(1)
Medical Sciences Division Northern Ontario School of Medicine West Campus, Lakehead University, Thunder Bay, Ontario, Canada
Key Topics
Challenges with pancreatic cancer (PanCa) screening
Screening for PanCa
Molecular pathology of PanCa
Circulating PanCa miRNA biomarkers
Circulating PanCa proteomic and metabolomic biomarkers
Circulating PanCa cells
Key Points
Pancreatic cancer (PanCa) has a high mortality rate primarily due to late diagnosis. The pancreas cannot easily be palpated and yet there are no validated noninvasive screening biomarkers, resulting in late stage presentations.
The established molecular pathologic changes in PanCa are detectable in ctDNA, and some including KRAS mutations are adjuncts to clinical decision-making in patient management.
Several circulating biomarkers including miRNAs, traditional serum proteins, and proteomic profiles as well as circulating PanCa cells are potential actionable clinical biomarkers.
9.1 Introduction
Pancreatic cancer (PanCa) is the tenth most diagnosed cancer in the United States and yet is the fourth cause of cancer-related fatalities. The 2012 global estimated incidence for PanCa was 337,760 with an associated mortality of 330,372. In the United States, similar dismal outcomes are consistently being reported with an estimated 53,070 new cases and 41,780 deaths in 2016. The global age-standardized incidence and mortality are 3.9/100,000 and 3.7/100,000, respectively. Thus, the incidence is similar to the mortality rate, and this has remained so for several decades. Indeed, mortalities from many cancers such as breast, prostate, and lung cancers are declining over the past decades, but those from PanCa have remained the same or are increasing, especially in the elderly (>70 years). The reason for this poor outcome is the absence of screening programs for the general population at low to medium risk, and thus many tumors are being diagnosed at an advanced stage, which are not amenable to surgery, and yet there is no effective alternative therapy. While there is no consensus on who, when, and how often screening should be done, several centers use their own discretion, with many urged to only screen those with a relative risk score of >10 (out of 14.3). Because many people at risk are excluded from screening, over 80 % of cases are detected at an advanced unresectable state, often associated with distant metastasis. In view of such late presentations, the majority (>74 %) of patients die within 12 months of initial diagnosis, and up to 94 % die within 5 years. However, the 5-year survival rate can be as high as 75 % when small tumors (<1 cm) are detected. Indeed, a screening program of high-risk individuals can potentially lead to early detection of all patients, with associated excellent cure rates. Thus, early detection of organ-confined small tumors should improve the outlook for PanCa patients. Additionally, PanCa is a latent disease that develops over several years after acquiring the initial genetic alterations. Modeling of sequencing data from primary and metastatic PanCa suggests it takes ~17 years for a cell to progress from initiation to metastasis. There is thus a wide window of opportunity for early detection, using molecular biomarkers in circulation. The need for noninvasive early detection biomarkers is thus eminent, and there are potential targets worthy of validation for product development.
9.2 Challenges with PanCa Screening
In spite of the rise in incidence and mortality, the screening for early detection of curable PanCa is far from optimum. The simplest reason is the lack of validated cost-effective serum biomarkers for population-based screening. Additionally, sensitive tissue-specific biomarkers detectable in pancreatic juice where they are enriched for cannot be applied to the entire population at risk (i.e., people over 30 years of age), because obtaining such fluid is invasive and expensive, requiring endoscopic retrograde cholangiopancreatography and sampling. A gastroduodenal capsule is an alternative minimally invasive means of obtaining pancreatic juice but will need to overcome some important hurdles including:
The sampling device will need validation.
A possible pharmacologic induction of pancreatic juice secretion.
This may require identification/validation of pancreatic cancer-specific biomarkers distinct from those of the hepatobiliary system.
Imaging modalities, especially molecular imaging, will offer a better noninvasive approach. However, molecular imaging is cost prohibitive for mass screening, and there are currently only experimental data with no biomarkers or probes developed for such utility. Thus, numerous efforts at biomarker identification for screening have focused on blood components. While this probably offers the best approach, there are inherent challenges with biomarker specificity.
