Pathogenesis of Diabetic Nephropathy
Gabriella Gruden
GianCarlo Viberti
The abnormal glycemic milieu of diabetes is closely related to the development of microvascular complications. However, the evidence of a straightforward causal relationship between hyperglycemia and renal disease is less compelling in humans than in animal models. Only about 30% of patients develop clinically overt nephropathy. The majority of subjects with diabetes escape renal failure, and although some histologic damage occurs in their kidneys, their renal function remains essentially normal until they die. It therefore appears that in humans hyperglycemia is necessary, but not sufficient, to cause the renal damage that leads to kidney failure. The risk of developing kidney disease is not linearly correlated to the duration of diabetes; indeed, the incidence peaks after 17 years of type 1 diabetes and then sharply declines, a phenomenon likely due to exhaustion of the subgroup of susceptible subjects (1).
The central question is thus why some diabetic patients are susceptible to and others are protected from renal disease. A diabetic sibling of a person with type 1 diabetes and nephropathy has approximately a 72% cumulative risk of developing renal disease, whereas a diabetic sibling of a person with type 1 diabetes but without nephropathy has only a 25% risk (2). This indicates that inherited factors play an important role in determining susceptibility to diabetic nephropathy but does not provide insight into the nature of these factors.
Family studies have shown that a familial predisposition to raised arterial pressure is an important determinant of susceptibility to renal disease in persons with diabetes. Blood pressure levels are significantly higher in the parents of diabetic subjects with proteinuria than in the parents of diabetic subjects without proteinuria (3). Among Pima Indians with type 2 diabetes, the prevalence of proteinuria is higher in those with hypertensive parents than in those with normotensive parents (4). It has been calculated that the risk of nephropathy among patients with type 1 diabetes is three times higher in those who have at least one parent with a history of hypertension (5). Furthermore, prospective studies have demonstrated that mean blood pressure levels are significantly higher in those patients who progress to microalbuminuria than in those who do not, indicating that high blood pressure plays an important role in the pathogenesis of diabetic kidney disease (6).
Diabetic nephropathy and hypertension are multifactorial disorders that result from interaction between both environmental and genetic factors, making the identification of what confers susceptibility to diabetic kidney disease at the genetic level both difficult and complex. Polymorphisms of genes potentially involved in the genetic predisposition to hypertension, vascular reactivity, and insulin resistance, such as those of the renin-angiotensin system, nitric oxide, aldose reductase, GLUT I, and lipoproteins, have all been investigated in relation to diabetic nephropathy, but studies of candidate genes have, by and large, been inconclusive or, at best, have shown weak associations (7). However, a strong association between a polymorphism in the 5′ end of the aldose reductase gene and the development of diabetic nephropathy in type 1 diabetes has been reported (8) and recently confirmed (9).
Studies of intermediate phenotypic markers of hypertension have provided some insight into the predisposition to diabetic renal disease. The red blood cell sodium-lithium countertransport system is a cell-membrane cation transport system whose activity is both genetically determined and increased in essential hypertension. Sodium-lithium countertransport activity is abnormally high in diabetic patients with proteinuria or microalbuminuria (10,11). High countertransport activity may thus constitute a suitable cell marker for identifying patients at risk of kidney disease. However, sodium-lithium countertransport is not operating in vivo; therefore, the pathophysiologic relevance of its abnormalities remains uncertain. The sodium-hydrogen antiport, a ubiquitous transmembrane protein that
catalyzes the electroneutral exchange of extracellular sodium for intracellular hydrogen, operates in vivo and regulates important cellular events, such as intracellular pH, cell volume, and cell proliferation. Furthermore, at the kidney level, the sodium-hydrogen antiport plays an important role in sodium reabsorption and may thus be involved in the pathogenesis of hypertension. Sodium-hydrogen antiport activity is increased in a number of cell types from patients with type 1 diabetes with microalbuminuria and proteinuria, as well as in subjects with hypertension (12,13). The activity of the sodium-hydrogen antiport is increased by high glucose concentrations in cells from patients with diabetic nephropathy but not in cells from diabetic patients without nephropathy or in cells from healthy controls (13). Furthermore, in prospective studies, the sodium-hydrogen activity of red blood cells predicts the development of proteinuria (14). Cultured skin fibroblasts from siblings of patients with type 1 diabetes show a very close concordance for the sodium-hydrogen antiport activity, indicating that antiport activity is by and large genetically determined (15). However, in both hypertension and diabetic nephropathy, the increased sodium-hydrogen antiport activity is not due to increased expression of the gene encoding for this protein. Alterations in the regulatory pathways of the sodium-hydrogen exchange are more likely to be implicated in its overactivity. Although the pathogenic role of the sodium-hydrogen antiport in diabetic nephropathy remains to be proven, together these studies of intermediate phenotype show that the increased susceptibility to diabetic nephropathy most likely resides in a genetically determined host-cell response to the diabetes environment (16).
