The myeloproliferative neoplasms include both chronic myeloproliferative disorders and overlap myelodysplastic/myeloproliferative syndromes. This chapter reviews the pathology of the myeloproliferative diseases, including pertinent clinical, laboratory, histologic, cytogenetic, and molecular genetic findings necessary for the classification of these disorders. Both myeloproliferative neoplasms (MPNs) and overlap myelodysplastic/myeloproliferative neoplasms (MDS/MPNs) are clonal neoplasms with typical increased marrow cellularity, maturation of cell lineages, and organomegaly. The MPNs also share effective hematopoiesis and varying amounts of marrow fibrosis, but generally differ by which myeloid cell lineage dominates hematopoiesis. The MDS/MPNs overlap myeloproliferative and myelodysplastic disorders, with varying degrees of effective hematopoiesis and myelodysplasia. A common theme of aberrant activation of tyrosine kinase signaling pathways has emerged among the myeloproliferative diseases1,2 and 3,4,5. Not only has this helped further our understanding of these complex disorders, but also identification of aberrant kinase signaling cascades has led to targeted small molecule tyrosine kinase inhibitors (TKIs), such as imatinib, being used successfully in the treatment of certain diseases. Thus, the diagnosis and classification of the MPNs and overlap disorders requires correlation of morphology with clinical, hematologic, and molecular genetic findings6. An abbreviated overview of select myeloid neoplasms including the myeloproliferative diseases (Table 80.1) correlates each disease with its corresponding identified aberrant tyrosine kinase or related gene involved in the pathogenesis of the disorder and corresponding molecular genetic findings. In categorizing these disorders, separation of the MPNs from the overlap MDS/MPNs is recognized by the presence of myelodysplasia in the latter syndromes. Of the MPNs, there are the four common disorders, recognized as chronic myelogenous leukemia (CML), with its characteristic 9;22 translocation and BCR-ABL1 fusion protein, and three non-CML MPNs: polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). These common non-CML MPNs (PV, ET, PMF) share a high incidence of the acquired point mutation (V617F) in the JAK2 kinase7,8,9 and 10, a cytoplasmic tyrosine kinase important in hematopoietic proliferation. This mutation is associated with constitutive Jak-STAT activation in cell lines and MPN-like disorders in mouse models3,11, and occurs in stem cells in humans with polycythemia vera predisposing toward erythroid hyperplasia12,13. Other JAK2 mutations have also been described in patients with PV14. Given the importance of JAK2 V617F, the 2008 revision of the World Health Organization (WHO) classification system now includes this mutation in the diagnostic criteria for the common non-CML MPNs15,16,17.
The MPNs also include a number of uncommon or atypical disorders. These uncommon MPNs include chronic eosinophilic leukemia not otherwise specified (CEL), systemic mastocytosis (SM), and chronic neutrophilic leukemia (CNL). Several atypical MPNs and MDS-MPNs with eosinophilia are now also recognized as sharing tyrosine kinase abnormalities, such as FIP1L1-PDGFRA in disorders previously considered to be CEL and SM with eosinophilia, ETV6-PDGFRB in cases with features of chronic myelomonocytic leukemia (CMML) with eosinophilia or atypical chronic myeloid leukemia (aCML), and FGFR1 rearrangements seen in a number of hematologic neoplasms (formerly classified as the 8p11 myeloproliferative syndrome). These disorders are now classified as myeloid neoplasms with eosinophilia and abnormalities of PDGFRA, PDGFRB, or FGFR118 rather than as subtypes of other MPNs. The presence of these genetic abnormalities is not merely academic, as patients with PDGFRA or PDGFRB rearrangements have shown responsiveness to TKIs. For example, MPNs with eosinophilia and FIP1L1-PDGFRA can contain proliferations of CD25 positive mast cells that are sensitive to imatinib, whereas SM with KIT D816V mutations (and dense proliferations of CD25 positive mast cells) is resistant to the same drug19,20. The latter patients may respond to second-generation TKIs initially developed for imatinib-resistant CML patients or FLT3 inhibitors developed for patients with acute myeloid leukemia, as these drugs have activity against multiple tyrosine kinases21,22. The development of new tyrosine kinase pathway-targeted small molecule inhibitors is likely to accelerate further the molecular classification of myeloproliferative neoplasms.
The overlap MDS/MPNs, according to the WHO classification, have traditionally included chronic myelomonocytic leukemia (CMML), juvenile myelomonocytic leukemia (JMML), and atypical CML (aCML). A provisional entity, refractory anemia with ringed sideroblasts and thrombocytosis (RARS-T), also fits within this overlap group, sharing features of dysplasia and MPN. These overlap syndromes also share tyrosine kinase pathways, including JAK2 and MPL W515K/L mutations in RARS-T. As with all classification schemes, not every patient’s findings are amenable to these guidelines, and it is preferred to term these “unclassifiable” until further information clarifies the disorder.
