GENETIC PROFILE OF PANCREATIC CARCINOMA
As one of the most commonly mutated genes in pancreatic cancer, Ras is an attractive target for the development of gene-specific therapies, and an understanding of the normal biology of the Ras protein should help in the development of these Ras targeted therapies. The Ras proteins require an attachment to the plasma membrane for activity. For many proteins, including Ras, a hydrophobic prenyl group is essential for the attachment. Either farnesyl (15-carbon) or geranylgeranyl (20-carbon) makes a covalent thioether linkage at a cysteine residue located near the C-terminal end of Ras proteins, termed the CAAX motif. Working mostly in artificial legacy models of the HRAS oncogene (rather than the more widely available but experimentally less tractable natural KRAS-mutant cancer cell lines), the farnesylation reaction was readily inhibited by various means; in these models, the Ras protein was rendered inactive and often accompanied by cytotoxicity limited to the mutant cells.
Although many types of compounds capable of blocking the farnesyltransferase enzyme were developed as drugs, they have not been successful anticancer agents. There are many reasons for this. Although Hras protein is linked predominantly through farnesyl groups, the Kras protein can be alternately prenylated by geranylgeranyl linkages. Unfortunately, the latter type of linkage is thought to be critical for a wider number of cellular proteins, and for fear of excessive toxicity, geranylgeranyl linkages have not usually been considered as an attractive drug target. Kras protein also appears to bind more tightly than Hras to the farnesyltransferase enzyme, requiring higher drug concentrations.16 Additionally, the artificial models usually employed the engineered overexpression of the Ras protein, a situation in which the unattached Ras proteins would serve as a dominant-negative inhibitor, binding the necessary interacting proteins and sequestering them in the cytoplasm to ensure the inactivation of all three Ras pathways. Such a concentration-driven mechanism would presumably not occur under the normal levels of Ras proteins present in human cancers.20 Indeed, it is proposed that the limited efficacy of farnesyltransferase inhibitors observed in some experimental models and in clinical trials may be attributable to a cellular target not yet identified.21 Attention has turned to compounds that target the downstream mediators, such as Raf and Mek protein kinase inhibitors.
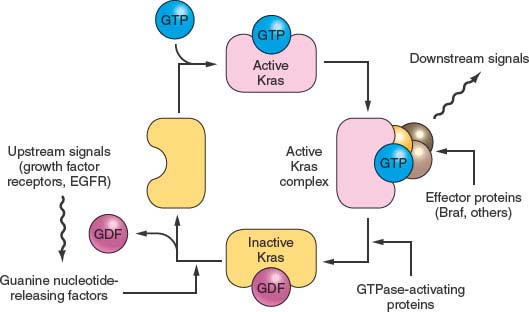
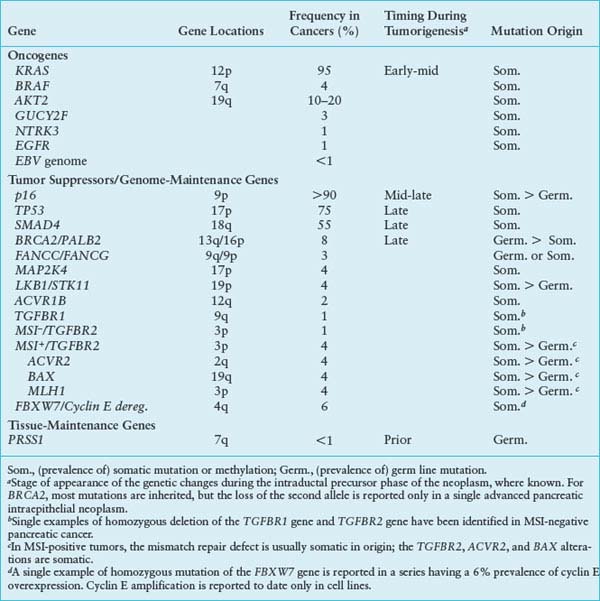
FIGURE 17.1 The KRAS pathway. KRAS normally integrates and regulates signals arising in the growth factor receptors that are passed to KRAS using the Grb2 and the Sos1 nucleotide exchange factor. The active GTP-bound form of KRAS recruits effector proteins such as Raf1 and Braf, in turn stimulating the downstream mitogen-activated protein kinases such as MEK and ERK and activating certain transcription factors. The EGF receptor can be overexpressed and occasionally mutated to provide inappropriately strong upstream signals, and the BRAF protein can be activated by point mutation, but more often in pancreatic cancer the Kras protein is mutated. These latter mutations impair the GAP (GTPase-activating protein)-stimulated reaction that normally returns Kras to the inactive state.
