(1)
Medical Sciences Division Northern Ontario School of Medicine West Campus, Lakehead University, Thunder Bay, Ontario, Canada
Key Topics
Colorectal cancer (CRC) screening
Molecular pathology of CRC
Circulating CRC biomarkers
Circulating SEPT9 methylation as CRC biomarker
Circulating CRC miRNA biomarkers
Circulating CRC cells
Key Points
CRC remains a disease of importance, being among the most commonly diagnosed adult cancers, and a major cause of cancer mortality due to late diagnosis. The dismal 8 % 5-year survival rate can be improved to even 100 % with detection of adenomas likely to progress.
As one of the major cancers with well-charted molecular pathologic progression model, there are valid biomarkers for use in clinical practice. These include mutations in KRAS, BRAF, and APC, as well as microsatellite alterations.
Noteworthy, these actionable biomarkers are detectable and measurable in body fluids and are paving the way for noninvasive CRC management. Additionally, novel circulating biomarkers such as miRNAs, protein/peptides, and circulating CRC cells should augment future management of CRC patients.
7.1 Introduction
Colorectal cancer (CRC) remains a challenge in oncology. First, they remain the most commonly diagnosed cancers, and second, they are the fourth cause of cancer-related mortality worldwide, in spite of the wealth of molecular genetic knowledge on this cancer. The estimated number diagnosed in 2012 was 1.36 million, and as many as 694,000 deaths were recorded for the same year. In the US alone, an estimated 134,490 people will be diagnosed, with 49,190 deaths in 2016. Whereas there are wide geographic variations in CRC incidences, the highest rate occurs in Australia and New Zealand, with the lowest in West Africa. However, the incidence is on the rise globally, and this could partly be accounted for by increased awareness and intensive screening efforts, especially in the more developed world. There are equally geographic variations in mortality rates, with the highest in Central and Eastern Europe and the lowest in West Africa.
The vast majority (up to 85 %) of CRCs are sporadic with no evidence of hereditary components. The risk factors for acquiring somatic gene alterations leading to the development of CRC include age (mean age at diagnosis is 66 years), consumption of diet rich in red meat and unsaturated fat, excessive alcohol use, sedentary lifestyle, and high-energy input. However, the pathophysiology may include a complex interplay between bioenergetics, inflammation, hormonal actions, and even the gut microbiota. These complex and dynamic interacting factors can cause epigenetic and genetic alterations in a vast majority of cells, creating an enlarged area of damaged mucosa consistent with the concept of field cancerization. Subsequently, clonal selection of a CRC cell to expand, grow, and form overt tumors occurs.
Improved surgical techniques coupled with adjuvant chemotherapies and novel biotherapies are making positive impacts on the 5-year survival rates for patients with CRC. However, this benefit is optimal mostly for patients with localized early stage disease (stage I disease patients have a 95 %, while stage II is 82 %). This survival rate drops to 61 % in patients with regional lymph node spread (stage III) and very dismal (8 %) for stage IV-disease patients with distant metastasis. Yet less than 40 % of all patients are diagnosed with early stage disease. The need for improved early detection and effective therapies is urgent and is actively being pursued.
While mostly sporadic, 15–30 % of CRCs have some hereditary components. Achievements made at delineating the molecular genetic alterations in hereditary CRC have helped in the elucidation of the molecular pathology of sporadic CRCs as well. Multiple genes altered in hereditary CRC and also in sporadic cancers include APC, AXIN2, MLH1, MSH2, MSH6, PMS2, GTBP, LKB1, STK11, MYH, PTEN, BMPR1a, and DPC4. Despite the plethora of authenticated genetic information on CRC, screening recommendations still rely on fecal occult blood test (FOBT), followed by colonoscopy to visualize and obtain biopsy samples for eventual histopathologic diagnosis. FOBT is a noninvasive assay but suffers from accuracy, while colonoscopy is invasive with possible serious complications. Therefore, validated biomarkers in body fluids should enhance CRC early detection and management. Thus, the recently FDA-approved methylated SEPT9 blood test is a step in the right direction that should augment CRC screening efforts.
7.2 Screening Recommendations for CRC
It is a general recommendation that CRC screening begin at age 50 and continue until age 75. However, individuals at higher risk (e.g., people with known hereditary components) than the general population should begin screening at a younger age. Screening of people between ages 76 and 85 should be done based on individual preferences, in consultations with their healthcare provider. There is no recommended screening for people over 85 where the harms outweigh any benefits. Screening should be carried out annually with high sensitive FOBT. Sigmoidoscopy and colonoscopy are offered every 5 and 10 years, respectively. High-risk individuals may need more frequent screening than these recommended timeframes. Newer tests and investigational modalities including fecal immunochemical test (FIT), fecal DNA test, and CT colonography are being considered for possible inclusion in the next recommendation. While laudable, there are compliance issues with CRC screening.