MYELOPROLIFERATIVE NEOPLASMS
The traditional classification of the common MPNs is based on which cell line is most proliferative (i.e., granulocytic, erythroid, and megakaryocytic) and the amount of marrow fibrosis, combined with clinical, laboratory, and cytogenetic/molecular genetic features (Table 80.2). As a consequence of excess cell proliferation with effective maturation, there is resulting leukocytosis, erythrocytosis, and/or thrombocytosis. Subsequent hepatosplenomegaly follows due to sequestration of excess cells, extramedullary hematopoiesis, or infiltration by neoplastic cells. Although this initial dysregulated proliferation may manifest as an indolent disorder, these clonal stem cell disorders all have the potential for evolution. This manifests as either a stepwise increase in fibrosis to an end-stage myelofibrosis or as an increase in blasts, transforming through an accelerated phase (10% to 19% blasts) to overt acute leukemia (20% or more blasts). The MPNs differ in the incidence of this evolution and transformation.
TABLE 80.1 MYELOID NEOPLASMS WITH ASSOCIATED TYROSINE KINASES AND GENETIC ABNORMALITIES
Myeloproliferative Neoplasms
CML
ABL1
t(9;22)(q34;q11); BCR/ABL1
Non-CML MPN
PV
JAK2
JAK2 V617F, JAK2 exon 12
ET
JAK2, MPL
JAK2 V617F, MPL W151L/K
PMF
JAK2, MPL
JAK2 V617F, MPL W151L/K
Uncommon non-CML MPNs and other myeloid neoplasms
Myeloid neoplasms with eosinophilia
PDGFRA
del(4q12); FIP1L1-PDGFRA
PDGFRB
t(5;12); ETV6-PDGFRB
FGFR1
8p11 abnormalities
SM
KIT
KIT D816V
Myelodysplastic/Myeloproliferative Syndromes
JMML
RAS, NF1, PTPN11
RARS-T
JAK2
JAK2 V617F
CML, chronic myelogenous leukemia; ET, essential thrombocythemia; JMML, juvenile myelomonocytic leukemia; MPN, myeloproliferative neoplasm; PMF, primary myelofibrosis; PV, polycythemia vera; RARS-T, refractory anemia with ring sideroblasts and thrombocytosis; SM, systemic mastocytosis.
Chronic Myelogenous Leukemia
Chronic myelogenous leukemia, BCR-ABL1 positive (CML)23,24,25, is the most common of the MPNs and can occur at any age, although it is uncommon in children. The average age at diagnosis is 50 to 60 years old with a slightly increased male-to-female ratio. CML is defined by the presence of the Philadelphia chromosome or molecular genetic evidence of the BCR-ABL1 fusion. In contrast to the t(9;22) of acute leukemias, which usually demonstrate the p190 BCR-ABL1 fusion protein, CML almost always demonstrates the p210 fusion protein of this translocation. The rare p230 fusion protein is typical of the rare neutrophilic form of CML, which is discussed later. CML is primarily a proliferation of granulocytic cells, although multiple cell lines demonstrate the Philadelphia chromosome. This expansion of myeloid cells typically involves the blood, bone marrow, spleen, and liver. Extramedullary involvement may be seen during the blast phase of the disease. The typical clinical course of CML is an indolent chronic phase followed by either blast crisis or progression to an accelerated phase with subsequent blast crisis.
TABLE 80.2 SELECT MOLECULAR GENETIC, MORPHOLOGIC, LABORATORY, AND CLINICAL FINDINGS IN THE COMMON MYELOPROLIFERATIVE NEOPLASMS
Disease
Molecular Findings
Blood Smear
Bone Marrow
Fibrosis
Splenomegaly
Other
CML
BCR-ABL1
Leukocytosis with immature granulocytes, basophilia
Marked myeloid hyperplasia with prominence of neutrophils and myelocytes
Variable
++
PV
JAK2
V617F
JAK2 exon 12
Normo- or hypochromic anemia, may have thrombocytosis and mild basophilia
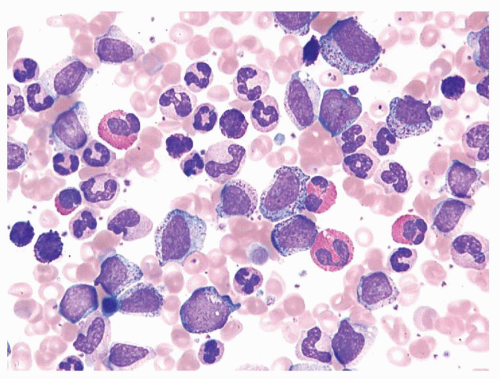
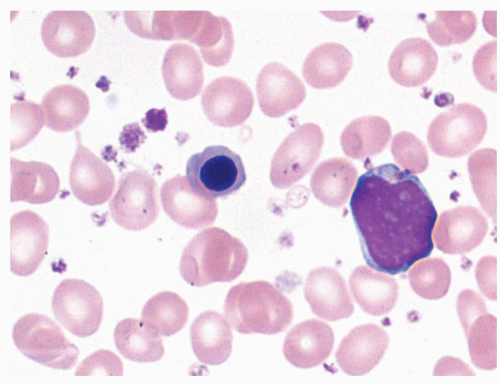
FIGURE 80.1. Chronic myelogenous leukemia. The peripheral blood demonstrates neutrophilia with prominent left shift, eosinophilia, and basophilia. Typically numerous myelocytes and segmented neutrophils predominate. Wright-Giemsa, 500×.