catalyzes the electroneutral exchange of extracellular sodium for intracellular hydrogen, operates in vivo and regulates important cellular events, such as intracellular pH, cell volume, and cell proliferation. Furthermore, at the kidney level, the sodium-hydrogen antiport plays an important role in sodium reabsorption and may thus be involved in the pathogenesis of hypertension. Sodium-hydrogen antiport activity is increased in a number of cell types from patients with type 1 diabetes with microalbuminuria and proteinuria, as well as in subjects with hypertension (12,13). The activity of the sodium-hydrogen antiport is increased by high glucose concentrations in cells from patients with diabetic nephropathy but not in cells from diabetic patients without nephropathy or in cells from healthy controls (13). Furthermore, in prospective studies, the sodium-hydrogen activity of red blood cells predicts the development of proteinuria (14). Cultured skin fibroblasts from siblings of patients with type 1 diabetes show a very close concordance for the sodium-hydrogen antiport activity, indicating that antiport activity is by and large genetically determined (15). However, in both hypertension and diabetic nephropathy, the increased sodium-hydrogen antiport activity is not due to increased expression of the gene encoding for this protein. Alterations in the regulatory pathways of the sodium-hydrogen exchange are more likely to be implicated in its overactivity. Although the pathogenic role of the sodium-hydrogen antiport in diabetic nephropathy remains to be proven, together these studies of intermediate phenotype show that the increased susceptibility to diabetic nephropathy most likely resides in a genetically determined host-cell response to the diabetes environment (16).
In patients with type 2 diabetes, the evidence of a genetically determined susceptibility to the development of diabetic nephropathy is less compelling than in patients with type 1 diabetes. However, there is evidence of a familial clustering of diabetic nephropathy in type 2 diabetes (17,18,19), and affected sib-pair linkage analyses have identified loci associated with diabetic nephropathy in type 2 diabetes (20,21,22). Moreover, in patients with type 2 diabetes, nephropathy is associated with cellular markers for hypertension (23), and in families with type 2 diabetes, albumin excretion rate is a heritable trait, with a heritability similar to that for blood pressure (24). Finally, in Pima Indians, blood pressure levels before the onset of diabetes predict the future risk of developing the complication (4).
THE INSULTS: HYPERGLYCEMIA, HYPERTENSION, AND PROTEINURIA
Diabetic nephropathy is characterized histologically by thickening of the glomerular basement membrane, increased fractional mesangial volume, and podocyte abnormalities. Expansion of the glomerular mesangium, which occurs at the expense of the glomerular capillary lumen and filtration surface area, correlates most closely with the decline in renal function and the development of proteinuria (25,26). Ex vivo studies have shown that the mesangial expansion is due to both increased production and reduced degradation of extracellular matrix proteins such as type IV and I collagen, laminin, and fibronectin (27).