9.3 Screening Recommendations for PanCa
Several risk factors are identified for the development of PanCa. Some, such as age, gender, race, smoking, alcohol, and obesity, have overlap risks with several other cancers. Pancreatitis and diabetes are associated with increased risk for PanCa. Allergies are putatively associated with 30–45 % reduced risk. Moreover, genetic polymorphisms especially non-O blood group type alleles increase the risk for PanCa. Also implicated is H. pylori infection. However, the highest risk is among people with familial syndromes (Table 9.1). But, only 3–16 % of all PanCas are familial or syndromic with well-established defined gene alterations. The majority are therefore sporadic cases.
Table 9.1
Hereditary syndromes associated with risk of PanCa
Hereditary syndrome | Genes involved | Relative risk | Extra-pancreatic involvement |
|---|---|---|---|
Familial pancreatic syndrome | Unknown | 4.6–32 | – |
Familial adenomatous polyposis | APC | 4 | Colorectum, small intestines, stomach |
Familial atypical multiple mole melanoma | CDKN2A | 9–22 | Skin (melanoma) |
Hereditary breast and ovarian syndrome | BRAC1, BRAC2, FANCC, FANCG, PALB2 | 3.5–10 | Breast, ovary, prostate |
Hereditary pancreatitis | PRSS1/TRY1, SPINK1 | ~50 | – |
Hereditary nonpolyposis colorectal carcinoma syndrome | MLH1, MSH2 | 8.6 | Colorectum, small intestines, endometrium |
Peutz–Jeghers syndrome | ST11/LKB1 | ~132 | Small intestines, colorectum, esophagus, stomach, bile duct, lungs, breast, ovary, uterus |
Li–Fraumeni syndrome | TP53 | Several |
Screening recommendation from the fourth International Symposium on Inherited Diseases of the Pancreas is for healthy people with at least three affected first-degree relatives or BRCA2 mutation carriers with at least one family member diagnosed with PanCa. Screening should begin at age 50 or at an age 10 years earlier than the age at which the youngest family member was diagnosed. Also consensus practice recommendation suggests an increased risk of tenfold should guide screening (but this tenfold cutoff implies several people with elevated risk are excluded from screening). Screening frequency is not established, and screening algorithms or procedures are not standardized. Currently, screening involves a multidisciplinary team of experts using various imaging techniques.
Unlike other cancers such as breast and prostate that can be detected by palpation on physical examination, and gastrointestinal tract tumors that can easily be visualized with an instrument by an expert for possible early detection, the retroperitoneal anatomic location of the pancreas precludes easy access. In view of this, and coupled with the various advances made in imaging modalities, imaging has been the primary tool for the clinical evaluation of pancreatic masses, and screening of those with elevated risk for developing PanCa. While endoscopic ultrasound (EUS) or magnetic resonance imaging (MRI) is initially used, and if abnormal other modalities are deployed, the eventual diagnosis requires EUS-guided fine needle aspiration and histopathologic visualization of cancer cells. Various other imaging modalities including computerized tomography (CT) and positron-emission tomography (PET) scans, endoscopic retrograde cholangiopancreatography (ERCP), and magnetic resonance cholangiopancreatography (MRCP) are used by various centers for screening. Effective screening algorithm with imaging, combined with genetic testing, proves useful for early detection, not only of PanCa but neoplastic lesions outside of the pancreas such as ovarian, papillary thyroid cancer, and carcinoid tumors. The best imaging modality is EUS. This is more accurate than the other modalities at viewing deep-seated masses. When combined with sampling of the mass by fine needle aspiration, EUS offers a high sensitivity of 92 % for PanCa detection. This imaging modality is superior to CT scans and MRI for detecting lesions <1 cm and intraductal papillary mucinous neoplasm (IPMN). EUS is also better at resolving lymph node metastasis and vascular infiltration compared to CT scans.