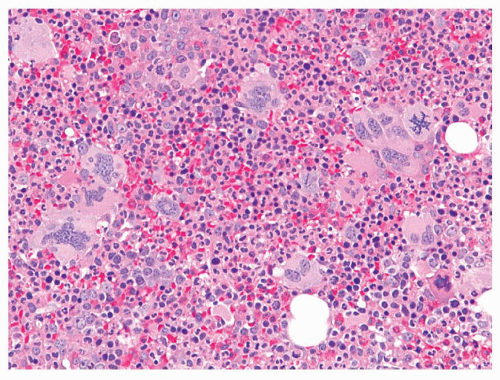
The peripheral blood findings are those of a leukocytosis with granulocytes at all stages of maturation (Fig. 80.1). Segmented neutrophils may show abnormal nuclear segmentation. Myelocytes and metamyelocytes are present in high numbers in the peripheral blood with concomitant basophilia and often eosinophilia. Abnormal basophils with “washed-out” granules may be identified. Platelets are usually elevated. An absolute monocytosis may also be present. The bone marrow is markedly hypercellular with cellularity approaching 100% in most untreated cases (Fig. 80.2). The myeloid-to-erythroid ratio of the bone marrow is increased, often >10:1. An atypical megakaryocytic hyperplasia is present, with clustering of megakaryocytes, which are small and contain hypolobated nuclei. Some degree of reticulin fibrosis is usually demonstrated in these cases, and marked increases in reticulin and collagen marrow fibrosis are associated with accelerated phase and decreased survival26. Pseudo-Gaucher histiocytes are common in CML, reportedly present in 37% to 70% of cases27. These histiocytes have abundant fibrillar birefringent cytoplasm, and their presence has been associated with an improved survival26. Leukemoid reactions, in patients with infectious or other reactive conditions, may demonstrate similar features to CML. These reactive proliferations, however, do not usually show the basophilia, degree of marrow cellularity, or megakaryocyte clustering characteristic of CML. However, cytogenetic or molecular genetic studies are indicated if the differential diagnosis includes CML.
FIGURE 80.2. Chronic myelogenous leukemia (CML). The bone marrow biopsy is hypercellular with myeloid and megakaryocytic hyperplasia. Note the clusters of small hypolobated megakaryocytes typical of CML. Hematoxylin and eosin, 500×.
Chronic myelogenous leukemia usually presents in chronic phase, which is essentially defined by the lack of features of accelerated phase or blast crisis. Marrow blasts are usually <5%, but may range up to 9%. The natural course of the disease is progression from chronic phase to a more aggressive phase of the disease within 3 to 5 years. Any one of a variety of parameters defines the accelerated phase of CML, but the criteria are inconsistent in the literature28,29. WHO criteria (Table 80.3) require the presence of either 10% to 19% blasts in the peripheral blood or bone marrow, the acquisition of additional chromosomal abnormalities, elevations of basophils to 20% or more of blood cells, persistent thrombocytopenia (<100 × 109/L), or either of the following that do not respond to conventional therapies: platelet elevations of >1,000 × 109/L or increasing splenomegaly associated with an increasing white blood cell (WBC) count30. In addition, megakaryocytic proliferation in sheets and clusters associated with marked fibrosis and/or marked granulocytic dysplasia has also been described as suggestive of an accelerated phase31. Blast phase, or blast crisis, of CML is defined by the presence of 20% or more blasts in the peripheral blood or bone marrow (Fig. 80.3)30. Large clusters of blasts on the bone marrow biopsy are also sufficient for a diagnosis of blast phase31. The development of extramedullary myeloid tumors (myeloid or granulocytic sarcomas, chloromas) is also sufficient for blast phase using the WHO criteria. Cytogenetic clonal evolution, which may be present at the time of transformation to either the accelerated phase or blast phase, typically includes an extra Philadelphia chromosome, trisomy 8, or i(17q)25,32,33.