The Smad pathway mediates signals initiated on the binding of the extracellular proteins TGFβ and activin to their receptors (Fig. 17.2). These signals are transmitted to the nucleus by the Smad family of related genes, including SMAD4 (DPC4).22 Smad protein complexes bind specific recognition sites on DNA and cause the transcription of certain genes.23 Mutations in the SMAD4 gene are found in nearly half of pancreatic carcinomas, including both homozygous deletions and intragenic mutations combined with loss of heterozygosity (LOH).24 Other Smad genes are also mutated occasionally.1
Homozygous deletions and mutation/LOH affecting the TGFβ receptor genes are seen in a few pancreatic cancers.25 A more common abnormality, in pancreatic as well as in other tumor types, is the underexpression of TGFβ receptors, which results in cellular resistance to the usual suppressive effects of the TGFβ ligand.26
The p16/RB1 pathway is a key control of the cell division cycle (Fig. 17.3). The retinoblastoma protein (Rb1) is a transcriptional regulator and regulates the entry of cells into S phase. A complex of cyclin D and a cyclin-dependent kinase (Cdk4 and Cdk6) phosphorylates and thereby regulates Rb1. The p16 protein is a Cdk-inhibitor that binds Cdk4 and Cdk6.27–29 Virtually all pancreatic carcinomas suffer a loss of p16 function, through homozygous deletions, mutation/LOH, or promoter methylation of the p16/CDKN2A gene associated with a lack of gene expression.30,31 In addition, inherited mutations of the p16/CDKN2A gene cause a familial melanoma/pancreatic cancer syndrome known as familial atypical multiple mole melanoma.32–36 Occasional pancreatic cancers have inactivating mutations of the RB1 gene.37
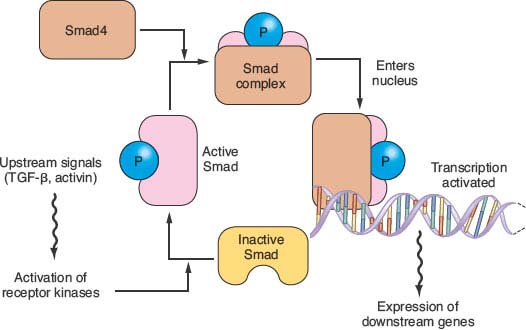
FIGURE 17.2 The transforming growth factor-beta (TGFβ)/Activin/Smad pathway. Dimeric kinase receptors of the TGFβ superfamily respond to extracellular ligands, causing phosphorylation of one or more of the receptor-associated Smad proteins and leading them to complex with the unphosphorylated common Smad, Smad4. This complex binds to specific DNA sequences and works with other transcription factors to stimulate gene expression. Mutations in pancreatic cancer can inactivate either partner of the dimeric receptors that respond to extracellular TGFβ or activin. More commonly, however, mutations and large deletions in the SMAD4 gene destabilize its protein product or ablate gene expression.
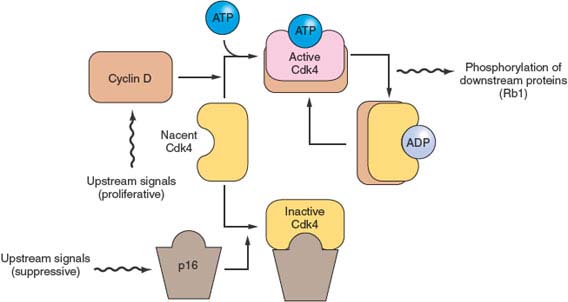
FIGURE 17.3 The p16/RB1 pathway. p16 binds to, inhibits, and thereby controls the availability of the cyclin-dependent kinases Cdk4 and Cdk6 (not shown). When activated by binding to cyclin D, these kinases phosphorylate and thereby inactivate the Rb1 tumor suppressor protein. The activity of p16 is controlled in a complex manner, through changes in gene expression and by displacement reactions involving other similar kinase inhibitor proteins. p16 mutations and deletions are nearly ubiquitous in pancreatic cancer, resulting in dysregulation of these cyclin-dependent kinases that regulate the cell division cycle.