While early detection with curative intervention strategies will obviously reduce mortalities from PanCa, screening is impractical at present for the entire asymptomatic population at risk due to cost prohibition, invasiveness of screening procedures that can be associated with complications such as pancreatitis, lack of proven sensitivities, and the generally low disease prevalence. The need for accurate noninvasive and cost-effective biomarkers for screening and triaging of asymptomatic individuals for more invasive assessment is therefore eminent. Such biomarkers in circulation will be ideal. While many are discovered, validation, product development, and widespread clinical translation are awaited.
9.4 Molecular Pathology of PanCa
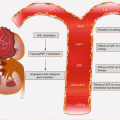
Many PanCas (~65 %) are localized to the head of the pancreas and may present early with signs of pancreatic ductal obstructions (Fig. 9.1). However, the remaining 35 % occur elsewhere in the pancreas. Putatively, the anatomic location may be associated with prognosis, with cancers at the tail being associated with increased in-hospital complications and mortality, probably due to late diagnosis of advanced diseases.
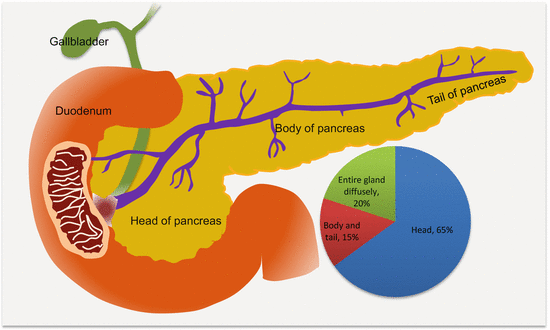
Fig. 9.1
Frequencies and anatomic locations of PanCas
9.4.1 Familial and Sporadic PanCa
While the majority of PanCas occur without any relationship to familial segregation (sporadic PanCa, SPC), up to 10 % of all PanCas have some hereditary components (familial PanCas, FPC). The genetic landscape of SPC is well elucidated; however, FPC is often associated with genes involved with other cancer and disease syndromes (Table 9.1). Familial PanCa is considered when at least a pair of first-degree relatives (two siblings or a sibling and a parent) is diagnosed with PanCa. It has been estimated that the risk of developing PanCa is 18-fold in healthy people from these families, and this increases to 56-fold when the disease has afflicted ≥3 family members.
Implicated in the pathogenesis of FPC are alterations in genes involved in the associated syndromes (Table 9.1), as well as PALLD, PALB2, and PHOX2B. It may also appear that the genetics of FPC cases of late onset have identical molecular pathology as SPC. Norris et al. used whole exome, whole genome, and RNA sequencing together with high-density SNP arrays to study cell lines and matched germline samples from a cohort of FPC patients and revealed that the four major driver genes of SPC, KRAS, TP53, CDKN2A/p16, and SMAD4/DPC4 were consistently altered in these samples as well [1].
9.4.2 Molecular Subtypes of PanCa
Work by Collisson and colleagues on gene expression analysis enabled the classification of PanCa into three distinct molecular subtypes, namely, classic, quasimesenchymal, and exocrine-like [2]. These subtypes also differ in terms of their response to treatment and clinical outcome:
The classic subtype overexpresses high levels of adhesion-associated and epithelial genes, including S100PBP, AGR2, CEACAM5, CEACAM6, and FXYD5. Tumors of this subtype are more sensitive to oral EGFR tyrosine kinase inhibitors such as erlotinib.
The quasimesenchymal tumors express high levels of mesenchyme-associated genes such as TWIST1, and S100A2. They are more sensitive to gemcitabine therapy and also have worse outcome than the classic subtype.
The exocrine-like PDACs express high levels of genes involved with digestion such as REG3A (PAP), PRSS1/2, SLC3A1, and CFTR. It has been demonstrated that tumors of this subtype that express CYP3A5 are resistant to TKIs [3].
9.4.3 Genetics of PanCa
There are marked geographic and racial variations in PanCa incidence, being high among African-American men in particular and also in Koreans, Czechs, Native Hawaiians, New Zealand Maoris, and Latvians. Globally, PanCa ranks 13th in incidence and 8th case mortality among cancers. However, the high incidence in the identified populations is not necessarily explained by known risk factors, making genetic factors the probable culprits.