TABLE 80.3 WHO CRITERIA FOR ACCELERATED PHASE OF CHRONIC MYELOGENOUS LEUKEMIAa
Blasts 10-19% in blood or marrow
Peripheral blood basophilia ≥ 20%
Cytogenetic clonal evolution
Persistent thrombocytopenia (<100 × 109/L)
Persistent thrombocytosis (>1,000 × 109/L) unresponsive to therapy
Increasing splenomegaly and/or increasing WBC count unresponsive to therapy
WBC, white blood cell; WHO, World Health Organization.
aThe diagnosis requires one or more of the listed criteria.
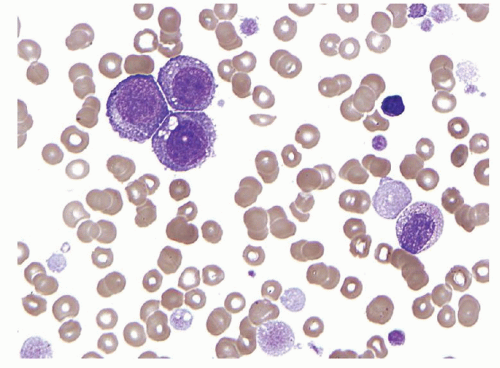
Detection of the t(9;22)(q34;q11) by karyotype analysis, fluorescent in situ hybridization (FISH), or reverse transcriptasepolymerase chain reaction (RT-PCR) is essential for the diagnosis of CML. Detection of this abnormality confirms the clonal or neoplastic nature of the proliferation and excludes reactive conditions and other MPNs that may mimic CML. Immunophenotyping studies add little in the chronic phase of CML, but are helpful in defining the blast cell population of accelerated and blast phases of this disease34,35. The majority of blast crisis cases will express myeloid-associated antigens without lymphoid markers and are easily classified as myeloid blast crisis by immunophenotyping studies; however, some myeloid blast crisis cases may be myeloperoxidase negative by cytochemistry. Although most blast transformations of CML are proliferations of myeloblasts, approximately one third of cases are lymphoid blast crises (Fig. 80.4). The vast majority of lymphoid blast crisis cases are of precursor B-cell lineage, but rare T-cell blast crisis cases occur. Lymphoid blast crisis is reported to have a better prognosis than myeloid blast crisis, and lymphoid blast crisis has traditionally been defined as a blast cell proliferation that is terminal deoxynucleotidyl transferase (TdT) positive. More detailed immunophenotyping of these cases usually demonstrates expression of other precursor B-cell markers, such as CD19 and CD10, but expression of myeloid-associated antigens, such as CD13 and CD33, is also common. Cases with a lymphoid immunophenotype lacking myeloperoxidase, irrespective of the expression of other myeloid antigens, have an improved survival35 and are probably best classified as lymphoid blast crises. A small proportion of CML blast crises are mixed phenotype, which must be distinguished from de novo mixed phenotype acute leukemia (MPAL), a subset of which also has the BCR-ABL1 gene rearrangement.
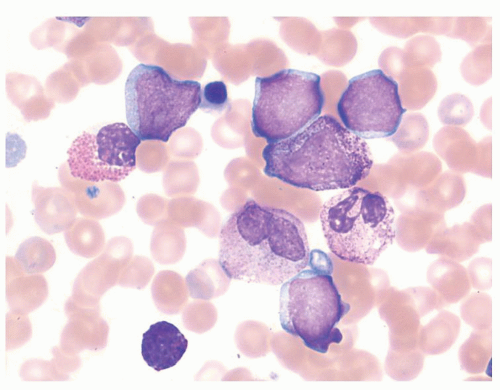
FIGURE 80.3. Myeloid blast crisis of chronic myelogenous leukemia (CML). Peripheral blood smear showing numerous agranular blasts on a background of neutrophils, immature granulocytes, eosinophils, and basophils. When blasts are the dominant cell present, the background changes of CML may be subtle or obscured. Wright-Giemsa, 1,000×.
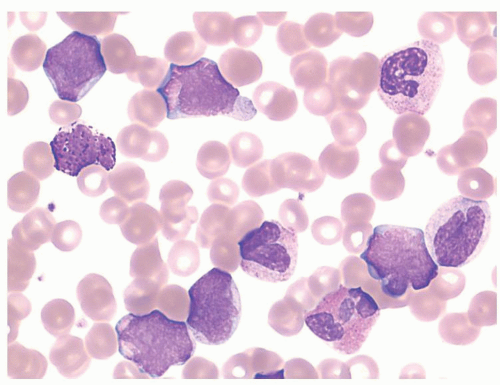
FIGURE 80.4. Lymphoid blast crisis of chronic myelogenous leukemia (CML). Peripheral blood smear shows intermediate-sized blasts with high nuclear-to-cytoplasmic ratios, a very small amount of basophilic cytoplasm, irregular and clefted nuclear contours, and immature chromatin. The background shows features of CML. Flow cytometry demonstrated a B-lymphoblastic immunophenotype. Wright-Giemsa, 1,000×.
