Fig. 1
The hepatitis B virus genome and the covalently closed circular DNA (cccDNA). The viral genome (left panel) is made of two linear DNA strands maintained in circular configuration by their cohesive ends. The minus strand is around 3.182 kb in length, while the plus strand has variable length with fixed 5′ end position and variable 3′ end. The broad arrows surrounding the viral genome represent four open reading frames that encode the viral proteins. After viral entry, the HBV genome is delivered into the nucleus where it is converted into cccDNA by the cellular machinery. The cccDNA is organized into a minichromosome with bound histone and non-histone proteins including HBx and the core protein. By recruiting different chromatin modifying factors such as CBP/p300, HBx plays a pivotal role in the modulation of epigenetic marks such as histone tail acetylation and methylation that results into active HBV transcription
Upon HBV infection, the partially double-stranded DNA genome is delivered to the nucleus where it is converted into a covalently closed circular DNA (cccDNA). The cccDNA serves as the template for transcription of all viral RNAs by the host RNA polymerase II (Fig. 1). The production of the major HBV transcripts of 3.5-, 2.4-, 2.1-, and 0.7-kb is controlled by 4 distinct viral promoters and by 2 enhancers, Enh I and Enh II (Doitsh and Shaul 2004; Moolla et al. 2002). Several binding sites for ubiquitous and liver-specific transcription factors that likely regulate HBV transcription in vivo have been identified in the cis-acting sequences of HBV promoters and enhancers (Fallot et al. 2012; Moolla et al. 2002). Moreover, it has been shown that HBV cccDNA transcription is regulated by epigenetic mechanisms such as DNA methylation and histone acetylation (Pollicino et al. 2006; Vivekanandan et al. 2009). Because persistence of the cccDNA might be responsible for the failure of most antiviral treatments to completely eradicate the virus, it represents an interesting target for effective therapy of chronic hepatitis B.
Following transport of HBV RNAs to the cytoplasm and their translation into viral proteins, the pregenomic RNA is selectively packaged with the polymerase into progeny capsids and reverse-transcribed into RC-DNA by the viral polymerase. The HBV polymerase harbors RNA-dependent DNA polymerase activity and sequence homology with the RTs of retroviruses and cauliflower mosaic virus (CaMV) (Toh et al. 1983). HBV replication is initiated by the binding of the viral RT to the encapsidation signal epsilon located at the 5′ end of pregenomic RNA (Bartenschlager and Schaller 1992). After asymmetrical replication of the two HBV DNA strands, capsids containing mature RC-DNA are then assembled with the viral envelope in the endoplasmic reticulum, and subsequent budding of HBV is thought to occur at intracellular membranes (Bruss 2007). Additionally, empty envelope particles are produced in large excess, behaving probably as decoys for the host immune system. A minor portion of mature capsids can re-cycle RC-DNA to the nucleus and contribute to the amplification of the nuclear cccDNA pool. The availability of envelope proteins in adequate amounts and their efficiency to package nucleocapsids are critical for coordinate regulation of virion morphogenesis and accumulation of cccDNA (Summers et al. 1991). While huge amounts of viral particles can be produced in healthy liver from asymptomatic HBV carriers, liver damage can result from disruption of the normal replication process leading to intrahepatic accumulation of the large envelope protein or excessive amounts of cccDNA (Wang et al. 2006).
3 HBx: A Promiscuous Transactivator
The HBx (or pX) protein encoded by the X gene is a small polypeptide of 154 amino acids (16.5 kDa) that is expressed at low levels in acute and chronic infections and induces humoral and cellular immune responses (Su et al. 1998). The X gene is conserved in mammalian hepadnaviruses, but curiously, it is absent from avian hepadnavirus genomes. Different domains associated with specific functions have been identified in this small protein (Fig. 2). However, little is known about the three-dimensional structure of HBx, as it has not been possible so far to produce sufficient amounts of soluble protein. HBx appears as an unstructured protein that might be folded and gain secondary structure through its interaction with partner proteins (Rui et al. 2005). Such flexibility could account for the large array of HBx activities.