9.4.3.1 Intragenic Point Mutations in PanCa
Almost all PanCas demonstrate intragenic mutations in genes involved in several signaling pathways. Oncogenes (KRAS2 and BRAF), TSGs (CDKN2A, TP53, and SMAD4), and caretaker genes (BRAC2, FANCC, and FANCG) are all mutated at various frequencies in a variety of PanCas. KRAS2 mutations occur in as many as 95 % of PDACs. Interestingly, cancers without KRAS2 mutations tend to harbor BRAF mutations, indicating an activation of the RAS/BRAF/MEK/ERK pathway in almost all PDACs. Other MAPK pathway mutations include STK11/LKb1 (of Peutz–Jeghers syndrome). TGFβ/activin pathway mutations involve SMAD4/DPC4, TGFβRI, TGFβRII, ACVR1B, and ACVR2. The Fanconi anemia pathway is involved by mutations in BRAC2, FANCC, and FANCG.
9.4.3.2 Important Oncogenes and Tumor Suppressor Genes in PanCa
KRAS Alterations in PanCa
The KRAS oncogene is mutated at a high frequency in PanCa. KRAS gene on chromosome 12 encodes KRAS protein, which is a member of the RAS family of GTP-binding proteins that control cellular proliferation, growth, cytoskeletal remodeling, motility, differentiation, and survival. KRAS is the most frequently mutated gene in PanCa, occurring in almost all (>95 %) PanCas. Mutations are early events in PDAC, being detected in PanIN1. Activating mutations in this oncogene, especially in codon 12, leads to constitutive KRAS activity, which includes activation of RAF family of serine threonine kinases and eventual downstream BRAF-MAPK and PI3K/AKT signaling. The final output of such a cell is acquisition of uncontrolled division and growth, evasion of death signals, and eventual development of invasive phenotype. The few (~5 %) of PanCas without KRAS mutations harbor BRAF V600E point mutations and are often microsatellite unstable.
CDKN2A Alterations in PanCa
The CDKN2A tumor suppressor is also inactivated in many (~95 %) PDACs. Located on chromosome 9, one product of this gene is the p16 protein that inhibits cyclin-dependent kinase 4- and 6-mediated phosphorylation of retinoblastoma (RB). This prevents entry into the S phase of the cell cycle (G1-S check point). Thus CDKN2A inactivation via promoter methylation, intragenic mutations, homozygous deletion, or LOH with subsequent loss of the other allele, leads to inappropriate cell cycle progression and unregulated cell proliferation and growth. Inactivation is an intermediate event in PDAC progression model, being detected in PanIN2.
TP53 Alterations in PanCa
The TP53 tumor suppressor is inactivated in 50–75 % of PDACs. Located on chromosome 17, TP53 encodes the p53 transcription factor that controls several genes involved in cell cycle progression, specifically, G2-M phase arrest and G1-S checkpoint. P53 also controls DNA repair and cell death processes. In cells with severe DNA damage, p53 inhibits cell cycle to allow for possible repair before cycle progression, else it will induce expression of genes involved in cell death (its cell cycle checkpoint functions). Inactivating intragenic mutations causes defective p53 protein production that loses its functional interactions with DNA and hence loss of cell cycle control. Thus, expansion of clones of cells with genomic instability occurs with increasing propensity for cancer formation.
SMAD4/DPC4 Alterations in PanCa
Also known as deleted in pancreatic cancer locus 4 (DPC4), SMAD4 is located on chromosome 18q and is inactivated by homozygous deletions and intragenic mutations with loss of the second allele, in as many as 55 % of PDACs. The encoded protein, SMAD4, plays an important role in the TGFβ signaling pathway that functions to inhibit cell growth. The interaction of TGFβ with its receptor (type I and type II serine/threonine kinase surface receptors) induces receptor dimerization and activation of type I receptor that phosphorylates SMAD2 and SMAD3. Phosphorylated SMAD2/3 complex binds to SMAD4, and they translocate into the nucleus where in association with cofactors induce expression of genes that control cell cycle, growth, and differentiation. Loss of SMAD4 causes loss of TGFβ-mediated cell growth control leading to unregulated cell proliferation. Loss of SMAD4 occurs in late stages of PanCa progression, being observed in PanIN3 and infiltrating adenocarcinomas, and is associated with poor prognosis.