Typically, patients with all phases of CML are treated with a TKI, imatinib, which directly blocks the effects of the BCR-ABL1 fusion protein36. Imatinib results in a clinical, morphologic, and at least partial cytogenetic remission in most patients, with reduction in marrow cellularity, normalization of the myeloid-to-erythroid ratio, and normalization of megakaryocyte number and morphology37,38,39,40,41. The peripheral blood is the first to respond to imatinib therapy with a normalization of the white blood cell and platelet counts, a decrease in basophils, and normal-appearing platelets occurring after about 2 months of therapy. During therapy, the hemoglobin level tends to decrease slightly, and a subset of patients may develop neutropenia or thrombocytopenia. Bone marrow hypercellularity gradually decreases and by 8 to 11 months the marrow is normocellular or hypocellular with a normal or decreased myeloid-to-erythroid ratio in most patients. Even in chronic phase, bone marrow blasts and megakaryocytes decrease, and the number of hypolobated megakaryocytes and the presence of megakaryocyte clustering become less common as the marrow cellularity decreases. This therapy has also been reported to gradually eliminate the marrow fibrosis that is prominent in some cases of CML37,38,42. Patients with accelerated or blast phases of CML show similar changes with rapid decreases in peripheral blood and bone marrow blast cell counts37. Two thirds of patients treated in chronic phase tolerate imatinib therapy well and experience an excellent and sustained remission40,41. More recently, the development of second-generation TKIs (dasatinib, nilotinib) have proven effective in many patients who fail or are not tolerant of imatinib, and these may become effective alternatives for front line therapy43,44 and 45. Whereas TKIs are now well established as front line therapy, stem cell transplantation (SCT) remains an effective curative option although the risks are not insignificant. SCT is therefore now employed as salvage therapy46.
Neutrophilic-chronic myelogenous leukemia appears to be a less aggressive variant of CML in which the t(9;22) codes for a 230-kD BCR-ABL1 fusion protein47. This disease is associated with a proliferation of more mature granulocytes, usually at the segmented neutrophil stage of development. These patients have less severe clinical symptoms and are slower to progress to a blastic stage. This CML variant has been shown in one study to have low levels of p230 BCR-ABL1 messenger RNA and undetectable protein product, which may explain the milder phenotype in these patients48.
Philadelphia chromosome-negative CML is not recognized in the 2008 WHO classification and such a diagnosis should be made with caution. Cryptic BCR-ABL1 translocations may occur that cannot be identified by routine karyotype analysis. When this is suspected, molecular genetic studies, such as FISH or RT-PCR analysis, are indicated. When these studies are negative, other diagnostic considerations must be entertained. Review of cytogenetic and molecular genetic Philadelphia chromosome-negative CML cases has resulted in most being reclassified as chronic myelomonocytic leukemia or atypical CML (described below)49.
Polycythemia Vera
Polycythemia vera is a clonal proliferation, usually occurring in elderly patients with a male predominance, that presents as an expansion of the red blood cell mass50,51,52. This expansion is secondary to increased red cell production from dysregulated erythropoiesis. An acquired JAK2 V617F mutation in exon 14 is detected in 95% to 97% of patients with PV53,54, with exon 12 mutations accounting for most JAK2 V617F negative cases14,55. The WHO 2008 criteria for PV now include JAK2 or functionally similar mutations as a major criterion for diagnosis (Table 80.4)15,17. These mutations produce a PV-like disease in mice14,55 and involve a stem cell in humans predisposing toward erythroid differentiation8. Typically, the spleen is enlarged and erythropoietin levels are decreased in this disease56. The presence of a JAK2 mutation allows for the exclusion of a reactive erythrocytosis, but is not diagnostic for PV, as JAK2 mutations occur in approximately one half of patients with ET and PMF, as well as involving atypical MPNs and overlap MDS/MPNs at lower levels53,57,58.
TABLE 80.4 WHO CRITERIA FOR THE DIAGNOSIS OF POLYCYTHEMIA VERAa
Major Criteria
Hemoglobin >18.5 g/dl in men, >16.5 g/dl in women or evidence of increased red cell volumeb
Presence of JAK2 mutation
Minor Criteria
Hypercellular bone marrow biopsy with panmyelosis with prominent erythroid, granulocytic, and megakaryocytic hyperplasia
Low serum erythropoietin level
Endogenous erythroid colony formation in vitro
WHO, World Health Organization.
aDiagnosis requires the presence of both major criteria and one minor criterion or the presence of the first major criterion and two minor criteria. The JAK2 mutation refers to the JAK2V617F or other functionally similar JAK2 mutation.
b Hemoglobin or hematocrit >99% of method-specific range for age, sex, and altitude of residence or hemoglobin >17 g/dl in men, >15 g/dl in women if associated with a sustained increase of at least 2 g/dl from an individual’s baseline value that cannot be attributed to correction of iron deficiency.