Although the glomerulus, particularly the mesangium, has been the major focus of interest, tubulointerstitial injury is also a feature of diabetic nephropathy and a predictor of renal dysfunction. Pathologic changes that have been described in association with diabetic nephropathy include thickening of the tubular basement membrane, tubular atrophy, interstitial fibrosis, and arteriosclerosis. Interstitial expansion correlates closely with renal dysfunction, albuminuria, and mesangial expansion. Moreover, the impact of interstitial expansion on renal dysfunction is additive to that of mesangial expansion, suggesting an independent effect (28).
If diabetes-induced abnormalities were sufficient to cause renal disease, all diabetic patients, given time, would develop overt renal disease, and the study of hyperglycemia-induced abnormalities would be sufficient to explain the pathogenesis of these functional or structural abnormalities. But this is not the case. To gain insights into the pathogenic mechanisms of diabetic kidney disease, it is therefore necessary to take into account other insults that either influence the host response to diabetes-induced environmental disturbances, thus conferring susceptibility, or accelerate the progression of kidney damage.
Hyperglycemia
There is no doubt that poor glycemic control is associated with diabetic nephropathy. Levels of hemoglobin A1c (HbA1c) are higher in patients with microalbuminuria and macroalbuminuria than in those with normoalbuminuria (29), and in two longitudinal studies, the glycemic control predicted the future development of microalbuminuria in normotensive type 1 diabetic patients with normoalbuminuria (6,30).
Intervention studies have demonstrated the renoprotective effect of optimal glycemic control in both human and experimental diabetes. In animals, structural glomerular changes and albuminuria can be prevented by the maintenance of normoglycemia by either islet cell transplantation or intensive insulin therapy or can be reversed by transplantation of an affected kidney into a nondiabetic animal (31,32,33). In humans, the Diabetes Control and Complications Trial (DCCT), a prospective multicenter randomized clinical trial comparing the effect of intensive and conventional insulin therapy on the risk of development and progression of diabetic chronic complications in 1,441 patients with type 1 diabetes, has demonstrated that a sustained improvement in HbA1c reduces the risk of development of diabetic nephropathy (34). Similarly, the United Kingdom Prospective Diabetes Study (UKPDS) has shown that improved glycemic control is effective in the prevention of microalbuminuria in patients with newly diagnosed type 2 diabetes (35). Finally, in a small study of eight patients with type 1 diabetes, pancreatic transplantation and near normoglycemia for 10 years reversed structural abnormalities in the kidney (36).
There is strong evidence that hyperglycemia is necessary in the pathogenesis of diabetic nephropathy, and some of the mechanisms that link hyperglycemia to the functional and structural abnormalities of diabetic kidney disease have been elucidated.
Extracellularly, glucose reacts nonenzymatically with primary amines of proteins, forming glycated compounds. Glucose can also be transported into cells mainly by glucose transporters and can be metabolized to sorbitol via the polyol pathway and to glucosamine in the hexosamine biosynthetic pathway. These biochemical pathways have been implicated in hyperglycemia-induced kidney damage. Furthermore, it has become increasingly clear that excess glucose can exert toxic effects directly by altering intracellular signaling pathways; currently, this is believed to be a major mechanism by which hyperglycemia results in kidney damage.
NONENZYMATIC GLYCOSYLATION
Sustained hyperglycemia leads to nonenzymatic protein glycation, a posttranslation modification that occurs physiologically but is greatly enhanced in hyperglycemic conditions. Glycation results from condensation of reducing sugars with ε-amino groups of lysine residue of proteins. The resulting Schiff base
undergoes rearrangement to form relatively stable ketoamines, the Amadori products. These glycated proteins undergo progressive dehydration, cyclization, oxidation, and rearrangement to form advanced glycation end-products (AGEs). Once these AGEs are formed, the reaction is not reversible, and they gradually accumulate over the lifetime of the protein (37).
undergoes rearrangement to form relatively stable ketoamines, the Amadori products. These glycated proteins undergo progressive dehydration, cyclization, oxidation, and rearrangement to form advanced glycation end-products (AGEs). Once these AGEs are formed, the reaction is not reversible, and they gradually accumulate over the lifetime of the protein (37).