7.3 Molecular Pathology of CRC
Visible outgrowths of the colorectal mucosa (polyps) are very common in middle aged and older people. Although the frequency of polyps in this demographic is as high as >30 %, only <1 % will progress to become cancerous. Pathologically, polyps are classified as nonneoplastic hamartomas (also known as juvenile polyps), hyperplastic polyps, or adenomatous polyps. The majority of CRCs develop from the latter category, which are established precursor lesions. While CRCs may develop in any part of the colorectum, they are more common in the sigmoid colon and rectum (with ~67 % of all cases) than the rest of the large bowel (Fig. 7.1). Grossly, right-sided tumors appear polypoid or fungating exophytic lesions, while the left-sided lesions tend to be annular, constricting the bowel and therefore may present with bowel obstruction or changes in bowel movement such as constipation or diarrhea.
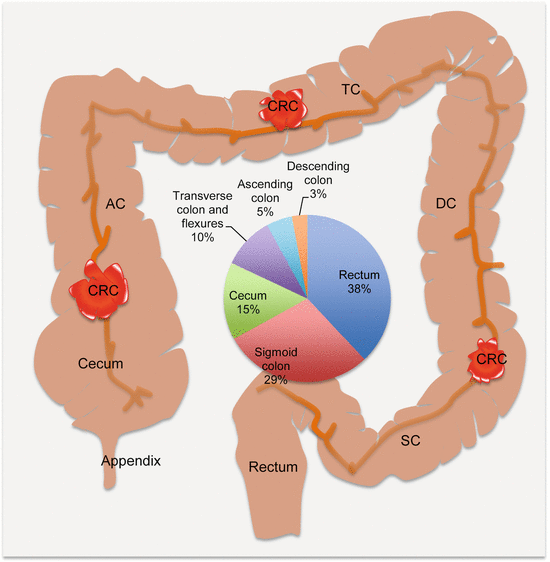
Fig. 7.1
Anatomic locations and frequencies of CRCs. AC ascending colon, TC transverse colon, DC descending colon, SC sigmoid colon
Colorectal cancer is one of the major cancers with well-characterized molecular pathology. The pathogenic mechanisms of hereditary, familial, and sporadic CRCs are initiated and driven by epigenetic and genetic alterations including chromosomal abnormalities involving established oncogenes and tumor suppressor genes. These genetic changes modulate various oncogenic signaling pathways to deregulate normal cellular processes leading to abnormalities in cell proliferation, growth, survival, death, and metabolism. The molecular alterations have given rise to numerous biomarkers for CRC diagnosis, prognosis, and therapy selection. Clinically available biomarkers of CRC include predictive KRAS codons 12 and 13 and BRAF V600E mutation testing, as well as microsatellite instability (MSI) analysis for disease prognostication. Numerous early stage biomarkers are at various phases of validation.
Colorectal cancers are classified genetically into three subtypes, namely, sporadic, familial, and hereditary CRCs. Sporadic cases occur in individuals with no family history of CRC or any evidence of hereditary (germline) CRC gene mutations. Familial cases occur in people who have at least one first- or second-degree relative, who has been diagnosed with CRC or adenoma but lack any germline mutations or clear evidence of Mendelian segregation. True hereditary CRC, however, by definition afflicts individuals who inherit germline mutations in cancer susceptibility genes, such as APC and MLH1.
Germline and somatic mutations, as well as chromosomal aberrations, underlie colorectal carcinogenesis. The vast majority (up to 90 %) of CRCs involve somatic gene mutations (or familial components without detectable germline mutations), while ~15 % harbor some hereditary component, meaning involvement of first- or second-degree relatives with evidence of germline mutations. Many of those with possible hereditary disease have hereditary nonpolyposis CRC (HNPCC) or Lynch syndrome and familial adenomatous polyposis (FAPs), with the rest being associated with other familial cancer syndromes where CRC is a component of the syndrome. While in the minority, knowledge about hereditary CRCs has provided direction for the study and uncovering of the numerous genetic alterations in sporadic CRCs.
HNPCC occurs in people with mutations in mismatch repair (MMR) genes leading to MSI that affects many tumor susceptibility genes. Cancers are mostly located at the right side (~70 % are proximal to the splenic flexure). The mean age at cancer diagnosis is 45. FAPs evolve in people with germline APC mutations. It is characterized by hundreds of adenomatous polyps. Less than 1 % will progress to develop CRC. “Attenuated FAPs” are associated with fewer polyps. Hamartomatous polyposis syndromes such as Peutz–Jeghers, juvenile polyposis, Cronkhite–Canada, and Cowden disease, among others, are associated with elevated risk of CRC.