From Swerdlow SH, Campo E, Harris NL, et al. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC, Lyon, 2008.
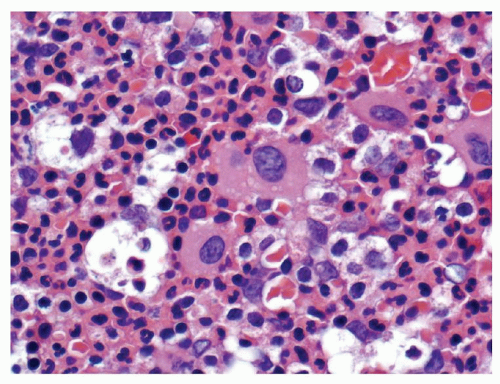
FIGURE 80.5. Polycythemia vera. The bone marrow biopsy from a patient with polycythemia vera is hypercellular, with panmyelosis, an increased number of erythroid precursors, and large atypical megakaryocytes in loose clusters. Hematoxylin and eosin, 200×.
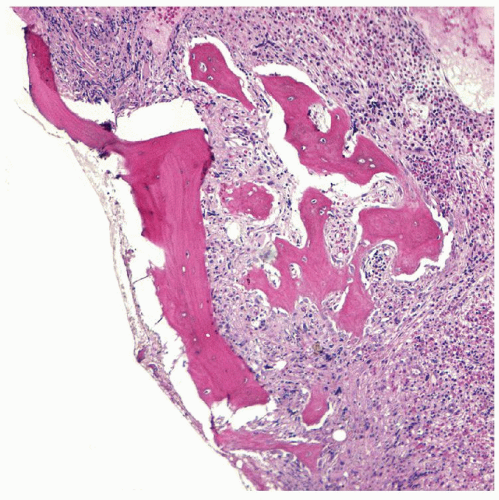
Two morphologic phases of PV are well described59. The proliferative or erythrocytotic phase typically shows elevations of red blood cells, white blood cells, and platelets. Slight elevations in the peripheral blood basophil count may be present, but are not as elevated as is usually seen in CML. A neutrophilia with left-shifted granulocytes is commonly seen. Platelet counts can exceed 600 × 109/L, which may cause confusion with essential thrombocythemia. It is not uncommon for patients to have associated iron deficiency with microcytic red blood cells; absent stainable iron on marrow examination is typical59. The bone marrow is usually moderately hypercellular with trilineage proliferation (Fig. 80.5). In contrast to the other MPNs, however, the erythroid series is relatively increased. Loose clusters of pleomorphic megakaryocytes are prominent, with very small and giant megakaryocytes adjacent to each other. Marrow fibrosis may be minimal in this stage of the disease60,61. Reactive lymphoid aggregates are also common62. The spent phase or post-polycythemic phase of PV is associated with marked marrow fibrosis and shows peripheral blood and bone marrow changes that are similar or identical to those seen in primary myelofibrosis with leukoerythroblastic peripheral blood changes, splenomegaly, and marrow fibrosis (Fig. 80.6). Differentiation between these two diseases may not be possible without a history of the earlier phase of PV (Fig. 80.7). Approximately 10% of PV patients will transform to acute myeloid leukemia within 15 years and up to half will develop acute leukemia over 20 years (Fig. 80.8)63. Rare cases of myelodysplastic transformation are also reported in the literature, which appear to be treatment related64.
FIGURE 80.6. Post-polycythemic phase of polycythemia vera. Bone marrow biopsy showing increased fibrosis and osteosclerosis. Hematoxylin and eosin, 400×.
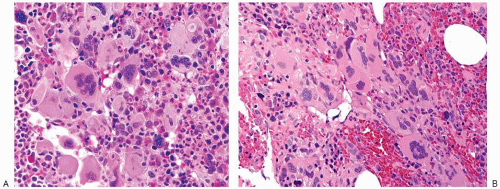
FIGURE 80.7. The post-polycythemic phase of polycythemia vera shows bone marrow changes (A) indistinguishable from those of the fibrotic stage of primary myelofibrosis (B). Both bone marrow biopsies are hypercellular with atypical megakaryocyte clustering, including hyperchromatic enlarged megakaryocytes amidst a fibrotic background. Hematoxylin and eosin, 400×.