The hypothesis that AGEs are involved in diabetic kidney disease is based on several observations. In both human and experimental diabetes, AGEs accumulate in renal glomeruli and tubules. This phenomenon is specific, as it does not occur in normal kidneys or in kidneys from patients with glomerulonephritis and without diabetes (38). In normal animals, injection of AGE-modified albumin results in overexpression of matrix molecules, albuminuria, and glomerulosclerosis (39). In diabetic animals, accumulation of AGEs in the kidney is paralleled by the development of albuminuria, mesangial expansion, and thickening of the glomerular basement membrane. Furthermore, administration of aminoguanidine (AGN), an inhibitor of AGE formation, attenuates renal accumulation of AGEs and reduces both albuminuria and mesangial expansion (40).
Ultrastructural and in vitro studies have cast light on the mechanisms whereby AGEs can lead to kidney damage. AGEs directly alter the structural and functional properties of extracellular matrix proteins. AGE-mediated protein cross-linking increases matrix rigidity and reduces the susceptibility of protein to enzymatic digestion, thus inducing accumulation and thickening, and favors trapping of plasma proteins, such as low-density lipoprotein (LDL) and immunoglobulin G (IgG). AGN normalizes the defect in matrix degradation that occurs when matrix is incubated in a medium high in glucose, suggesting that the AGN effect in preventing mesangial expansion is in part due to its capacity to normalize mesangial matrix digestion (41). In addition, AGEs can elicit a variety of cellular responses by binding to AGE-specific receptors present on many cell types, including mesangial cells and tubular epithelial cells (42). Interaction of AGE-modified proteins with the AGE receptors serves to degrade AGE proteins but also induces the synthesis and release of cytokines, such as transforming growth factor (TGF-β1), platelet-derived growth factor (PDGF), and insulin-like growth factor (IGF), and results in enhanced production of collagen, laminin, and fibronectin (43,44).
The importance of AGE receptors has recently been given further support by the observation that diabetic animals overexpressing the AGE receptor (RAGE) rapidly develop glomerular lesions similar to those seen in advanced diabetic nephropathy (45). RAGEs are also expressed on tubular epithelial cells, and it has recently been reported that RAGE activation can induce tubular epithelial cell transdifferentiation to myofibroblasts, a key event in the development of tubulointerstitial fibrosis (46).
THE POLYOL PATHWAY
In the polyol pathway, glucose is reduced to sorbitol by the enzyme aldose reductase. In many tissues, the physiologic significance of aldose reductase is difficult to define, but in the renal medullary cells, the primary role of aldose reductase seems to be the formation of sorbitol, an organic osmolyte, in response to the high salinity in the medullary interstitium. It has been argued that, in tissues in which glucose entry into cells is insulin independent, more glucose becomes available for reduction by aldose reductase, resulting in an increased concentration of sorbitol and/or a reduced intracellular concentration of myoinositol. These changes might contribute to diabetic complications via an upset of cellular osmoregulation (47,48). Enhanced activity of the polyol pathway has been demonstrated in diabetic glomeruli in humans (49). However, the initial hypothesis that accumulation of sorbitol and reduction of myoinositol cause tissue damage is unlikely to operate in the kidney because compensatory mechanisms prevent the depletion of inositol in kidney cells (50).
More recently, enhanced AGE production and oxidative stress have been proposed as alternative mechanisms linking the polyol pathway to kidney injury in diabetes. In the polyol pathway, excess sorbitol is oxidized to fructose by the enzyme fructose dehydrogenase. The increased ratio of NADH/NAD (reduced form of nicotinamide adenine dinucleotide/oxidized form of nicotinamide adenine dinucleotide) coupled to the oxidation of sorbitol to fructose can result in cellular oxidative stress. Furthermore, fructose is a reactive sugar that can lead to AGE production (51,52). Recent experiments in cultured bovine aortic endothelial cells have demonstrated that high glucose concentrations increase the production of reactive oxygen species. Further, blockade of the electron transport chain complex II normalizes mitochondrial reactive oxygen species and prevents glucose-induced activation of protein kinase C (PKC), formation of AGEs, accumulation of sorbitol, and activation of the transcription factor nuclear factor-κB (NF-κB) (53).