7.3.1 Molecular Pathology of Multistep Colorectal Carcinogenesis
Colorectal cancer develops in a sequential fashion, with evidence of field cancerization of the colorectal epithelium. Many small polyps (<5 mm in size) are often hyperplastic polyps, which are not major precursors of CRC. However, adenomatous polyps or adenomas can progress to invasive cancers. These adenomas originate from glandular epithelium and are dysplastic lesions with abnormal epithelial differentiation. Their prevalence is about 25 % by the age of 50 but increases to about 59 % by age 70. Adenomas can be prevented from progression by surgical extirpation or polypectomy. Large or advanced adenomas harbor foci of carcinomas, and CRCs tend to have adenomatous lesions within them on histopathologic examination. The genetic condition, FAP, increases the risk of adenomas progressing to CRC, often occurring in early ages (third to fourth decades) if prophylactic colectomy is not performed.
Epigenetic and genetic defects drive adenomas toward overt cancers. The genetic events drive transformation of normal epithelia into benign neoplastic lesions (adenomas), then into invasive cancers, and finally into metastatic cancers with adverse outcomes. The genetic events involved in these steps were initially uncovered and proposed by Vogelstein’s group and has been referred to as the classic CRC progression model. This model pertains to progression of tubular and tubulovillous adenomas. With further elucidation of the molecular progression of CRC, alternative pathways have been uncovered. Serrated polyps (both sessile and traditional serrated polyps) can progress to invasive cancers (these occur primarily in the proximal colon).
The placement of genes in the sequence is not invariant but reflects the frequencies of events at the specific step in tumor evolution. Of note, all the alterations described in the sequence are harbored by only a few CRCs, but at any step in the sequence, the allotted specific genetic event is detectable in many CRCs at a high frequency at that stage, enabling the placement of the gene in that order. The classic tubular adenoma pathway is molecularly characterized by tumors with biallelic loss of APC, followed by KRAS mutations, loss of TP53, and often harbors chromosomal instability (CIN). The serrated adenoma pathway on the other hand displays MSI, CpG island methylator phenotype (CIMP), and BRAF V600E mutations. Tumor progression is further augmented by inactivation of genes with repetitive elements (MSI sites) in their coding sequences (e.g., BAX, ACVR2, and TGFβR2). Figure 7.2a–c summarizes the knowledge of these events in the different pathways. This knowledge is very important as it reflects on biomarker detection in body fluids used for cancer management.
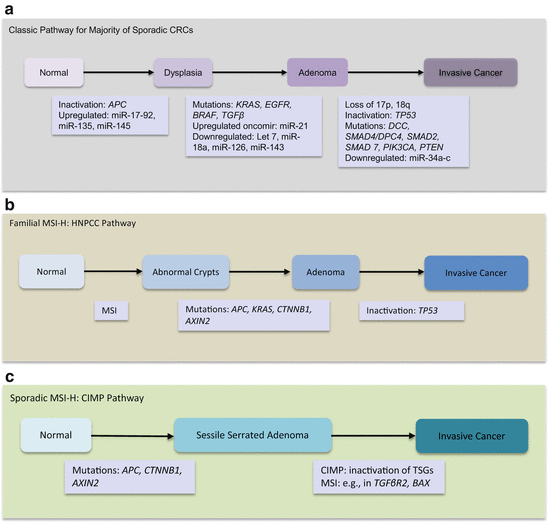
Fig. 7.2
Multistep CRC progression models. (a) The classical pathway involving majority of CRCs. (b) Familial MSI-H HNPCC pathway. (c) Sporadic MSI-H/CIMP pathway
7.3.2 Epigenetic Alterations in CRC
When compared to normal colonocytes, CRC and adenomas exhibit generalized CpG hypomethylation, which is tightly associated with CIN tumors. This general finding contrasts with CpG islands at promoter regions that are often hypermethylated leading to gene silencing. Indeed, many CRC cells exhibit promoter hypermethylation of tumor suppressor genes such as the WNT/β-catenin pathway antagonist, SFRP, and hypermethylated in cancer 1 (HIC-1). CpG island methylator phenotype (CIMP), while without consensus definition, involves the methylation of multiple gene loci in a particular tumor. The common definition used is promoter hypermethylation of at least three loci from a panel of five markers. Making it even more worrisome is the lack of standardized markers, thus hindering study comparison. A group of sporadic CRCs with CIMP harbors hypermethylation of mismatch repair (MMR) genes (e.g., MLH1) constituting the sporadic MSI-H category. This group of tumors harbors BRAF mutations and histopathologically are sessile serrated adenomas.
7.3.3 Genetic Alterations in CRC
7.3.3.1 Chromosomal Alterations in CRC
CIN in CRC is characterized by both numerical (aneuploidy) and structural chromosomal changes. Chromosomal changes in CRC include losses at 5q, 8p, 17p, and 18q and gains at 8q, 13q, and 20q. MSI-H tumors harbor very few or no allelic losses (about 15 % of CRCs without CIN are MSI-H tumors). While the role of CIN in CRC progression is unclear, it may be involved in establishing clonal cell diversity. However, the presence of CIN is a hallmark of poor prognosis. Loss of 18q is demonstrated in ~70 % of CRCs, with frequency distribution associated with tumor size. About half of all large and late-state adenomas harbor deletions, while these are present in only 10 % of small early stage adenomas. This 18q locus harbors important genes implicated in CRC, especially SMAD4, as well as SMAD2 and SMAD7 involved in the TGFβ pathway. Also at this locus is deleted in colorectal cancer (DCC), a netrin 1 receptor involved in axon guidance. Often, DCC is silenced either by epigenetic mechanisms or loss of 18q21. Credible data suggests DCC is a tumor suppressor. Loss of DCC is not involved in tumor initiation but has been suggested to promote CRC growth once the tumor is established. The predictive relevance of 18q loss in CRC is under clinical trial NCT00217737 (ECOG 5202 study).