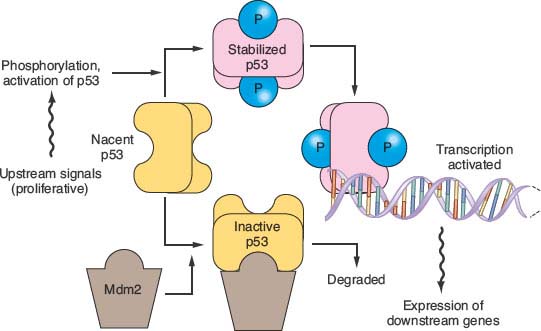
FIGURE 17.4 The p53 pathway. Many modes of control affect p53 activity, one of which is shown in the diagram. Stresses such as DNA damage result in phosphorylation of p53, preventing its degradation by an Mdm2-directed pathway. When stabilized, p53 binds to specific DNA sequences and activates the transcription of many genes, including Mdm2 as part of a negative feedback loop. When p53 is mutated, it fails to bind effectively to DNA to activate transcription. Because Mdm2 then lacks its transcriptional stimulus from p53, mutant but inactive p53 proteins are usually expressed at very high levels.
The protein product of the TP53 gene binds to specific sites of DNA and activates the transcription of certain genes that control the cell division cycle and apoptosis.38,39 Tp53 protein, normally a short-lived protein, becomes phosphorylated and stabilized after DNA damage and other cellular stresses (Fig. 17.4). In about 75% of pancreatic cancers the TP53 gene has point mutations that inhibit the ability of p53 to bind DNA, or occasionally other types of inactivating mutation.40–42
Most human carcinomas have chromosomal instability (CIN), which produces changes in chromosomal copy numbers or aneuploidy.43 Most pancreatic cancers have complex karyotypes including deletions of whole chromosomes and subchromosomal regions.44–46 CIN is the process that causes most of the tumor deletions (LOH).47 A few percent of pancreatic carcinomas, however, do not have significant gross or numerical chromosomal changes and instead have a different form of genetic instability; they have defects in DNA mismatch repair, producing high mutation rates at sites of simple repetitive sequences termed microsatellites.8,9,48–51 The pattern of genetic damage in these carcinomas differs considerably from the pattern in carcinomas with CIN. The type II TGFβ and activin receptors (TGFBR2) and (ACVR2), as well as the BAX gene have a repetitive sequence within their coding regions, and biallelic inactivating mutations of these sequences are seen in many microsatellite instability (MIN) pancreatic cancers.25,51–54
There are also alterations in pancreatic carcinomas, some probably being important to tumorigenesis, that are not attributed to genetic mutations. These include expression of telomerase,13,14 underexpression of TGFβ receptors,26 overexpression of the growth-stimulating HER2/neu cell surface receptor,55–58 a constitutive elevation of RelA and hedgehog pathway activities.55,56 Some of these activities are proposed to be attractive as therapeutic targets, although supportive clinical evidence is not yet available. Pancreatic carcinomas also have reproducible alterations in gene expression, such as overexpression of the proteins mesothelin and prostate stem cell antigen that currently can serve as diagnostic aids in the histopathologic interpretation of biopsies and surgical resections.57–59 The epigenetic patterns of gene hypermethylation in pancreatic cancers are considered promising for developing additional diagnostic markers for analysis of pancreatic secretions and for noninvasive diagnostic screening.59
The dominating genetic patterns previously described are altered among other diagnostic categories of pancreatic neoplasia. The precursor lesions, termed PanIN (pancreatic intraepithelial neoplasia), in their most advanced stage, closely resemble the genetic patterns of the conventional invasive ductal carcinomas. The intraductal papillary mucinous neoplasms, the mucinous cystic neoplasms, acinar cell carcinomas, neuroendocrine carcinomas, pancreatoblastomas, and solid pseudopapillary neoplasms, however, diverge significantly from the patterns of PanINs and typical invasive ductal carcinomas. Notable differences are the PIK3CA mutations present in some intraductal papillary mucinous neoplasms and the colloid carcinomas that can derive from them,60 the CTNNB1 (beta-catenin) mutations present in virtually all solid pseudopapillary neoplasms61 and pancreatoblastomas,62 and the MEN1 mutations in many neuroendocrine neoplasms.63
LOW-FREQUENCY GENETIC CHANGES
The causative genes of Fanconi anemia play a role in human tumorigenesis. The BRCA2 gene represents Fanconi complementation group D1 and is thought to aid DNA strand repair.64 Because of this function, it is perhaps best to categorize BRCA2 as a genome-maintenance gene rather than a standard tumor suppressor. As much as 7% of apparently “sporadic” pancreatic cancers (more, in instances of familial aggregation) harbor an inactivating intragenic inherited mutation of one copy of the BRCA2 gene, accompanied by LOH.1,2,66 The PALB2 gene represents Fanconi group N, and its protein product functions by binding the Brca2 protein.65 Three percent of familial pancreatic cancers had a germ line inactivating mutation of PALB2, and in a tumor studied in depth, the other copy was inactivated by a somatic mutation,1,66,67 The FANCC and FANCG genes have somatic or germ line mutations in some pancreatic cancer patients, again with loss of the wild type allele in the cancer.68 The known hypersensitivity of Fanconi cells to interstrand DNA-crosslinking agents, such as cisplatin, melphalan, and mitomycin C, has led to the hypothesis that pancreatic cancers with Fanconi pathway genetic defects should be especially susceptible to treatment with such agents.69–72 Occasional complete remissions of pancreatic cancer have been reported with therapies that included DNA cross-linkers,73–77 and there are recent reports of prolonged responses using such agents in patients having BRCA2 mutations.78,79 Cells made experimentally deficient for Fanconi genes are also hypersensitive to certain nongenotoxic compounds,80 and patients having BRCA2-mutant cancers other than pancreatic cancer are reported to respond to therapeutic drug inhibition of the poly (ADP-ribose) polymerase enzyme, which normally becomes activated to facilitate DNA strand repair.81 These opportunities are beginning to be explored clinically in pancreatic cancer.