Various pathophysiologic mechanisms linking activation of the polyol pathway to kidney damage can be postulated. However, to prove that activation of the polyol pathway is not merely an epiphenomenon but a mechanism of renal injury, it is important to have in vivo evidence that inhibition of the polyol pathway is beneficial in diabetic nephropathy. To gain this evidence, a series of studies of aldose reductase inhibitors (ARIs) have been carried out in both experimental and human diabetes. Early studies using the ARIs were inconclusive because of dubious compound specificity and drug toxicity. More recently, treatment of diabetic rats for 6 months with the ARI tolrestat resulted in a slight reduction in albumin excretion rate (54), but to date no convincing effect of aldose reductase inhibitors has been reported in controlled studies in humans. This indicates that activation of the polyol pathway is more likely to be an epiphenomenon and that other, more central mechanisms are operating in the pathogenesis of diabetic nephropathy.
HEXOSAMINE BIOSYNTHETIC PATHWAY
Approximately 5% of the glucose entering the cell is metabolized via the hexosamine biosynthetic pathway (HBSP), which converts glucose-6-phosphate into hexosamine-6-phosphate. Glutamine:fructose-6-phosphate aminotransferase (GFAT) is the first and the rate-limiting enzyme of this pathway. In both mesangial cells and NIH-3T3 fibroblasts, GFAT overexpression, which increases the flux through the HBSP, leads to enhanced expression of TGF-β and fibronectin (55,56). Furthermore, high glucose-induced production of TGF-β1 and matrix appears, at least in part, to be mediated by the HBSP because such production is significantly reduced by the GFAT inhibitor azaserine (57). A very low level of expression of GFAT usually is found in glomerular cells, but such expression is significantly enhanced in the glomerular cells of patients with diabetic nephropathy (58), suggesting a potential in vivo relevance of the in vitro findings. Both high glucose and angiotensin II activate the GFAT promoter in mesangial cells (59), and this provides a potential molecular mechanism of GFAT overexpression in the diabetic glomeruli.
GLUCOTOXICITY
Consistent with the finding in clinical studies that hyperglycemia per se plays a key role in the development of diabetic kidney disease, studies on both kidney cells and isolated glomeruli have confirmed that high glucose concentrations directly alter extracellular matrix deposition.
In mesangial cells, high glucose concentrations induce cell hypertrophy and increase gene expression and protein secretion of extracellular matrix components, such as collagen, laminin, and fibronectin (60). Similarly, levels of type IV and I collagen messenger RNA (mRNA) are enhanced in tubular epithelial cells exposed to high glucose concentrations (28). An additional mechanism whereby high glucose leads to exaggerated matrix deposition is by reducing the activity of metalloproteases, enzymes responsible for extracellular-matrix degradation (61).
Renal cells, similar to cells at other sites of diabetic complications, do not have an absolute requirement of insulin for glucose uptake, so that the intracellular levels of glucose more directly reflect its plasma concentration. The importance of excess glucose entry into mesangial cells is underscored by the observation that glucose-induced effects can be mimicked in normal glucose concentrations by overexpression of the cellular glucose transporter GLUT 1, thus increasing basal glucose uptake (62). This finding also indicates that factors regulating expression of glucose transporter and/or activity can influence glucose uptake and thus glucotoxicity. High glucose concentrations would be expected to reduce glucose transporter expression/activity in order to protect cells from excess glucose entry. However, in vitro studies of cultured mesangial cells suggest that exposure to elevated glucose concentrations actually enhances GLUT 1 expression by mesangial cells, triggering a positive feedback mechanism that may result in progressive damage (63). Inhibition of GLUT 1 overexpression prevents overproduction of extracellular matrix molecules in mesangial cells exposed to high glucose (64), providing evidence of a key role of GLUT1 in glucotoxicity (64)
The cellular and intracellular mechanisms whereby high glucose can lead to matrix deposition have been extensively studied since the 1980s, and will be reviewed in depth below.