7.3.3.2 Microsatellite Alterations in CRC
Mononucleotide and/or dinucleotide tract MSI accounts for ~15 % of all CRCs. The NCI consensus markers BAT-25, BAT-26, NR-21, NR-24, MONO-27 (these are selected because of their more accurate performances in CRC detection) have been used for CRC MSI detection. MSI was first demonstrated in patients with HNPCC, occurring in all tumors from these patients. Tumors that have gene sequences characterized by >40 % mononucleotide or dinucleotide unstable repeats are classified as MSI-H (or simply MSI+), and tumors without MSI loci are designated as microsatellite stable (MSS). Some tumors, however, harbor 10–29 % unstable MS loci, and these are often designated as MSI-low (MSI-L). While MSI-L tumors have distinct features from MSI-H and MSS tumors, it is not generally accepted whether these are distinct classes of tumors.
In HNPCC, the MMR genes MSH and MLH1 are mutated in ~70 % of tumors. This accounts for the MSI-H phenotype. In sporadic CRCs, loss of MLH1 expression through promoter hypermethylation accounts for MSI-H tumors. MSI-L tumors have no MMR gene mutations (mechanism unclear). Arguably, MSI-L tumors may not differ from MSS tumors. MSI-H tumors have more favorable prognosis than CIN tumors, and MSI-H and MSS tumors have distinct responses to chemotherapy. Sporadic MSI-H tumors progress via the serrated adenoma pathway and often harbor the BRAF V600E mutation. In contrast, for people with germline MMR gene mutations (as in HNPCC/Lynch syndrome) and MSI-H tumors, disease progression may be determined by KRAS but not BRAF mutations.
7.3.4 Important Signaling Pathways in CRC
7.3.4.1 WNT/β-Catenin Pathway Alterations in CRC
APC mutations are etiologic factors in familial adenomatous polyposis (FAP) predisposition to cancer. Over 95 % of APC mutations in FAP are frameshift or nonsense mutations (more frameshift than nonsense mutations at a ratio of 3:1). These germline alterations are mostly clustered at the 5′ end of the gene, with codons 1061 and 1309 accounting for ~35 % of all mutations. APC mutations are early events in 70–80 % of sporadic colorectal adenomas and cancer. Almost all somatic inactivating mutations cause premature truncation of APC. These somatic mutations cluster at codons 1309 and 1450 and are found in early adenomas and may be the drivers of adenoma formation. Apparently, both alleles are inactivated in colorectal neoplastic lesions in both FAP and sporadic colorectal adenomas and cancers.
A number of CRCs without APC mutations harbor mutations in other members of the WNT/β-catenin pathway. For example, activating mutations in CTNNB1 (encodes β-catenin) are more associated with colorectal adenomas (12.5 %) than carcinomas (1.4 %). These somatic mutations affecting critical amino acids in the N-terminal phosphorylation and ubiquitination motifs disrupt APC-mediated degradation of β-catenin. Mutations in AXIN1 and AXIN2 characterize many CRCs, although their relevance is yet to be determined. AXIN1 mutations are predominantly missense substitutions, while AXIN2 tends to be premature truncating mutations leading to loss of the C-terminal domain. TCF4 inactivating mutations are found in some CRCs as well.
7.3.4.2 PI3K/AKT Pathway Alterations in CRC
Members of the PI3K/AKT pathway are mutated in >40 % of CRCs. Most frequently affected gene is PIK3CA. Specifically, mutations occur in the p110α catalytic subunit. These mutations promote adenoma progression to carcinoma and are found in up to 32 % of CRCs. The negative regulator of the PI3K pathway, PTEN, is mutated in 10 % of CRCs, but PTEN protein expression is lost in 15–20 % of CRCs. Mutations are found in ~30 % of MSI-H and in 9 % of CIN CRCs. PIK3CA and PTEN mutations may have predictive utility in anti-EGFR therapy, because of the cross talk between the EGFR and PI3K pathways.