9.4.4 Precursor Lesions of PanCa
Approximately 95 % of PanCas originate from the exocrine pancreas, with the remaining 5 % from the endocrine portion. Cancers arising from the pancreatic duct, pancreatic ductal adenocarcinoma, account for ~80 % of all exocrine PanCas, with the remaining being intraductal papillary mucinous tumors, acinar cell carcinomas, mucinous cystic tumors, and serous cystic tumors. Three histologic types of precursor lesions are described for PanCa: pancreatic intraepithelial neoplasia (PanIN), intraductal papillary mucinous neoplasm (IPMN), and mucinous cystic neoplasm (MCN) (Table 9.2). Many pancreatic cancers, however, develop from PanIN that usually arises from the small terminal pancreatic ducts.
Table 9.2
Molecular pathology of pancreatic precursor lesions and cancer
Lesion | Altered genes |
|---|---|
Precursor lesions | |
PanIN1 | Telomere shortening (90 %) KRAS2 mutations (45 %) |
PanIN2 | CDKN2A/p16 |
PanIN3 | TP53, SMAD4/DPC4, BRAC2 |
IPMN | KRAS2 (50 %), TP53, CDKN2A/p16 |
MCN | KRAS, SMAD4 |
Pancreatic cancer | |
Adenocarcinoma | CDKN2A/p16, SMAD4, TP53 |
Adenosquamous carcinoma | CDKN2A/p16, SMAD4, TP53, P63 |
Medullary carcinoma | MLH1, MSH2 |
Colloid carcinoma | MUC2, CDX2 |
Undifferentiated carcinoma | Loss of CDH1 (L1CAM, Cox2, EGFR) |
9.4.4.1 PanIN
Based on the degree of cytological and architectural atypia, three types of PanIN are recognized – PanIN1 (A/B), PanIN2, and PanIN3. The genetics of these lesions are fairly well characterized, and their roles in signaling pathways that mediate disease progression are described in the section on progression model.
9.4.4.2 IPMN
IPMNs are macroscopically visible neoplasms that arise in the mucin-producing main pancreatic duct or their branches. They grow and distend the duct and are associated with excessive mucin production. The genetic alterations of IPMN resemble those of PanIN to some degree. About 50 % of low-grade IPMN harbors KRAS mutations, and the mutation frequency increases with the degree of dysplasia. High-grade IPMN lesions acquire TP53 and CDKN2A mutations as well. However, unlike PanIN, SMAD4 mutations and loss of expression are infrequent in IPMN (occurring in just ~3 % of cases), but invasive cancers that arise from IPMN may harbor these mutations. However, inactivating mutations and loss of tumor suppressor gene functions of the serine/threonine kinase STK11/LKB1 occur in IPMN, and these are not seen in PanIN. Additionally, activating mutations and AKT signaling are observed in ~10 % of IPMN.
9.4.4.3 MCN
MCNs are more frequent in women. They are also macroscopically visible but are typically not associated with the ductal system (tend to be intraparenchymal). They have mucinous epithelial lining and ovarian-like stroma. While the molecular pathology is not well established, mutations in TP53 and KRAS are noted with increasing frequencies mirroring increasing dysplasia. Mutations and loss of SMAD4 are observed in infiltrating MCN.