BRCA1 gene mutations are not found in unselected pancreatic cancers or pancreatic cancer families.82 Nonetheless, pancreatic cancers do occur in carriers of BRCA1 inactivating mutations.83,84 In these persons, the relatively high rate of LOH affecting the other BRCA1 copy indicates that a loss of BRCA1 function likely plays a role in tumorigenesis within these patients.83
The mitochondrial genome is mutated in a majority of pancreatic cancers.85–87 These mutations most likely represent genetic drift, and perhaps do not directly contribute to the process of tumorigenesis.87 Such mutations, however, could potentially serve as a diagnostic target because of the large number of copies of the mitochondrial genome in human carcinoma cells.86,87
The MAP2K4 (MKK4) gene participates in a stress-activated protein kinase pathway.81,82 It is stimulated by various influences, including chemotherapy, and its downstream effects include apoptosis and cellular differentiation. The MKK4 gene is inactivated by homozygous deletions or mutation/LOH in about 4% of pancreatic cancers.88,89 Experimental loss of one or both copies of the MKK4 gene in cancer cells reduces Jun kinase activation and JUN expression; this is a rare example of a tumor-suppressor pathway affected by gene dosage. Such gene dose-dependent effects could rationalize the high rate of loss of chromosomal arm 17p affecting 90% of pancreatic cancers and more than half of the TP53-wild type cancers.90
Germ line mutations of the STK11 (LKB1) gene, a serine-threonine kinase, are responsible for the Peutz-Jeghers syndrome (PJS).91,92 PJS was anecdotally associated with pancreatic cancer.93 A follow-up study examined lifetime risk, finding nearly a third of PJS patients to develop pancreatic cancer.94 Sporadic pancreatic cancers, independent of PJS, also lose the gene by homozygous deletion or by somatic mutation/LOH in about 4% of cases.95
Kinase oncogenes are mutated at low frequency, including the GUCY2F, EGFR, and NTRK3 genes.91,92 This class of mutations is important in that these mutations can be targeted with antikinase drugs.93
Gene amplification also occurs in pancreatic cancer. Amplified regions include the AKT2 gene within an amplicon on chromosome 19q, involving about 10% to 20% of cases studied.94–96 About 6% of pancreatic cancers overexpress the oncogene, CCNE1 (cyclin E). Two mechanisms have been demonstrated, cyclin E gene amplification and the genetic inactivation of the FBXW7 (AGO) gene, which normally serves to degrade cyclin E during the normal phases of the cell division cycle.1,96
The patterns of chromosomal deletion in pancreatic cancer are complex. In one study, from 1.5% to 32% of all tested loci, in different cancers, had a deletion.97 For most lost regions, we know of no particular tumor-suppressor genes targeted by the deletions. Conversely, in some regions known to harbor tumor-suppressor genes, the known mutated genes do not justify the high observed prevalence rates of LOH unless gene dose-dependent effects are postulated (90). Individual homozygous deletions are found at some additional genetic locations, again without a definitive target gene yet identified for most of these events.98
Defects in DNA mismatch repair (MIN) are seen in some pancreatic cancers. These cancers typically have a medullary histologic phenotype8 and mutations of the type II TGFβ (TGFBR2) and activin (ACVR2) receptor genes.25,52,53 They can also have mutations of the proapoptotic BAX gene51 and of the growth factor pathway mediator BRAF gene (affecting the same pathway, presumably, as mutations of the KRAS gene).8,9,51,96 The MIN tumors do not have the propensity for large chromosomal alterations and gross aneuploidy.15,99 In a study of four cases of pancreatic cancers having MIN, all lacked expression of the Mlh1 protein.9 Not all cancers with a medullary phenotype have MIN. Yet, medullary pancreatic carcinomas as a whole have a number of clinical and genetic differences as compared to those with conventional histologic appearance; the carcinomas have pushing rather than infiltrative borders, the KRAS gene often is wild type, and there is often a family history of malignancy.5,6,47 A reported case of Epstein-Barr virus (EBV)–associated pancreatic cancer9 had a medullary phenotype with heavy lymphocytic infiltration. Because of its distinctive features, it is advisable to separately designate the medullary category in the reporting of all clinical, genetic, and pathologic studies of pancreatic cancer.