7.3.4.3 TGFβ Pathway Alterations in CRC
The TGFβ pathway is deregulated in a vast majority of CRCs. Inactivating mutations are observed in receptor genes and intracellular pathway components. Importantly, loss of SMAD4 occurs in ~50 % of CRCs and is implicated in early stages of CRC development. Thus, it mediates the development of adenomas and drives adenoma progression to carcinoma. Loss of SMAD4 correlates with lymph node involvement and demonstrates both prognostic and predictive relevance. Other members of the SMAD family members are altered as well. SMAD2, also located on 18q, is lost in about ~5 % of CRCs, while SMAD3, located on chromosome 15, is inactivated in similar proportions of CRC. Risk alleles (e.g., SNP rs7229639) are putatively found in the SMAD7 locus (18q21). Inactivating mutations in TGFβR2 occur in as many as 30 % of CRCs. Functional studies indicate TGFβR2 mutations drive late adenomas to invasive cancer. While mutations occur in MSI-H cancers, they are observed in only ~15 % of MSS tumors. The coding region of TGFβR2 has a long mononucleotide tract of adenines that are destabilized by biallelic insertions or deletions in a vast majority (>90 %) of MSH-H tumors. Similarly, ACVR2A that encodes a type II pathway receptor is somatically mutated at a polyA repeat in exon 10 in ~85 % of MSI-H tumors and is associated with poor prognosis.
7.3.4.4 TP53 Pathway Alterations in CRC
TP53 shows LOH in ~70 % of CRCs. Mutations in the other allele, observed in many CRCs, lead to TP53 inactivation. Many (~85 %) of the mutations are missense, affecting primarily codons 175, 245, 248, 273, and 282. Nonsense and frameshift mutations are also observed. CRCs without chromosome 17 LOH tend to have mutations affecting both alleles. While mutations are rare in adenomas, they are often observed in later stages in the adenoma–carcinoma sequence. Evidence suggests that TP53 loss drives adenoma to carcinomas because the occurrence of these mutations followed by LOH of the other wild-type allele coincides with the development of invasive cancers from adenomas.
7.3.4.5 MAPK Pathway Alterations in CRC
Mutations in KRAS occur in about 40 % of CRCs. The most frequent hotspots are codons 12 and 13. While these mutations are early events in CRC adenoma–carcinoma sequence, they are preceded by APC mutations. The early mutations are maintained throughout tumor progression as they are easily observed in metastatic deposits from the primary tumor. This is important in metastatic CRC patient management where subsequent samples are needed for KRAS mutation analysis, because other samples such as body fluids or even archived material will suffice such analysis. KRAS mutations also occur at the same frequency (40 %) in adenomas, but the frequency depends on the size of the adenomas. About 58 % of adenomas >1 cm in size harbor KRAS mutations, which is similar to the frequency of mutations found in focal adenomas associated with invasive cancers. This finding contrasts with the frequency of 9 % observed in small adenomas <1 cm. Because of their increase in frequency in advanced adenomas, KRAS mutations appear to drive CRC growth. Another important gene mutated in CRC is BRAF. This gene is mutated in 10–15 % of CRCs. While other mutations occur, the majority is the observed codon 600 single base substitution of glutamic acid for valine (BRAF V600E ). Similar to other cancers (e.g., melanoma and thyroid cancer), BRAF mutations are mutually exclusive to KRAS mutations, in support of their dominance or sufficiency in activating the EGF pathway. These mutations occur more often in sporadic MSI-H and CIMP tumors and drive the sessile serrated neoplastic pathway of multistep colorectal carcinogenesis (Fig. 7.2). Consistently, ~50 % of MSI cancers have BRAF mutations compared to 5 % of MSS tumors. However, their clinical relevance in CRC risk stratification, prognosis, and treatment predictions is under investigation.
7.4 Circulating CRC Biomarkers
Biomarkers in circulation and stool have been extensively investigated as noninvasive approach to CRC management. While several of these biomarkers remain to be validated for their intended uses, a few including Epi proColon have been launched for CRC risk assessment and diagnosis.
7.4.1 Circulating Cell-Free Nucleic Acid Content as CRC Biomarkers
Multiple studies confirm the value of ccfDNA in the clinical management of CRC patients [1–3]. While not in routine clinical practice, the complementary utility with other serum biomarkers in CRC detection is compelling needing coordinated validation studies.
Serum DII of ALU repeats was much significantly higher in stage I/II (p = 0.002) and III/IV (p = 0.006) CRC patients than healthy controls. Accuracy as a diagnostic revealed an AUROCC of 0.79 [4]. At least this assay shows potential for CRC detection in a noninvasive manner. Mouliere et al. used atomic force microscopy to visualize ccfDNA in plasma from patients and controls and concluded that the majority of ccfDNA (>80 %) in CRC patients are below 145 bp in size [2]. Using allele-specific blocker qPCR, this group developed a method that could target the smaller ccfDNA fragments simultaneously at multiple levels (concentration, DII, point mutations, and proportion of mutant alleles). This assay provides a powerful noninvasive analysis of ctDNA in cancer patients. Thus, DII defined by ALU247/115 ratio has uncovered a diagnostic potential for ccfDNA in CRC patients [3]. The median absolute serum levels of ALU115 and DII were significantly higher in primary CRC patients than those with polyps and healthy controls (p < 0.0001). Similarly, patients with metastatic and recurrent diseases harbored significantly much higher levels than those with primary CRC. The preoperative elevated levels decreased postoperatively. These biomarkers were complementary to serum CEA in CRC detection. Yoruker et al. provide corroborative and confirmatory evidence for the high degree of fragmented DNA in circulation of CRC patients [1]. Two DIIs (ACTB384/106 and ALU247/115) were assessed, and data correlated with circulating nucleosomes. While DII differed between cancer patients and controls, shorter DNA fragments correlated more with circulating nucleosomes, suggesting the presence of highly fragmented nucleosomes (mononucleosomes) in CRC patients.
