not respond to standard therapies, or if they suffer a relapse, the prognosis is universally grim. Despite intensified therapies, the survival for most children with relapsed cancer has not improved in decades. In large part, this is due to the toxicities of such regimens as we have likely reached the tolerable limit with most chemotherapeutic agents. Thus, as is the case for adults with cancer, there has been a focus on understanding the underlying biology of the disease to find targetable lesions and to develop novel agents and treatment regimens to improve survival in children with relapse. However, there are multiple challenges we face. First, the spectrum of pediatric cancers is distinctly different from adults (Fig. 46-1), with acute lymphoblastic leukemia (ALL), medulloblastoma and gliomas, neuroblastoma, Hodgkin and non-Hodgkin lymphomas, and sarcomas the most common cancers in children as opposed to the most common carcinomas of the prostate, breast, lung, colon, and so on in adults. Second, the relative rarity of these cancers results in less preclinical research to find and develop potential therapeutic targets and limited patient numbers to enroll and test novel therapeutic strategies.
One of our approaches has been to study the pediatric cancers in parallel with similar adult tumors and to enroll patients on early adult trials to give children access to promising agents and to provide some data on safety and efficacy to support the development of pediatric-specific trials. In this chapter, we present a few examples of tumor types seen commonly in children and describe some of the therapeutic advances and promising strategies for treating children with relapsed cancers.
SALVAGE STRATEGIES
Acute lymphoblastic leukemia is the most common cancer diagnosis in children, accounting for 25% of cancer diagnoses in children 15 years old or less. An estimated 3,000 new cases of childhood ALL are diagnosed yearly in the United States. After a peak incidence of 90 cases per million per year at age 2 to 3 years, ALL incidence rates decrease steadily into adolescence. Initial complete remission (CR) rates are 95%, and survival in childhood ALL is approaching 90% through the application of reliable prognostic factors that permit use of risk-oriented treatment protocols. However, relapse occurs in approximately 20%, with higher rates of relapse in adolescents and young adults as well as children less than 1 year of age (ie, “infants”). Despite excellent outcomes overall, relapsed patients with ALL outnumber nearly all other childhood malignancies. With traditional intensive combination chemotherapy and allogeneic hematopoietic stem cell transplantation, 30% to 40% of all children with relapsed ALL can be cured. The factors that effect salvage rate are timing of relapse (18-36 months from diagnosis), site of relapse (bone marrow, central nervous system [CNS]/testicular, combined), and immunophenotype (T cell vs B cell). Unfortunately, most children still die from relapsed ALL despite aggressive chemoradiotherapy approaches, including transplantation, and novel salvage regimens are needed (1).
For children with first relapse of childhood ALL, the mitoxantrone-based Medical Research Council (MRC) ALL R3 relapsed protocol has relatively better outcomes than other common regimens and is currently our standard reinduction regimen (2). Promising CR2 rates have also been seen with the addition of the proteasome inhibitor bortezomib to the MRC ALL R3 backbone, with 80% (16/20) CR/CRi in B-ALL patients (3), as well as the addition of bortezomib to the more traditional four-drug reinduction regimen in COG AALL07P1, with 64% (63/99) of B-ALL patients achieving CR2. Surprisingly, AALL07P1 showed similar CR2 rates of 68% (15/22) in T-ALL patients (4). The current first relapse regimen under study in the COG is testing the substitution of the CD19-targeting bispecific engaging antibody blinatumomab to the MRC ALL R3 relapsed protocol.
With attainment of CR2 in 70% to 85% (<1% minimal residual disease by flow cytometry), we offer transplant to most children with relapsed ALL, with very late relapses and isolated CNS relapses as typical exceptions. We offer a variety of transplant trials, including cord blood and haploidentical, as well as addition of anti-CD19 chimeric antigen receptor (CAR) T cells, natural killer (NK) cells, and so on. Failure to respond adequately to first-relapse regimens leads to a variety of second-line regimens. We commonly offer clofarabine/cyclophosphamide/etoposide (CR2 rates of 44% [5]), mitoxantrone/cytarabine (CR2 rate of 57% [6]) or one of several targeted agents with the institutional adult ALL regimen hyper-CVAD (7), or promising single agents when available. With recently published complete responses in children (8), we are testing the combination of anti-CD22 immunotoxin inotuzumab with hyper-CVAD (without anthracycline), with some dramatic responses in children. In addition, the combination of the mTOR inhibitor everolimus and Hyper-CVAD is being tested. For T-cell ALL specifically, we have used nelarabine in combination with hyper-CVAD (9), a trial testing a 5-day continuous infusion of nelarabine (NCT01094860), and a single agent Notch-inhibiting gamma secretase inhibitor BMS-906024 phase I trial (NCT01363817), based on multiple responses in adults (10).
Targeting ALL blasts with antibodies, both unconjugated and conjugated to toxins, has been shown to be an effective therapeutic strategy. Monoclonal antibodies to surface antigens such as CD19, CD20, CD22, and CD52 have been used in unconjugated form (eg, rituximab and epratuzumab), conjugated to immunotoxins or chemotherapeutic drugs (eg, moxetumomab, inotuzumab ozogamicin), or in the form of bispecific antibodies (eg, blinatumomab). The incorporation of rituximab (CD20) into the hyper-CVAD regimen showed improved outcome in adults younger than 60 years with more than 5% CD20-positive, BCR-ABL1–negative B lymphoblastic leukemia (11). Recent significant success with the anti-CD19–anti-CD3 bispecific T-cell engaging (BiTE) antibody blinatumomab has led to responses in about 50% of patients with relapsed pediatric B-ALL.