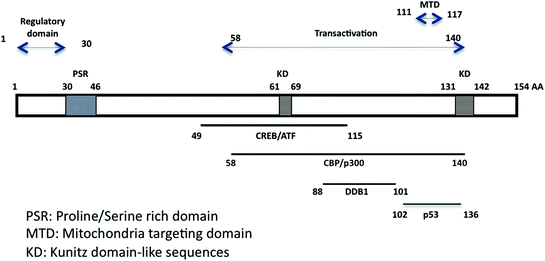
Fig. 2
Schematic representation of the small HBx protein with important functional domains. The N-terminal domain encodes a regulatory region (Murakami 1999) and a proline/serine-rich region. The domain carrying most HBx transcriptional transactivation activity has been mapped by Kumar and colleagues (Kumar et al. 1996). It includes the mitochondrial targeting region (Li et al. 2008). Binding domains with cellular partners are shown at the bottom part, including CREB/ATF (Maguire et al. 1991), CBP/p300 (Cougot et al. 2007), DDB1 (Becker et al. 1998; Lin-Marq et al. 2001), and p53 (Lin et al. 1997)
An essential role of HBx is virus replication in vivo, but this role is far from being completely understood, and it is likely multifactorial. Indeed, HBx has been endowed with multiple activities. The HBx protein is described as a promiscuous transactivator of viral and cellular genes, acting both from cytoplasmic and nuclear locations. HBx activates the transcription from cellular and viral promoters including HBV promoters, and it subverts different cellular functions and signal transduction pathways through modulation of cytoplasmic calcium, cell proliferation, and apoptosis. While these cellular activities modulated by HBx likely contribute to increase virus replication, evidence for a more direct role of HBx in stimulating HBV transcription has been provided by different studies in cell lines and murine models. HBx strongly activates HBV replication in HBV transgenic mice and in mice hydrodynamically injected with a plasmid encoding the HBV genome, and this effect is less pronounced in hepatoma cell lines (Keasler et al. 2007; Leupin et al. 2005; Tang et al. 2005). HBx is also required to initiate and maintain HBV replication as a key regulator during natural HBV infection (Lucifora et al. 2011). It has been shown recently that HBx interferes with epigenetic regulation of the HBV minichromosome by increasing the active chromatin marks and antagonizing the inhibitory factors (Belloni et al. 2009; Cougot et al. 2012).
HBx exerts its activities by interacting with a large number of cellular partners that are located either in the cytoplasm or in the nucleus, in agreement with the dual location of HBx. Consistent with its role in transcription, HBx has been shown to interact with components of the basal transcriptional machinery (RPB5, TFIIB, TBP, TFIIH), with transcription factors (ATF/CREB, c/EBP, NF-IL6, RXR receptor) as well as coactivators (CBP/P300, ASC-2) or repressor (DNMT) (reviewed in Benhenda et al. 2009a). We have shown that HBx is recruited to the promoters of CREB target genes and increases the recruitment of CBP/P300 on CREB, thereby activating transcription (Cougot et al. 2007). Thus, HBx could be directly involved in assembly of enhancer–transcription factor complexes. HBx clearly plays a direct role in the epigenetic regulation of HBV transcription, by modulating a network of transcription regulatory factors within large molecular weight protein complexes (Belloni et al. 2009; Cougot et al. 2012).
4 HBx and E3 Ubiquitin Ligases: A Link Beyond Protein Stability?
Manipulation of protein degradation machineries is a common strategy used by viruses to provide a favorable environment for their replication and escape protective mechanisms raised by the host cell (Barry and Fruh 2006). Interaction between HBx and damage-specific DNA binding protein 1 (DDB1), an adaptor subunit of the Cul4A-based ubiquitin E3 ligase complex, has been extensively documented (Lee et al. 1995; Leupin et al. 2003; Sitterlin et al. 1997). DDB1 is a highly conserved protein that links multiple WD40 proteins to the CUL4– ROC1– E2 catalytic core and that is implicated in DNA repair via its association with DDB2 (Jackson and Xiong 2009). The minimal DDB1 binding domain on HBx (amino acids 88–100) contains a sequence evoking the DDB1-binding WD40 (DWD) motifs shared by cellular DCAFs (He et al. 2006). Several point mutations in this region impede HBx binding to DDB1 (Bergametti et al. 2002; Hodgson et al. 2012) (Table 1). High-resolution crystal structure demonstrated that a peptide consisting of the HBx amino acids 88–100 could directly bind to DDB1 (Li et al. 2010). Moreover, the specific region on DDB1 that is bound by HBx is the common binding site of most DCAFs. These data suggest that HBx shares similar properties with cellular DCAFs and might displace other DCAFs, thereby modifying the profile of Cul4A complex substrates. So far, however, a role of HBx in regulating the stability or localization of such substrates has not been identified.