7.4.2 Circulating CRC Epigenetic Biomarkers
Exploration of epigenetic changes in circulation of CRC patients has mostly focused on known cancer susceptibility genes including CDKN2A, APC, and KRAS and MMR genes such as hMLH1. While the clinical importance of circulating promoter hypermethylation of CDKN2A is consistent, the reported frequencies of detection vary due to different detection techniques and methodological sensitivities. Promoter methylation of CDKN2A in serum as diagnostic or predictive biomarker has been investigated in CRC tissues and matched sera. CDKN2A was methylated in 38 % of CRC tissue samples. Serum from 70 % of the positive cases also harbored methylation, and this methylation status was associated with late Dukes’ stage. Another series uncovered CDKN2A methylation in 47 % of cancer tissue samples and 30 % of matched sera. In this study, however, methylation in circulation was stage independent, suggestive of its possible use in early detection [5]. Another study by this group found serum CDKN2A promoter methylation could detect 69 % of patients with recurrent disease [6]. Later, this group used an MSP to generate numerical methylation scores for circulating CDKN2A in CRC patients. The score showed significant correlation with tumor stage, increasing from stage I to IV. Additionally, high scores were significantly associated with lymph node involvement, intravascular invasion, and poor survival [7]. They subsequently developed a more sensitive method for methylation detection in serum, referred to as limiting dilution MSP (LD-MSP). This new assay increased methylation detection sensitivity up to tenfold. Thus, the initial sensitivity of 30 % by MSP was enhanced by the LD-MSP method to 68 % coverage [8]. Further clinical utility of CDKN2A methylation in serum a day before surgery, and on follow-up after surgery as a prognostic biomarker, was investigated. Methylation was demonstrated in eight serum samples out of 13 positive tumors and decreased 2 weeks after surgery (two patients with potential for recurrence retained serum methylation). Recurrence a month after surgery was associated with dramatic increases in serum CDKN2A methylation. [9]. The prognostic value of ccfDNA in CRC was questioned using dual makers, KRAS codons 12 and 13 mutations and CDKN2A promoter hypermethylation [10]. KRAS was mutated in 38 % of tumors and in 45 % of corresponding plasma samples. The methylation of CDKN2A was in 53 % of tumors and 68 % of corresponding plasma samples. An alteration of one or both genes was in 39 tumor samples, of which 37 plasma samples were available, and 70 % of these samples were positive for gene alterations. The 2-year survival was significantly reduced in patients with ctDNA compared to those without, and this prognostic relevance included risk of tumor recurrence.
Other relevant genes with promoter methylation in CRC include MLH1, HLTF, APC, HPP1, THBD, and DAPK. The hMLH1 and HLTF genes have been examined in circulation of CRC patients with potential clinical relevance. Pretreatment sera from CRC patients and controls were assayed for methylation in APC, hMLH1, and HLTF. The serum concentrations of methylated HLTF and hMLH1 significantly differed between cancer and controls (but not for APC). Methylation in at least one of these genes gave a sensitivity of 57 % and specificity of 90 % in CRC detection. Methylation in two markers occurred in patients with advanced stage disease [11]. Promoter hypermethylation and gene silencing of hMLH1 are associated with sporadic microsatellite unstable CRCs. Detection of this methylation in serum achieved a sensitivity of 33 % and a specificity of 100 % for diagnosis of CRCs with MSI [12]. In an evaluation set of sera from controls and patients with localized and metastatic cancer, HPP1/TPEF, HLTF, and hMLH1 were identified as useful serum prognostic biomarkers. These genes were then tested on sera from 104 CRC patients. Methylation of the three genes significantly correlated with tumor size. But methylation of HPP1/TPEF and HLTF was significantly associated with metastasis and tumor stage. Examination of pretreatment sera from 77 patients revealed that methylation of HPP1/TPEF and HLTF conferred poor prognosis. Multivariate analysis identified methylation of these two genes in serum to be independent predictors of poor outcome with an RR of death of 3.4 [13].