Recent therapeutic advances in ALL have also included the use of adoptive immunotherapies. Anti-CD19 CAR-expressing T cells have shown dramatic success in patients with relapsed B-ALL, with up to 90% remission rates reported (12). We have developed a unique nonretroviral approach utilizing the Sleeping Beauty transposon system to transfect T cells with CAR constructs, opening this technology to rapid translation of targetable tumor markers (13).
Patients with BCR-ABL1–positive ALL had poor prognoses. When tyrosine kinase inhibitors targeting ABL1 are added to multidrug chemotherapy, CR rates are more than 90%, and event-free survival (EFS) is superior to that in historical controls. Recent work has discovered that a subset (~10%) of children with BCR-ABL1–negative ALL have gene expression patterns similar to those with BCR-ABL1–positive ALL. Most children and adults with this “Ph-like” ALL harbor a range of novel kinase translocations (including ABL1, ABL2, PDGFRB, EPOR, JAK2, JAK1, etc.), some of which are targetable clinically. Thus, we and others have begun to screen for these lesions and to target these cases with appropriate kinase inhibitors (dasatinib, ruxolitinib) and chemotherapy in the clinical trial setting (14,15).
Hodgkin lymphoma (HL) is one of the first malignancies cured by a combination of chemotherapy and radiation therapy (RT). With appropriate staging and evaluation of treatment response using positron emission tomographic (PET) scanning, current treatment for HL achieves a 5-year EFS of 80% and 5-year OS of more than 90% (16). The standard of care for patients with relapsed or refractory HL is salvage chemotherapy followed by autologous stem cell transplantation (SCT), which can induce long-term remissions in approximately 50% of patients. The malignant Hodgkin’s Reed-Sternberg cells of classical HL are characterized by the expression of CD30, a member of the tumor necrosis factor T cells, and eosinophils; it represents an ideal target for monoclonal antibody therapy. Brentuximab vedotin (SGN-35) is an antibody-drug conjugate (ADC) comprising an anti-CD30 antibody conjugated by a protease-cleavable linker to the potent antimicrotubule agent monomethyl auristatin E (MMAE). Binding of the ADC to CD30 on the cell surface initiates internalization of the ADC-CD30 complex, which then disrupts the microtubule network, induces cell cycle arrest, and results in apoptotic death of the CD30-expressing tumor cell. Brentuximab vedotin is currently approved by the Food and Drug Administration (FDA) for HL after failure of autologous SCT or failure of two chemotherapy regimens with patients who are not candidates for autologous SCT (17). Use of brentuximab vedotin at an earlier treatment time is under investigation with multiple clinical trials. Phase I study of brentuximab vedotin with standard chemotherapy of doxorubicin, bleomycin, vincristine, and decarbazine (ABVD) and modified standard chemotherapy with doxorubicin, vincrisitine, and decarbazine (AVD) as first-line treatment for advanced-stage HL showed significantly increased pulmonary toxicity associated with the combination of brentuximab vedotin and bleomycin. This study confirmed the dose of brentuximab vedotin was safely escalated to 1.2 mg/kg and showed a remarkable response rate of 95% to 96% to the therapy (18).
Osteosarcoma is the most common malignant bone tumor of childhood. Osteosarcoma typically affects pubertal adolescents, with a peak incidence in childhood of 12 years. Risk stratification can be made using tumor stage, location, and response to therapy. Survival of patients with nonmetastatic disease at the time of diagnosis has improved dramatically over the past 30 years due to advances in chemotherapy and surgery, with 60% to 65% of patients surviving more than 10 years. However, patients who present with metastatic disease at the diagnosis or those who have recurrent disease have a poor prognosis, with OS rates of less than 20% (19).
Treatment of metastatic or relapsed osteosarcoma is challenging, although multiple novel approaches are being tested. One approach under investigation is targeting the bone-forming behavior of this tumor through treatment with radioactive samarium 153–EDTMP or more recently radium 223 dichloride, which concentrated in bone-producing osteosarcoma (20). As another approach, we have introduced immunemodulators into osteosarcoma therapy. Based on its potent immune-stimulatory properties, liposomal muramyl tripeptide phosphatidylethanolamine (L-MTP-PE) was added to standard osteosarcoma chemotherapy. Although the statistical benefit to survival was not clear in the largest US trial, the European Medicines Evaluation Agency approved this therapy for patients with pulmonary metastases of osteosarcoma; however, it is not currently available in the United States. More recently, we have used aerosolized interleukin (IL) 2 to enhance local immune responses against pulmonary metastases. Preclinical evidence demonstrates that aerosolized gemcitabine upregulates Fas expression in pulmonary metastases, leading to immune sensitivity (21).
Desmoplastic small round cell tumor (DSRCT) is an extremely rare undifferentiated mesechymal tumor that affects about 100 children, adolescents, and young adults annually in the United States. It is an aggressive tumor that may show a response to multimodal frontline therapy, although OS rates remain below 30%. The t(11;22) EWS-WT1 translocation is seen in the majority of cases, confirming the unique pathobiology of this disease.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