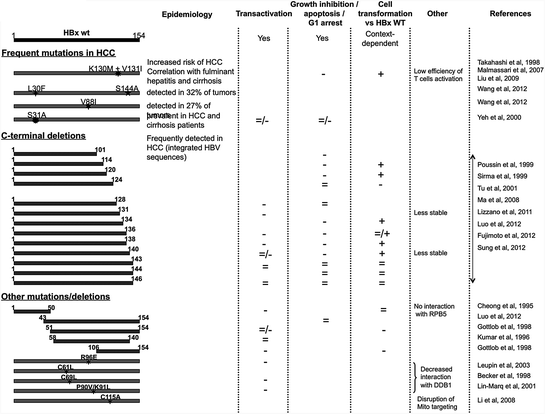
Table 1
Survey of recurrent HBx mutants detected in hepatocellular carcinoma and synthetic mutants used for demonstrating specific HBx functions. For each HBx mutant, reported activities such as transcriptional transactivation, deregulation of cell cycle and viability, and transformation are shown. Increased activity of mutant as compared to wild-type HBx is shown by +, equal activity by =, and reduced activity by −. Divergent data from 2 different papers are represented as double symbols (=/+ or =/−). Corresponding references can be found in the reference section
It has been shown that the HBx/DDB1 interaction is conserved among mammalian hepadnaviruses and plays essential role in virus replication and in the stability and functions of HBx (Bontron et al. 2002; Sitterlin et al. 2000a). In particular, transcriptional and pro-apoptotic properties of HBx are dependent upon its interaction with DDB1 (Lin-Marq et al. 2001; Sitterlin et al. 2000b). Mutant HBx that has lost DDB1 binding capacity also loses concomitant cytotoxicity, but regains activity when directly fused to DDB1. These studies indicate that HBx does not inhibit the normal DDB1 functions, but confers apoptotic potential to DDB1 (Bontron et al. 2002). However, in other studies using HBx mutants that no longer bind to DDB1, the transcriptional activity of HBx has been dissociated from DDB1 binding (Wentz et al. 2000) (see Table 1). Moreover, recent studies of HBx-mediated activation of HBV transcription and replication have confirmed the importance of HBx-DDB1 interaction, but also pointed to the requirement for the HBx carboxy-terminal region for maximal stimulation of HBV transcription (Hodgson et al. 2012; Luo et al. 2012).
DDB1 also belongs to the UV-DDB complex implicated in repair of UV-damaged DNA, and HBx has been shown to inhibit nucleotide excision repair (NER) (Mathonnet et al. 2004). Further investigations of the effects of HBx-DDB1 interaction on DNA repair have shown that HBx induces mitotic abnormalities indirectly, by interfering with S-phase, and that an altered S-phase can result in abnormal centrosome duplication. HBx induces lagging chromosomes in cells dividing with a normal bipolar spindle, followed by the formation of multinucleated cells with abnormal centrosome numbers and multipolar spindles, and these effects are strictly dependent upon correct binding of HBx to DDB1 (Martin-Lluesma et al. 2008). This interaction is therefore of crucial importance for the pathogenesis of chronic hepatitis B.
5 HBx and Malignant Transformation of Liver Cells
The regulatory hepatitis B virus X protein is thought to be involved in oncogenesis. Although HBx does not behave as a strong oncogene per se, it can transform SV40-immortilized murine hepatocytes and induce liver tumors or accelerate carcinogenesis induced by carcinogens or by c-Myc in HBx transgenic mice. (Höhne et al. 1990; Kim et al. 1991; Madden et al. 2001; Terradillos et al. 1997). Transactivation of cellular oncogenes and growth factors, and deregulation of cell cycle progression are two mechanisms that might account for the weak oncogenicity of this viral protein (reviewed in (Bouchard and Schneider 2004). Indeed, in other human oncogenic viruses such as HTLV-1, EBV, and HPV-16 and -18, transforming capacity is associated with the transcriptional transactivation activity of viral gene products. Comparative analyses of the different viral transactivators may help guide future work in this field. Additionally, the ability of HBx to induce aberrant activation of several signaling pathways, including the Src, Ras, MAP kinase, and CREB pathways, might lead to accumulation of cellular dysfunctions that favor malignant transformation (reviewed in Andrisani and Barnabas 1999).