Methylation of other genes including DAPK, THBD, and C9orf50 has been assayed in circulation of CRC patients. DAPK methylation frequency was determined in CRC and paired serum samples. Methylation was in 55 % of primary tumors, and positive signal was in 21 % of serum samples [14]. Genome-scale CRC methylation biomarker discovery approach was used on pretreatment plasma/serum from 107 CRC patients and 98 controls to verify selected candidate markers. THBD and C9orf50 were eventually selected for analysis, and both outperformed CEA with high sensitivity and specificity for CRC detection. But THBD hypermethylation showed much promise in clinical samples warranting further studies [15].
7.4.2.1 Circulating SEPT9 Methylation as CRC Biomarker
A gene that has been well studied for noninvasive screening of CRC is the methylation of circulating SEPT9 gene. Septin 9 (SEPT9) located on chromosome 17q25 is a member of the evolutionary conserved septin gene family. Hartwell’s work on cell division led to the identification of the septins in yeast. These genes are highly conserved in eukaryotes. They are GTP-binding proteins involved in cytokinesis and also form part of the cytoskeleton. Additionally, septins are involved in cytoskeletal, microtubule, and chromosomal dynamics, vesicle trafficking, cell motility, exocytosis, phagocytosis, cell polarity, apoptosis, spermatogenesis, and platelet functions.
Septins are implicated in neurodegenerative diseases and cancer. They are found associated with protein complexes in Parkinson and Alzheimer’s diseases. They are also observed as mixed lineage leukemia (MLL) fusion proteins involved in leukemogenesis. Mutations in SEPT9, especially Arg88Trp (R88W) and gene duplications, are associated with hereditary neuralgic amyotrophy (muscle wasting disease with episodes of severe pains). Also, altered expression is associated with several cancers, including cancers of the thyroid, lung, breast, esophagus, liver, pancreas, kidneys, and ovaries. But promoter hypermethylation is specifically associated with CRC and is measurable in plasma and serum for diagnostic purposes.
Stringent sieving criteria involving multiple selection steps in tissue and normal body fluids for analysis enabled three methylated genes to be identified as good candidates for CRC detection in plasma samples. Promoter methylations of these genes (TMEFF2, NGFR, and SEPT9) were tested in 133 CRC and 179 healthy controls. The three genes performed independently at a sensitivity and specificity of 65 % and 69 % for TMEFF2, 51 % and 84 % for NGFR, and 69 % and 86 % for SEPT9 [16]. Two large-scale case–control studies of methylated SEPT9 DNA (mSEPT9) in plasma performed later yielded a detection rate of 48 % of CRCs with positive detection in 7 % of the control population in the training set. Blinded validation confirmed this result with sensitivity of 58 % for CRC detection and 10 % in control population. The assay performed at increased sensitivity of 72 % and specificity of 90 % when replicate additional measurements were incorporated. Polyps were detected at a rate of 20 % [17]. An improved mSEPT9 assay was developed by deVos et al., and this performed at a sensitivity of 72 % and specificity of 93 % in the training set with performance being identical (sensitivity of 68 % and specificity of 89 %) in the test or validation cohort [18].
Another group demonstrated the potential use of mSEPT9 for screening precancerous lesions [19]. Methylation of SEPT9 alone and in combination with ALX4 was investigated in plasma samples. Methylated SEPT9 was present in 73 % and 29 % of plasma samples from patients with CRC and precancerous lesions, respectively, and 9 % of the control group. When ALX4 methylation was included, the detection rate of CRC (60 %) and in controls (5 %) was similar to only mSEPT9 but that of precancerous lesions improved to 37 %. A 2011 study using the improved mSEPT9 assay increased the sensitivity to 90 % at a specificity of 88 % for CRC detection [20]. This assay identified early stage disease at a rate of 87 %. Cancers of all stages and locations were detected. Twelve percent of adenomas were also detected in a prospective study. A Hungarian study confirmed Warren et al.’s work that detection is unlimited to anatomic site of the cancer. They compared mSEPT9 to conventional gFOBT and serum CEA assays. Methylated SEPT9 outperformed both conventional tests. The mSEPT9 assay achieved a sensitivity of 95.6 % in detecting stage II–IV cancers and 84 % of stage I. Both left-sided (96.4 %) and right-sided (94.4 %) cancers were detected at the same rate [21].
The PRESPT clinical study evaluated the utility of mSEPT9 in plasma for CRC screening in asymptomatic targeted (50+) population [22]. This multicenter (32 centers) US and German study included 7941 participants of which 53 developed CRC. Samples were blindly assayed in three laboratories with primary focus on sensitivity and specificity for CRC and adenoma detection. This study yielded a sensitivity of 48.2 % for stages I–IV cancers and 11.2 % for advanced adenomas. Specificity was however as high as 91.5 %. Because of the low sensitivity, especially for early stage disease and precancerous lesions, this assay requires some improvement prior to clinical use as a screening tool for targeted population. However, because of the multiple successful case–control and clinical trial studies, this assay (known as Epi proColon) has been approved by the FDA for CRC screening in the average-risk (50–85 year) population.