9.4.5 Molecular Progression Model of PanCa
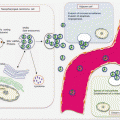
The multistep and field cancerization model of carcinogenesis has been fairly well established for PanCa (Fig. 9.2). While many genetic alterations occur to influence or alter several signaling pathways to drive PanCa progression, three distinct genetic alterations are well proven in pancreatic carcinogenesis. Initial events that occur in PanIN1 are telomere shortening and KRAS mutations. Observed mostly in PanIN2 are CDKN2A mutations, while late lesions tend to harbor TP53, SMAD4, and BRCA2 alterations. Knowledge on the progression of IPMN and MCN through various degrees of dysplasia to cancer is becoming available. This classic model of PDAC development has been supported by genome-wide profiling studies that reveal several mutations relevant to PanCa. The most frequently mutated genes were classified as “high-frequency driver genes,” and they included early genes such as KRAS, intermediate genes such as CDKN2A, and late genes inclusive of TP53 and SMAD4. Complementing the effects of these high-frequency genes are mutations in “low-frequency driver genes” such as SMARC4A, ID1, CDH1, EPHA3, NF1, EGFR, and FBXW7.
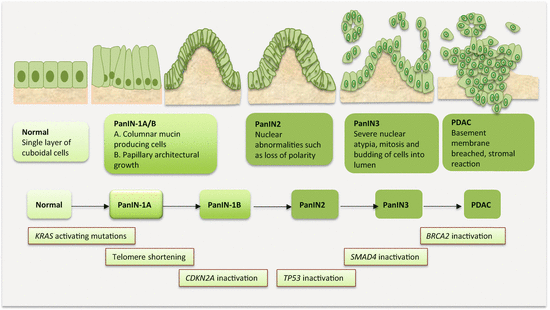
Fig. 9.2
Molecular pathology of multistep pancreatic carcinogenesis
9.5 Circulating PanCa Biomarkers
The obvious need for noninvasive PanCa biomarkers has led to the discovery of several. The genetic alterations in PanCa (e.g., CDKN2A methylation and KRAS mutations) are detectable in ctDNA. Also a number of miRNAs show differential levels between patients and control. Traditional serum proteins remain useful, but proteomic approaches have been extensively employed for discovery studies. The potential diagnostic, prognostic, and predictive roles of circulating PanCa cells require careful study.
9.5.1 Circulating Cell-Free Nucleic Acid Content as PanCa Biomarkers
Circulating cell-free DNA (ccfDNA) in PanCa patients has been analyzed primarily from the standpoint of gene mutations associated with the diagnostic, prognostic, and disease monitoring relevance. However, it also appears the absolute quantitative levels of circulating tumor DNA (ctDNA) have prognostic and treatment monitoring utility in PanCa patients. Elevated plasma DNA > 62 ng/ml was significantly associated with shorter OS (p = 0.002), presence of vascular encasement (p = 0.03), and metastasis (p = 0.001) [4]. Using KRAS mutations as a surrogate for ctDNA, 71 % of patients were positive prior to chemotherapy, and many of these patients experienced progressive disease. Importantly, pretreatment ctDNA level was a significant predictor of both PFS and OS. For many of these patients, changes in ctDNA levels corresponded with changes in radiographic imaging and CA19-9 levels on follow-up [5].
Circulating cell-free nucleosomes (ccfnucleosomes) as biomarkers of cancer have been demonstrated and have shown promise for early detection of PanCa. Intact ccfnucleosomes with specific histone modifications and variants were detected using a novel Nucleosome® ELISA platform in circulating blood from PanCa patients [6]. Using multivariate analysis, a panel of five ccfnucleosome biomarkers achieved an AUROCC of 0.95 for PanCa detection. Noteworthy, these patients had resectable tumors. A combination of CA19-9 with a panel of four ccfnucleosome biomarkers achieved the best diagnostic accuracy with AUROCC of 0.98, at a sensitivity of 92 % and specificity of 90 %.
9.5.2 Circulating PanCa Epigenetic Biomarkers
The epigenome is altered in PanCa, with DNA methylation being an early event in PanCa development. CpG island hypermethylation, genomic hypomethylation, as well as miRNA expressional changes underlie pancreatic carcinogenesis. Indeed, multiple genes are silenced or repressed by promoter hypermethylation of CpG islands in ~60 % of PanCas. DNMT1, an enzyme that controls the generational transfer of methylation patterns, is overexpressed in ~80 % of PanCas. Several TSGs, MMR genes, are silenced by promoter hypermethylation in PanCa. Given that PanCa has a favorable cure rate when detected early, such early detection biomarkers assayed in a noninvasive fashion hold tremendous potential for disease curtailment. However, only a few studies have addressed the clinical utility of circulating PanCa-specific methylation biomarkers.