Another relevant pro-carcinogenic activity of HBx has been shown to target the mitotic cell cycle. Interaction of HBx with different cellular partners implicated in centrosome formation might result in defects in centrosome dynamics and mitotic spindle formation. HBx has been shown to bind and partially inactivate BubR1, a mitotic kinase effector that specifies microtubule attachments and checkpoint functions (Kim et al. 2008). HBx also binds HBXIP, a major regulator of centrosome duplication, required for bipolar spindle formation and cytokinesis (Wen et al. 2008). Interestingly, DNA re-replication induced by HBx has been causally related to partial polyploidy, a condition known to be associated with cancer (Rakotomalala et al. 2008). Accordingly, HBx has been implicated in induction of chromosomal instability (CIN), a cancer hallmark. CIN cells with extra centrosomes display an increased frequency of lagging chromosomes during anaphase, leading to chromosome missegregation. In cells expressing HBx, prolongation of S-phase has been associated with aberrant centrosome duplication, multipolar spindle formation, chromosome segregation defects, and appearance of multinucleated cells (Studach et al. 2009, 2010). HBx-expressing polyploid cells are prone to oncogenic transformation mediated by the mitotic entry factor polo-like kinase 1 (Plk1), and in turn, Plk1 downregulates ZNF198 and SUZ12 that modulate HBV transcription and replication through epigenetic mechanisms (Wang et al. 2011). Others have implicated the interaction between HBx and DDB1 in increasing genetic instability (Li et al. 2010; Martin-Lluesma et al. 2008). Altogether, these data provide a strong link between HBx activities and chromosomal instability that could account for the high rate of chromosomal defects observed in tumors related to HBV infection compared with other risk factors (Boyault et al. 2007).
The question of whether HBx is involved in cancer progression has been raised by the observation that HBx expression is usually retained in human HCCs, correlating with HBV methylome studies that showed global hypermethylation of the HBV genome in HCC, except for the X gene (Fernandez et al. 2009).
6 Integration of HBV DNA and Mutated X Genes in HCC
Integrated HBV DNA sequences have been detected in a majority of HBV-related HCCs (Bréchot et al. 1980; Bonilla Guerrero and Roberts 2005). Recent analysis of viral insertion sites using various PCR-based methods has demonstrated that integration occurs not only in repetitive elements and fragile sites, but also frequently in actively transcribed regions, nearby or within cellular genes (Murakami et al. 2005). Evidence for oncogenic potential of the rearranged viral-host sequences has been provided conclusively in few cases in which production of chimeric HBV/cellular transcripts resulted in hybrid viral/cellular proteins. Besides “historical” targets such as the retinoic acid receptor a (RARα) and cyclin A2, a number of HBV insertions have been shown to target key regulators of cell proliferation and cell death. HBx fusion proteins have been described in many cases, including fusion with the SERCA1 gene and the MLL2, MLL3, and MLL4 genes (Chami et al. 2000; Saigo et al. 2008; Tamori et al. 2005).