Evaluation of red cell mass (RCM), which previously was recommended for the diagnosis of PV, has now largely been supplanted by JAK2 testing, bone marrow histology, and serum erythropoietin levels65. However, given the overlap in features between PV and other MPNs, some cases may only be resolved by RCM studies, although these studies have their own challenges65. An absolute erythrocytosis is the feature that separates PV from other MPNs, and RCM may be decreased in PV patients with concurrent iron deficiency; thus, studies may have to be repeated after iron therapy. Reactive or secondary polycythemias must also be excluded and these may be related to smoking, lung and renal disease, erythropoietin-producing tumors, congenital conditions causing erythropoietin overproduction, and exogenous administration of erythropoietin. Although many of these secondary polycythemias will lack JAK2 mutations, not all idiopathic erythrocytoses that meet neither criterion for PV or secondary polycythemia are well understood66. Some of these idiopathic erythrocytoses represent recently described exon 12 or 14 JAK2 mutations, whereas others are still being investigated14,55. Studies have emphasized the value of bone marrow histology in distinguishing between early-stage PV and reactive polycythemias. The atypical megakaryocytic hyperplasia of PV is not seen in secondary polycythemias. These reactive conditions tend to show only a borderline increase in cellularity, with an altered interstitial compartment containing increased deposition of cellular debris within histiocytic cells, hemosiderin-laden macrophages, and perivascular plasmacytosis60,61.
FIGURE 80.8. Transformation of polycythemia vera. Peripheral blood smear shows numerous giant platelets, including hypogranular forms, a myelocyte, a lymphocyte, and a cluster of three blasts. Wright-Giemsa, 600×.
Other karyotypic abnormalities may be detected in up to half of cases of PV, with chromosome 20q deletions being the most common67,68. These abnormalities, however, are not specific for PV69. Various point mutations occur in association with polycythemias, particularly in the congenital or familial forms70. Clonogenic stem cell assays are a minor diagnostic criterion used in the diagnosis of PV. Progenitor cells from patients with PV are hypersensitive to several growth factors, and in vitro detection of endogenous erythroid colonies occurs in PV. Although the predictive value of clonogenic stem cell assays in certain defined settings may be high (97%)71, their availability is limited and these assays are difficult to standardize. Thus, these assays are now infrequently used in the routine diagnosis of PV72.
Primary Myelofibrosis
Primary myelofibrosis, also known as chronic idiopathic myelofibrosis, myelofibrosis with myeloid metaplasia, and agnogenic myeloid metaplasia, occurs in elderly patients and usually presents with a leukoerythroblastic peripheral blood smear, massive splenomegaly, and marrow fibrosis73,74. The peripheral blood changes include the presence of large teardrop-shaped red blood cells (dacrocytes), a granulocyte left-shift that often includes rare myeloblasts, and thrombocytosis with giant platelets that are larger than a red blood cell (Fig. 80.9). Basophilia may be present and bare megakaryocyte nuclei are often seen in the blood and bone marrow. Although spleen enlargement occurs with many myeloproliferative neoplasms, the splenomegaly of PMF is striking, and may cause severe discomfort and wasting syndromes. The bone marrow may be hypercellular, particularly early in the disease when marrow fibrosis is less prominent, in the prefibrotic phase of PMF. The myeloid-to-erythroid ratio is slightly increased and megakaryocytes are increased. Atypical megakaryocyte clustering is prominent, with clusters of medium-sized to giant megakaryocytes often adjacent to sinuses and bony trabeculae. The megakaryocytes are atypical with hyperchromatic megakaryocytic nuclei and coarse lobulations75,76,77 and 78. The differential diagnosis between the prefibrotic stage of PMF and ET can be quite difficult, but abnormal megakaryopoiesis is most helpful in establishing the diagnosis of PMF, although other features including bone marrow cellularity (increased markedly in the cellular phase of PMF) and left-shifted myeloid hyperplasia (usual in PMF) are also useful79,80,81 and 82,83,84. Most patients are diagnosed in the fibrotic stage and show marked marrow fibrosis (Fig. 80.10), which may include collagen fibrosis. Interestingly, this accompanying fibrosis is reactive, whereas clonal studies have shown that the trilineage hematopoietic proliferation is monoclonal85. Clusters of atypical megakaryocytes remain prominent in association with the fibrosis, and megakaryocyte clusters in sinusoids may be evident. The sinuses are often dilated with intrasinusoidal hematopoiesis. Sclerosis of bone trabeculae also occurs in many patients with broad irregular trabeculae, which can occupy much of the marrow biopsy (Fig. 80.10D). Lymphoid aggregates, of predominantly T-cells, occur commonly in association with PMF.
FIGURE 80.9. Primary myelofibrosis. The peripheral blood changes of myelofibrosis show the characteristic findings of leukoerythroblastosis with rare blasts, nucleated red blood cells, large platelets, and teardrop-shaped red cells (dacrocytes). Wright-Giemsa, 1,000×.