Preproenkephalin gene promoter methylation was demonstrated in 30 % of plasma samples from PanCa patients, while CDKN2A methylated sequences were in 25 % of this cohort [7]. Park et al. observed that methylation of NPTX2 in plasma could differentiate PanCa patients from those with benign conditions at a sensitivity of 80 % and a specificity of 76 % [8]. Melnikov et al. demonstrated the ability of five methylated genes (VHL, CCND2, SOCS1, THBS2, and PLAU) in stratifying cancer cases from controls at a sensitivity and specificity of 76 % and 59 %, respectively [9]. In a follow-up study by this group, microarray was used to analyze ccfDNA, and this enabled the identification of 17-gene promoter methylation of relevance to PanCa detection. This gene signature could differentiate chronic pancreatitis patients from controls at a sensitivity of 81.7 % and specific of 78 %. Of even more importance in the detection of PanCa, the performance of these methylated gene panel had a sensitivity of 91.2 %, specificity of 90.8 %, and an overall accuracy of over 90 % in stratifying PanCa patients from those with chronic pancreatitis [10].
9.5.3 Circulating PanCa Genetic Biomarkers
The detection of KRAS mutations in circulation of PanCa patients has shown low sensitivity but has proven very specific for cancer detection. Some mutations found in plasma are not associated with PanCa, because circulating KRAS mutations are not isolated mutations of PanCa and could also predate the development of cancer. It should however be noted that a single mutation is probably insufficient to cause cancer, and hence such KRAS-mutation-positive individuals without cancer ought to be monitored closely for possible early PanCa detection, especially in the presence of risk factors. Indeed, KRAS mutations have been detected in plasma of healthy people who were diagnosed with PanCa 5–10 months later.
Mutations in KRAS codon 12 have been examined in plasma for their diagnostic and prognostic value in patients who were followed for various clinical outcomes. Detection of mutation in plasma was at 27 % in one series [11]. Plasma KRAS mutation was related to tumor size, distant metastasis, and shorter survival time, and this was an independent prognostic factor. Plasma KRAS mutant genomes were however not that sensitive (27 %) but very specific (100 %) in detecting cancers among patients with pancreatic masses [11]. Dabritz et al. also analyzed KRAS codon 12 mutation in plasma samples, and the frequency of detection was 28 %, with valine mutation being commonly (83 %) detected at this location [12]. In a follow-up study, circulating KRAS mutations in plasma were found in 36 % of PanCa patients but not in individuals with pancreatitis. Some patients with low to moderate CA19-9 levels were diagnosed early using plasma KRAS mutations (35 % of cases). When combined with CA19-9, plasma KRAS mutations achieved a sensitivity of 91 % in the detection of PanCa [13]. Moreover, mutant KRAS in plasma samples corresponded to CT imaging findings in some patients [14]. Plasma KRAS codon 12 mutation as well as promoter methylation of CDKN2A and preproenkephalin (PPENK) were examined and data related to smoking status among PanCa patients. KRAS mutation was detected at a frequency of 32.5 % compared to promoter methylations of PPENK (29.3 %) and CDKN2A (24.6 %). However, when considering alterations of at least one of these biomarkers, the detection rate of PanCa was 63 %. Smoking was associated with KRAS codon 12 mutations, especially G to A, more so in heavy smokers, but no association was found for promoter methylations and smoking [7].