On the other side, integrated HBx sequences are usually mutated, and they have been endowed with peculiar properties, leading to the notion of “transacting” oncogenic mechanisms. Because the 3′ end of the X gene coincides with the recombination-prone region of the HBV genome located between the direct repeats DR1 and DR2, frequent C-terminal truncations of the X gene have been detected in HCCs (Luo et al. 2012) (Table 1). Functional studies have outlined diverse properties of tumor-derived HBx proteins compared to wild type. In some cases, these sequences retained transactivating capacity and the ability to bind p53 and block p53-mediated apoptosis, which may provide a growth advantage for neoplastic cells (Huo et al. 2001). Moreover, in vitro and in vivo studies showed that the truncated rather than the full-length HBx could effectively transform immortalized liver cells and modulate expression of key genes implicated in the control of cell cycle and apoptosis (Ma et al. 2008). Recent studies have demonstrated that the C-terminus of HBx impacts the stability of the protein, its transactivation activity and its ability to stimulate HBV replication (Lizzano et al. 2011). By contrast, in other cases, the truncated X sequences harbored a number of missense mutations and were less effective in transcriptional transactivation, apoptosis induction, and in supporting HBV replication (Sirma et al. 1999). Collectively, recent studies of the HBx protein functions have contributed to better delineate the importance of the X gene product at different steps of the malignant process induced by persistent HBV infection.
7 Conclusions and Future Prospects
HCC is among the most frequent cancers in many countries, and its incidence has been rising during the last years (Ferlay et al. 2010). Like other human cancers, the tumorigenic process in HBV carriers evolves through multiple steps resulting in the accumulation of genetic and epigenetic lesions (Fig. 3). In the absence of direct transforming capacity of the virus in cell lines and transgenic mice, and because HCC develops after a long latency period following primary HBV infection, the question may be raised of whether the hepatitis B virus is oncogenic. In a simple view, immune responses against infected hepatocytes might trigger a pro-carcinogenic inflammatory process associated with continuous necrosis and cell regeneration, fostering the accumulation of genetic and epigenetic defects. In this model, the long-term toxic effects of viral gene products during productive HBV infection might potentiate the action of exogenous carcinogenic factors, such as Aflatoxin B1 and alcohol. However, it is worth noticing that chronic HBV infection has been assigned a strong and independent causative role by extensive epidemiological studies worldwide and that evidence is increasing for oncogenic potential of the regulatory protein HBx and surface glycoproteins in native or mutant state. Thus, HBV shares a number of basic strategies with other human oncogenic viruses. It might also be speculated that HCC latency period depends on the occurrence of a decisive HBV integration event that would promote genetic instability or lead to cis- or transactivation of relevant genes. Identifying the cellular effectors connecting HBV infection and liver cell transformation is of upmost importance.
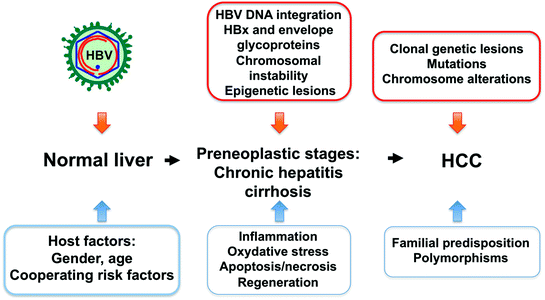
Fig. 3
Multistep process of HBV-induced liver tumorigenesis. Chronic HBV infection, alone or in combination with cofactors, triggers a cascade of events ultimately leading to transformation. At preneoplastic stages, the immune response induces a necroinflammatory disease that creates a suitable environment for the occurrence of genetic and epigenetic lesions. Integration of HBV DNA sequences into the host genome as well as long-term production of the viral HBx and surface proteins may contribute to the tumoral process via multiple mechanisms
The question of whether HBV could trigger distinct tumorigenic pathways compared to other risk factors can now be addressed via novel technologies for genome-wide scans of gene expression, genetic and epigenetic alterations, and proteomics. Thus, despite substantial heterogeneity, HBV-related tumors have been recently classified into subgroups that display high chromosomal instability, stemness features, increased frequency of p53 mutation, and low rate of ß-catenin mutation (Boyault et al. 2007; Guichard et al. 2012). It will now be important to apply these technologies to the analysis of chronic hepatitis B at different stages, in order to identify suitable prognostic markers and therapeutic targets. In addition, there is need to better define the role of HBV in the occurrence of epigenetic alterations such as methylation-associated gene silencing and abnormal expression of microRNAs that play crucial part in malignant transformation. Ongoing advances in this field will undoubtedly provide adapted tools for improving the management of chronic hepatitis B and liver cancer.
References
Andrisani OM, Barnabas S (1999) The transcriptional function of the hepatitis B virus X protein and its role in hepatocarcinogenesis. Int J Oncol 15:373–379PubMed
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