7.4.3 Circulating CRC Genetic Biomarkers
There are established genetic alterations in CRC initiation and progression. Some of these genes, notably KRAS, APC, CDKN2A, and TP53, have been explored in circulation of cancer patients for possible noninvasive translation.
Kopreski et al. first demonstrated the clinical potential of circulating KRAS mutations in CRC patients [23]. They detected mutant KRAS in plasma or serum from patients with advanced stage disease. To ascertain its potential clinical utility, a high concordance (83 %) between KRAS codon 12 mutations in ccfDNA and tissue samples from CRC patients was later reported in a large study [24]. KRAS mutations were detected in plasma of some individuals without colonoscopic evidence of cancer. However, these individuals had high-risk indices for developing CRC, suggesting its relevance as an early risk prediction biomarker. Mutations detected in plasma, coupled with CA19-9 assay, enabled a detection sensitivity of 90 % for CRC. For adenomas, KRAS mutations had a low detection sensitivity of 35 % [24].
The prognostic role of KRAS (codons 12 and 13) mutations and CDKN2A promoter hypermethylation in plasma was examined by Lecomte et al. using mutant allele-specific amplification and MSP [10]. KRAS mutations were detected at a rate of 45 %, while CDKN2A hypermethylation was observed in 68 % of plasma samples. Of the 39 tissue samples assayed in this study that showed either one or both alterations, 37 plasma samples (~95 %) were informative as well. Seventy percent of these samples showed tumor-specific alterations in plasma. Circulating tumor DNA correlated with poor OS and recurrence-free survival.
Circulating KRAS mutations as prediction of disease relapse have been explored with important clinical implications. The relevance of persistent KRAS mutation in circulation in postoperative patients was noted by Lindforss et al. [25]. Plasma KRAS mutation could be detected in pre- and early post-operative samples in all stages of CRC. While persistent circulating mutant KRAS was not associated with recurrence in this series, the authors still suggested its possible use for postoperative follow-up and early detection of recurrences. Thus, Frattini et al. targeted recurrences on follow-up, using ccfDNA, which decreased in tumor-free patients after surgery but remained elevated in those with recurrences or metastatic disease [26]. This data was supported by the detection of KRAS mutations and CDKN2A promoter hypermethylation in these samples as well. This group later examined KRAS mutations and CDKN2A promoter hypermethylation in primary CRC and matched multiple plasma samples at time of surgery and follow-up [27]. Expectedly, total plasma DNA levels were higher in CRC patient samples than in controls. In tumors with KRAS mutations and CDKN2A promoter hypermethylation, similar findings were observed in their plasma. Plasma DNA levels decreased after surgery but were high in patients with recurrences or relapse. Additionally, KRAS and CDKN2A alterations were associated with relapse following surgery. This study suggests combined qualitative and quantitative analysis for CRC diagnosis, monitoring for disease-free status, and possible relapse after surgery has potential. There is known acquired resistance to anti-EGFR antibodies during treatment of patients with wild-type KRAS genotype. KRAS mutation screening of sera from CRC patients on panitumumab (anti-EGFR antibody) monotherapy revealed that 38 % of patients with initial tumor wild-type KRAS developed KRAS mutations that were detectable in serum samples 5–6 months after treatment. But, mathematical modeling indicated these subclones harbored those mutations prior to the start of therapy [28]. This finding suggests the need for analytically sensitive technologies to uncover such clones prior to therapy.
Gocke et al. first detected mutations in APC and TP53 in plasma from patients with CRC and adenomas [29]. Hsieh et al. focused their examination on mutations in genes involved with the progressive model of CRC, i.e., APC, KRAS, and TP53 in 118 CRC patients and related these to prognostic parameters [30]. Mutations in APC and TP53 correlated with lymph node metastasis and TNM stage. Positive presence of at least one mutation was associated with depth of tumor invasion, lymph node metastasis, and TNM stage. These biomarkers were also related to postoperative metastasis. Mutations in TP53 were detected at a frequency of 14 % in patient sera using fluorescence-based PCR and SSCP assays. However, these circulating TP53 mutations were associated with advanced stage tumors with liver metastasis [31]. In another series, patients with advanced stage CRC had detectable mutant APC molecules in plasma (median APC molecules in plasma were 47,800/ml of plasma with 8 % being mutant molecules). In over 60 % of early disease states, mutant APC was found at lower levels of 0.01–1.7 % of total APC molecules, suggesting increasing release with progressive disease. Importantly, this study provides evidence of using sensitive technologies for early detection and disease tracking [32]. Diehl et al. compared the detection of mutant DNA molecules in stool and plasma using the same technology (BEAMing) [33]. The detection rate was 92 % in stool compared to 50 % of plasma samples, suggesting stool is superior to plasma for CRC mutation detection. Tumor mutations were mostly in small (150 bp) DNA fragments. The ability to reliably detect tumor mutation in circulating cell-free DNA suggests such DNA measurements are useful for monitoring tumor dynamics following surgery or chemotherapy [33].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