Circulating tumor DNA as detected using KRAS mutation has clinical utility in PanCa patients. KRAS somatic mutations (detected at a rate of 29.2 %) and somatic copy number alterations and gene amplifications were detectable by targeted deep sequencing and other genetic approaches [15]. The prognostic relevance of KRAS mutation detection in ctDNA is being established. Codon 12 mutations at G12V, G12D, and G12R were detected in 37.3 %, 29.3 %, and 8.0 %, respectively, in tumor tissues, and at 34.6 %, 38.6 %, and 5.3 %, respectively, in corresponding ctDNA, with a concordance of 77.3 % [16]. While KRAS mutations in tissue samples had no prognostic prediction, patients with circulating KRAS mutations had significantly shorter survival. Earl et al. arrived at a similar conclusion [17]. While the overall detection rate of KRAS mutations in circulation was low (26 %), OS was 60 days for KRAS mutation-positive compared to 772 days for KRAS mutation-negative patients. CTCs detected by CELLSEARCH® system were positive in 20 % of these patients and was equally associated with poor survival. However, compared to ctDNA that was positive in both patients with resectable and advanced diseases, CTCs were mostly detected in patients with metastatic disease.
Detection of other genetic alterations in ctDNA has diagnostic and prognostic associations. A next-generation sequencing approach enabled detection of pancreatobiliary tumor mutations in 90.3 % of ccfDNA that achieved diagnostic sensitivity, specificity, and accuracy of 92.3 %, 100 %, and 97.7 %, respectively. Importantly, these mutations can be detected without the need for knowledge of tumor genotype [18]. A whole gene sequencing of tumor tissue uncovered mutations in MLL, MLL2, MLL3, and ARID1A in 20 % of patients. As an early detection biomarker, 43 % of patients with localized tumors had detectable ctDNA at diagnosis. The presence of ctDNA after tumor resection was associated with poor outcome, and relapse, which was detectable 6 months before imaging detection [19].
9.5.4 Circulating PanCa Coding RNA Biomarkers
The circulating transcriptome has not been a focus of PanCa studies. However, Harsha et al. catalogued extensive expression data on pancreatic ductal adenocarcinoma (PDAC) and were able to identify 2516 differentially expressed genes between cancer and controls. Of these, 930 were detectable in body fluids, and 162 encode secreted proteins. [20]. The largest digital gene expression study of PanCa used serial analysis of gene expression coupled with massively parallel sequencing by synthesis. Five hundred and forty-one (541) genes showed differential expression over tenfold in >90 % of PDAC compared to normal controls. A good proportion of these genes also encode secreted proteins, including S100P that is already implicated in PDAC [21]. The findings from these expressional studies suggest the needed effort to identify the best candidates that can perform optimally for development of a noninvasive screening test.
9.5.5 Circulating PanCa Noncoding RNA Biomarkers
9.5.5.1 Functional Role of PanCa miRNAs
Oncomirs and tumor suppressormirs have been identified in PanCa. Pancreatic cancer oncomirs, which are upregulated in tissue samples, include miR-27a, miR-132, miR-155, miR-194, miR-200b, miR-212, miR-214, miR-220c, miR-310a, miR-421, miR-429, and miR-483-3p. The RB protein controls the cell cycle by sequestering E2F transcription factor. Retinoblastoma is a target of miR-132 and miR-212. By degrading RB, these miRNAs permit uncontrolled cell cycle progression in PDAC cells. Oncogenic PanCa miR-421 and miR-483-3p target SMAD4/DPC4, an inhibitor of cell growth receptor tyrosine kinase. MiR-27a decreases expression of SPRY2 leading to increased PDAC cell growth and migration. The EP300 protein suppresses tumor growth and metastasis. MiR-194, miR-200b, miR-220c, and miR-429 are highly expressed in metastatic than nonmetastatic PDAC cells, and EP300 is a target of these PDAC oncomirs. Another overexpressed miRNA in PDAC tissues is miR-155, and this targets and degrades tumor protein 53-induced nuclear protein 1 (TP53INP1) tumor suppressor. The NF-κβ pathway is activated by miR-310a via its repressive activity on NF-κβ-repressing factor (NKRF) in PDAC. As a molecular amplification positive feedback loop, activated NF-κβ signaling induces expression of miR-310a. Finally, miR-214 reduces sensitivity of PDAC cells to gemcitabine and also targets the inhibitor of growth protein 4 (ING4).
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