Hypertension
There is evidence that hypertension plays a critical role in the progression of diabetic nephropathy. Indeed the development of proteinuria is paralleled in most cases by a gradual rise in systemic blood pressure, and there is a significant correlation between the blood pressure levels and the rate of decline in glomerular filtration rate (65). Furthermore, intervention studies in both animals and humans have demonstrated significant renoprotective and antiproteinuric effects of antihypertensive therapy (66,67,68).
In diabetic nephropathy, hypertension is not merely the result of relentless kidney damage; there is considerable clinical evidence that the elevated arterial pressure is also important in the genesis of the glomerular lesion. In Pima Indians, higher mean blood pressure prior to the onset of diabetes actually predicts an abnormal albumin excretion rate after the diagnosis of diabetes (4). Further, prospective studies in both patients with type 1 and type 2 diabetes and normal albumin excretion have demonstrated that mean arterial pressure levels are significantly higher in those patients who progress to microalbuminuria than in those who do not progress (6,69).
The difference in arterial pressure levels of diabetic patients who develop renal complications and those who do not is numerically small, and the levels in those who develop complications are often in the “normotensive range.” However, this difference becomes biologically relevant in the presence of diabetes, which induces loss of autoregulation of glomerular capillary pressure. Under normal conditions, intraglomerular capillary pressure is tightly regulated by precise adjustments in afferent and efferent arteriolar resistance. Hyperglycemia induces vasodilatation, and in diabetes there is a marked reduction in afferent and a lesser reduction in efferent arteriolar resistance. This leads to an increase in levels of glomerular capillary pressure and moreover allows ready transmission of any increase in systemic blood pressure to the glomerular capillary network (70). The metabolic and the hemodynamic insults are thus intimately intertwined in determining altered glomerular hemodynamics.
The notion that enhanced glomerular capillary pressure is involved in diabetic glomerulosclerosis is based on several observations. Conditions known to increase glomerular capillary pressures, such as unilateral nephrectomy, Goldblatt hypertension, and high-protein diet, significantly enhance the extent of glomerular injury in rats with streptozotocin-induced diabetes. Conversely, amelioration of glomerular hypertension slows the rate of progression of the glomerular injury (71,72). Consistent with these animal studies, autopsy studies of diabetic patients with unilateral renal artery stenosis show nodular glomerulosclerosis lesions to be confined to the kidney with the patent renal artery (73). Finally, the greater efficacy of angiotensin-converting enzyme (ACE) inhibitors in their antiproteinuric and renoprotective action as compared with other blood pressure-lowering agents has been in part ascribed to the capacity of ACE inhibitors to reduce glomerular capillary pressure by removing the tonic constrictor effect of angiotensin II on the efferent arteriole (74).
Although increased glomerular capillary pressure has long been associated with the deposition of extracellular matrix, the mechanisms by which the mechanical insult of altered hemodynamics translates into kidney damage have only recently been elucidated. This has followed the demonstration of the unique elastic properties of the glomerular structure and the response of mesangial cells to mechanical stretch in culture.
Glomeruli are highly compliant structures, and when the intraglomerular pressure rises to levels approximating those observed in the diabetic and remnant kidney, glomerular volume increases by about 30%. Volume changes reach their maximum within 3 to 4 seconds following alteration in intraglomerular pressure, thus responding to the most transient variations in intraglomerular pressure. Glomerular expansion is associated with the stretching of its structural components, including the extracellular matrix and the cellular components. Because of the central location of the mesangial regions within the glomerular lobule, mesangial cells experience substantial mechanical stretching. Detailed morphologic studies have demonstrated how numerous cytoplasmic projections emerging from the mesangial cell body extend between adjacent capillaries and firmly attach to the perimesangial regions of the glomerular basement membrane. Therefore, the centrifugal displacement of these regions during glomerular expansion results in marked tridimensional stretching of mesangial cells (75).