Inherited mutations of the cationic trypsinogen (PRSS1) gene prevent the inactivation of prematurely activated trypsin within the ducts, causing a familial form of severe early-onset acute pancreatitis.100 Some affected kindred have a cumulative risk of pancreatic cancer that approaches 40% by the time the affected individuals reach 60 years of age.101 This cancer diathesis falls in a unique category of cancer susceptibility, in that the predisposition emanates from genetic alterations of a tissue-maintenance gene, one that is neither an oncogene, a tumor- suppressor gene, nor a genome-maintenance gene.
In summary, pancreatic cancer is fundamentally a genetic disease. An understanding of the genes altered in pancreatic cancer has led to a better understanding of the familial aggregation of pancreatic cancer, and, it is hoped, will lead to novel gene-specific targeted therapies for this deadly form of cancer.
Selected References
The full list of references for this chapter appears in the online version.
1. Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321:1801.
4. Goggins M, Schutte M, Lu J, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res 1996;56:5360.
6. Murphy KM, Brune KA, Griffin C, et al. Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: deleterious BRCA2 mutations in 17%. Cancer Res 2002;62:3789.
8. Goggins M, Offerhaus GJA, Hilgers W, et al. Adenocarcinomas of the pancreas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology: poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am J Pathol 1998;152:1501.
10. Hruban RH, Wilentz R, Kern SE. Genetic progression in the pancreatic ducts. Am J Pathol 2000;156:1821.
13. Hiyama E, Kodama T, Shinbara K, et al. Telomerase activity is detected in pancreatic cancer but not in benign tumors. Cancer Res 1997;57:326.
17. Almoguera C, Shibata D, Forrester K, et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988;53:549.
18. Caldas C, Hahn SA, Hruban RH, et al. Detection of K-ras mutations in the stool of patients with pancreatic adenocarcinoma and pancreatic ductal hyperplasia. Cancer Res 1994;54:3568.
24. Hahn SA, Schutte M, Hoque ATMS, et al. DPC4, a candidate tumor-suppressor gene at 18q21.1. Science 1996;271:350.
26. Baldwin RL, Friess H, Yokoyama M, et al. Attenuated ALK5 receptor expression in human pancreatic cancer: correlation with resistance to growth inhibition. Int J Cancer 1996;67:283.
30. Caldas C, Hahn SA, da Costa LT, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nature Genetics 1994;8:27.
34. Whelan AJ, Bartsch D, Goodfellow PJ. Brief report: a familial syndrome of pancreatic cancer and melanoma with a mutation in the CDKN2 tumor-suppressor gene. N Engl J Med 1995;333:975.
41. Redston MS, Caldas C, Seymour AB, et al. p53 mutations in pancreatic carcinoma and evidence of common involvement of homocopolymer tracts in DNA microdeletions. Cancer Res 1994;54:3025.
47. Hahn SA, Seymour AB, Hoque ATMS, et al. Allelotype of pancreatic adenocarcinoma using a xenograft model. Cancer Res 1995;55:4670.
48. Ionov Y, Peinado MA, Malkhosyan S, et al. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993;363:558.
66. Jones S, Hruban RH, Kamiyama M, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 2009;324:217.
81. Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009;361:123.
83. Al-Sukhni W, Rothenmund H, Borgida AE, et al. Germline BRCA1 mutations predispose to pancreatic adenocarcinoma. Hum Genet 2008;124:271.
88. Su GH, Hilgers W, Shekher M, et al. Alterations in pancreatic, biliary, and breast carcinomas support MKK4 as a genetically targeted tumor-suppressor gene. Cancer Res 1998;58:2339.
94. Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000;119:1447.
95. Su GH, Hruban RH, Bova GS, et al. Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am J Pathol 1999;154:1835.
101. Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst 1997;89:442.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