Ancillary studies are required to exclude the Philadelphia chromosome of CML, and approximately one half of PMF patients will have an acquired JAK2 mutation53. An MPL mutation, MPL W515L, has also been described in a subset of patients with PMF; some of these patients lacked the JAK2 mutation whereas others occurred concurrently86,87. Studies report that 35% to 61% of patients will demonstrate a cytogenetic abnormality, with deletions of chromosomal arms 20q and 13q most common, as well as der(6)t(1;6) (q21-23;p21.3)74,88,89,90 and 91. Other ancillary studies are of limited utility with the exception of immunophenotyping of blasts in cases that undergo blastic transformation.
The Italian criteria for myelofibrosis focused on the fibrotic phase of the disease, requiring diffuse fibrosis of the marrow, among other features92. The 2008 WHO criteria for PMF require three major and two of four minor criteria15. The first major criterion addresses bone marrow histology and allows diagnosis in either the fibrotic or prefibrotic phase by focusing on characteristic megakaryocytic proliferation and atypia accompanied either by significant fibrosis or a hypercellular marrow with granulocytic proliferation. The two additional required major criteria are the demonstration of the JAK2 V617F mutation or a similar clonal marker (such as MPL W515K/L), and the exclusion of other MPNs, MDS, and BCR-ABL1-positive CML (Table 80.5). Two of four minor criteria must also be met, including (a) leukoerythroblastosis, (b) elevated serum lactate dehydrogenase levels, (c) anemia, or (d) splenomegaly. As described above, the megakaryocytic atypia is quite marked with loose to tight clusters of megakaryocytes, including a wide variation in size with hyperchromatic, irregularly folded or bulbous nuclei, as well as abnormal nuclear-to-cytoplasmic ratios78,93.
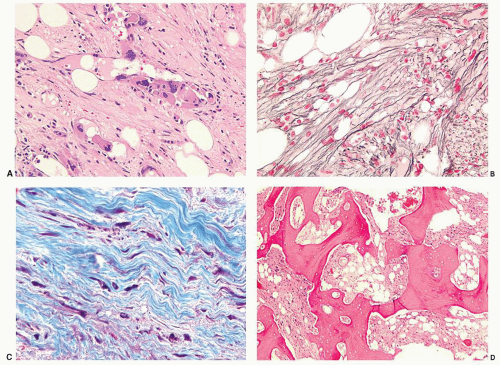
FIGURE 80.10. Bone marrow changes of fibrotic phase primary myelofibrosis. Fibrosis and intrasinusoidal clusters of atypical megakaryocytes are evident in (A). Hematoxylin and eosin, 200×. Diffusely increased coarse reticulin fibrosis is shown in (B). Reticulin stain, 500×. Coarse collagen fibrils are demonstrated in (C). Masson trichrome, 500×. Late-stage osteosclerotic changes are noted in (D), Hematoxylin and eosin, 50×.
Median survival for patients with PMF varies from 17.5 to 1.8 years for low- to high-risk subgroups94, using the Dynamic International Prognostic Scoring System (DIPSS)-plus with the following eight risk factors: age > 65 years, constitutional symptoms, RBC transfusion need, hemoglobin <10 g/dL, leukocyte count >25 × 109/L, circulating blasts ≥1%, platelet count <100 × 109/L, and an unfavorable karyotype95. The most frequent causes of death are complications of marrow failure (22%), including anemia, infection, and hemorrhage; transformation to AML (15%); and complications related to massive splenomegaly (11%)88. At present, there are few therapeutic options for PMF96. Allogeneic SCT provides a chance for cure, but the morbidity and mortality in PMF can be significant97.
Essential Thrombocythemia
Essential thrombocythemia is a bone marrow proliferation characterized primarily by an elevation in peripheral blood platelets, usually over 1,000 × 109/L. The 2008 WHO classification specifies four criteria for diagnosis: a sustained platelet count ≥450 × 109/L; bone marrow biopsy showing megakaryocyte proliferation with enlarged mature megakaryocytes and with no significant increase or left shift of granulopoiesis or erythropoiesis; the exclusion of PV, PMF, CML, MDS, or another myeloid neoplasm; and either the presence of JAK2 V617F or another clonal marker or the exclusion of reactive thrombocytosis (Table 80.6)98. Causes of reactive thrombocytosis include iron deficiency, splenectomy, surgery, infection, inflammation, connective tissue disease, metastatic cancer, and lymphoproliferative diseases. JAK2 mutations are found in ˜50% of patients with ET, and the MPL W515K/L in 3%, with both mutations found in a few patients58,99.
ET probably represents two different diseases, one clonal and one reactive, even using the listed criteria100. Patients with the nonclonal form of the disease may include those with abnormalities of the thrombopoietin gene, and appear to be at a decreased risk of developing thrombosis100,101. Both types show similar peripheral blood and bone marrow features.
Only gold members can continue reading. Log In or Register to continue