Mesangial cells exposed to mechanical stretch in culture undergo alterations in morphology and synthetic activity. Cyclical stretch stimulates the synthesis and deposition of matrix components, such as collagen (I, III, and IV), fibronectin, and laminin, in a manner proportional to the intensity of cellular stretch (76,77,78). Interestingly, although cyclical stretch stimulates collagen synthesis at all glucose concentrations, net collagen accumulation in the medium can be demonstrated only at high glucose levels (79), the result of catabolic rates that are insufficient to match the increased synthesis in a high-glucose environment. Therefore, the cellular response to a hemodynamic insult can be influenced and exaggerated by high glucose concentration in the milieu. In addition, GLUT1 is overexpressed in an animal model of glomerular hypertension in vivo, and stretch enhances glucose uptake by inducing GLUT1 expression in human mesangial cells in vitro (80). This suggests that glomerular hypertension may enhance glucotoxicity.
Proteinuria
In diabetic nephropathy and other progressive glomerulopathies, proteinuria is a strong and independent predictor of decline in renal function (81). Excessive protein overload appears to induce tubulointerstitial damage and hence to contribute to the disease progression (82). Specifically, the excessive tubular reabsorption of proteins and the consequent accumulation of proteins in tubular epithelial cells induce the release of vasoactive and inflammatory cytokines, such as endothelin-1, osteopontin, and monocyte chemoattractant protein-1 (MCP-1). These factors in turn lead to overexpression of proinflammatory and fibrotic cytokines and infiltration of mononuclear cells, causing injury of the tubulointerstitium and, ultimately, renal scarring and insufficiency (28,81,83). A vicious cycle is then established in which changes in renal hemodynamics, either primary or in response to nephron loss, induce further proteinuria, perpetuating a mechanism of interstitial scarring and loss of more nephrons. The tubular toxicity of protein raises the possibility that the beneficial effects of ACE inhibitors in diabetic renal disease may reflect their potent antiproteinuric action in addition to the reduction of angiotensin II-mediated effects on growth-factor activation and glomerular hemodynamics (28,81). Limiting protein excretion and the consequent activation of tubular epithelial cell prosclerotic signals thus appears instrumental in protecting the kidney from further damage (Fig. 51.1).
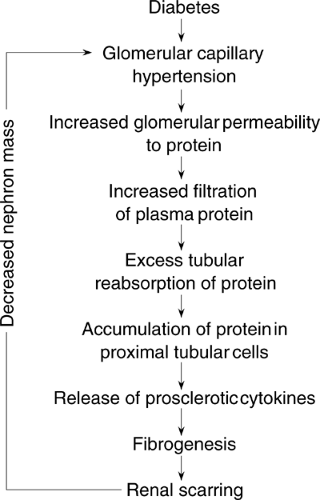 Figure 51.1. Potential mechanisms of proteinuria-induced renal damage. (Modified from Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med 1998;12:1448–1456.) |
MOLECULAR MEDIATORS
To enhance understanding of the pathogenesis of diabetic nephropathy, a number of studies have examined the cellular and molecular mechanisms of kidney damage. These studies have established the critical concept that the insults of hyperglycemia, high blood pressure, and protein overload converge at the cellular level by using similar molecular signaling pathways and influencing the expression of common cytokines (Fig. 51.2).
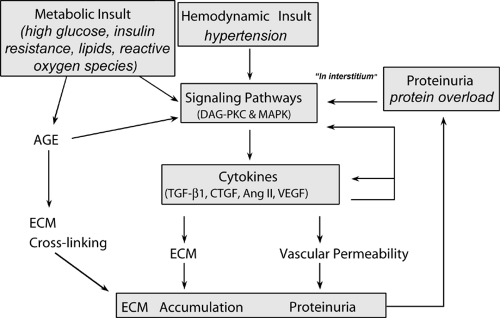 Figure 51.2. Schematic representation of the interaction, at the cellular and intracellular levels, of three insults operating in diabetic nephropathy. AGE, advanced glycation end-product; ECM, extracellular matrix; TGF-β1, transforming growth factor-1β; Ang II, angiotensin II; CTGF, connective tissue growth factor; VEGF, vascular endothelial growth factor; MAPK, mitogen-activated protein kinases; PKC, protein kinase C; DAG, diacylglycerol. |
Transforming Growth Factor-β1
TGF-β is a ubiquitous cytokine that regulates a variety of cellular processes. It exists in three isoforms—TGF-β1, TGF-β2, and TGF-β3—of which TGF-β1 is the best characterized and most highly expressed in the kidney. Glomerular and proximal tubule cells produce TGF-β1 and express both type I and type II TGF-β receptors, which bind to all TGF-β isoforms (84,85).
TGF-β1 has important prosclerotic properties and is a potent inducer of cell hypertrophy and apoptosis. In vitro, both glomerular mesangial cells and tubular epithelial cells increase their synthesis of collagen, fibronectin, and laminin in response to TGF-β1. Furthermore, TGF-β1 inhibits the synthesis of collagenases and stimulates production of metalloprotease inhibitors. This results in reduced degradation of extracellular matrix and further contributes to accumulation of matrix (85). Finally, in vivo, TGF-β1 induces glomerulosclerosis and proteinuria in healthy animals (86,87,88).
Numerous studies have explored the role of TGF-β1 in diabetic glomerulosclerosis, and at present there is overwhelming evidence implicating overexpression of TGF-β1 in the pathogenesis of diabetic nephropathy. In both human and experimental diabetes, TGF-β1 gene expression and protein secretion are increased in the glomeruli and tubules (89,90). This effect is sustained in time and closely correlates with the degree of mesangial matrix expansion and with HbA1c levels. The TGF-β type II receptor is also overexpressed in diabetic animals (91), suggesting that the diabetic kidney is hyperresponsive to TGF-β. Finally, and of the greatest importance, in diabetic mice TGF-β blockade significantly reduces overexpression of both type IV collagen and fibronectin and prevents glomerular hypertrophy, glomerulosclerosis, and renal insufficiency (92,93).
The importance of TGF-β1 is further highlighted by in vivo and in vitro studies showing that the three key insults implicated in the pathogenesis of diabetic nephropathy promote the expression of this cytokine. These studies also suggest that TGF-β1 may have a role in the interaction between metabolic and hemodynamic factors in mediating accumulation of extracellular matrix in the diabetic kidney.
High glucose concentrations induce the expression of TGF- β1 in both mesangial and tubular epithelial cells. Further, inhibition of TGF-β1 prevents glucose-induced hypertrophy of mesangial cells and production of extracellular matrix, indicating that these effects are mediated by TGF-β1 via an autocrine mechanism (94). Exposure of mesangial cells to high glucose concentrations induces overexpression of TGF-β receptors, suggesting that high glucose concentrations may also enhance the response to TGF-β1 (95). On the other hand, TGF-β1 induces the GLUT1 transporter in mesangial cells and can thereby enhance glucotoxicity (96). In vitro, AGEs induce TGF-β1 gene expression and protein secretion by mesangial cells (44), and in vivo administration of AGEs upregulates TGF-β1 in the kidney (97).
The link between hyperglycemia, TGF-β1, and kidney sclerosis is thus well established.
The link between hyperglycemia, TGF-β1, and kidney sclerosis is thus well established.
Recent studies in glomerular epithelial cells have shown that high glucose induces the TGF-β type II receptor without affecting TGF-β1 production. TGF-β mediates high glucose-induced production of fibronectin and collagen type IV (α3 chain) by glomerular epithelial cells, suggesting a role of TGF-β1 in the thickening of the glomerular basement membrane (98,99).
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


