Oncogenes
Marco A. Pierotti, PhD  Milo Frattini, MD, PhD
Milo Frattini, MD, PhD  Francesca Molinari, PhD
Francesca Molinari, PhD  Gabriella Sozzi, PhD
Gabriella Sozzi, PhD  Carlo M. Croce, MD
Carlo M. Croce, MD
Overview
The initiation and progression of human neoplastic diseases is a multistep process involving the accumulation of genetic changes in somatic cells. These genetic changes typically consist of the activation of cooperating oncogenes and the inactivation of tumor suppressor genes, which both appear necessary for a complete neoplastic phenotype. Oncogenes are altered versions of normal cellular genes called proto-oncogenes, usually involved in the regulation of cell growth and activated by mutation, chromosomal rearrangement, or gene amplification. In this chapter, at first we will describe the methods that have been applied by the researchers for the discovery and the identification of oncogenes. Then we will present the genetic mechanisms of proto-oncogenes activation (point mutations, gene amplifications, chromosomal rearrangements) with several examples, and we will describe the role played by oncogenes in the initiation and progression of various cancers.
The identification of oncogene abnormalities has provided tools for the molecular diagnosis and monitoring of cancer. Most important, oncogenes represent potential targets for new types of cancer therapies, which are continuously discovered and tested in clinical trials. The goal of these new drugs is to kill cancer cells selectively while sparing normal cells. These new therapies display an evident benefit for the treatment of several neoplastic diseases that were, before targeted therapies development, very hard to be treated and cured. However, they are not able to kill 100% of neoplastic cells, essentially due to the occurrence of mechanisms of secondary resistance. In the last part of the chapter, we will review all the genes for which a targeted therapy has been developed.
Since the early proposal of Boveri more than a century ago, multiple experimental evidence have confirmed that, at the molecular level, cancer is due to lesions in the cellular DNA. First, it has been observed that a cancer cell transmits to its daughter cells the phenotypic features characterizing the “cancerous” state. Second, most of the recognized mutagenic compounds are also carcinogenic. Finally, the karyotyping of several types of human tumors, particularly those belonging to the hematopoietic system, led to the identification of recurrent chromosomal aberrations, reflecting pathologic rearrangements of the cellular genome. Taken together, these observations suggest that the molecular pathogenesis of human cancer is due to structural and/or functional alterations of specific genes whose normal function is to control cellular growth and differentiation or, in different terms, cell birth and cell death.1, 2
The identification and characterization of the genetic elements playing a role in the scenario of human cancer pathogenesis have been made possible by the development of DNA recombinant techniques during the past decades, enhanced by the possibility to sequence even the whole genome. One milestone was the identification of the cellular origin of the “viral oncogenes” followed by the possibility to transfer the tumorigenic properties to the RNA tumor viruses also known as retroviruses.3, 4 Lastly, it was discovered that most cellular transforming genes do not have a viral counterpart. Besides the source of their original identification, viral or cellular genome, these transforming genetic elements have been designated as proto-oncogene in their normal physiologic version and oncogene when altered in cancer.5 A second relevant experimental approach has regarded the identification and characterization of clonal and recurrent cytogenetic abnormalities (including translocations and inversions) in cancer cells, especially those derived from the hematopoietic system. Additional oncogenes have been identified through the analysis of anomalously stained chromosomal regions (homogeneously staining regions (HSRs), representing gene amplification.6 Furthermore, the detection of chromosome deletions has been instrumental in the process of identification and cloning of a second class of cancer-associated genes, the tumor suppressors (which act in the normal cell as negative controllers of cell growth and are inactive in tumor cells).5, 6 Lastly, using automated sequencing instruments, it has been demonstrated that also point mutations are a frequent mechanism of oncogene activation.7 In the last subgroup, we can also number those obtained by the analysis of the protein kinases (kinome)8 or phosphatases (phosphatome)9 of the human genome or of several isoforms of a relevant protein involved in cancer development (such as PI3K).10
In this chapter, the methods by which oncogenes were discovered will be first briefly described. The various functions of cellular proto-oncogenes will then be presented, and the genetic mechanisms of proto-oncogene activation will be summarized. Then, the role of specific oncogenes in the initiation and progression of human tumors will be discussed. Lastly, the discovery that oncogenes may represent relevant target for new drugs will be described.
Discovery and identification of oncogenes
The first oncogenes were discovered through the study of retroviruses, RNA tumor viruses whose genomes are reverse transcribed into DNA in infected animal cells.11 During the course of infection, retroviral DNA is inserted into the chromosomes of host cells. The integrated retroviral DNA, called the provirus, replicates along with the cellular DNA of the host, leading to the production of viral progeny that bud through the host cell membrane to infect other cells.11 Acutely transforming retroviruses can rapidly cause tumors within days after injection. Chronic or weakly oncogenic retroviruses can cause tissue-specific tumors in susceptible strains of experimental animals after a latency period of many months.
Retroviral oncogenes are altered versions of host cellular proto-oncogenes that have been incorporated into the retroviral genome by recombination with host DNA, a process known as retroviral transduction.5 This surprising discovery was made through study of the Rous sarcoma virus (RSV) (Figure 1), revealing that the transforming gene of RSV was not required for viral replication.12 Molecular hybridization studies then showed that the RSV transforming gene (designated v-src) was homologous to a host cellular gene (c-src) that was widely conserved in eukaryotic species.13 Studies of many other acutely transforming retroviruses from fowl, rodent, feline, and nonhuman primate species have led to the discovery of dozens of different retroviral oncogenes (Table 1). In every case, these retroviral oncogenes are responsible for the rapid tumor formation and efficient in vitro transformation activity, characteristic of acutely transforming retroviruses.
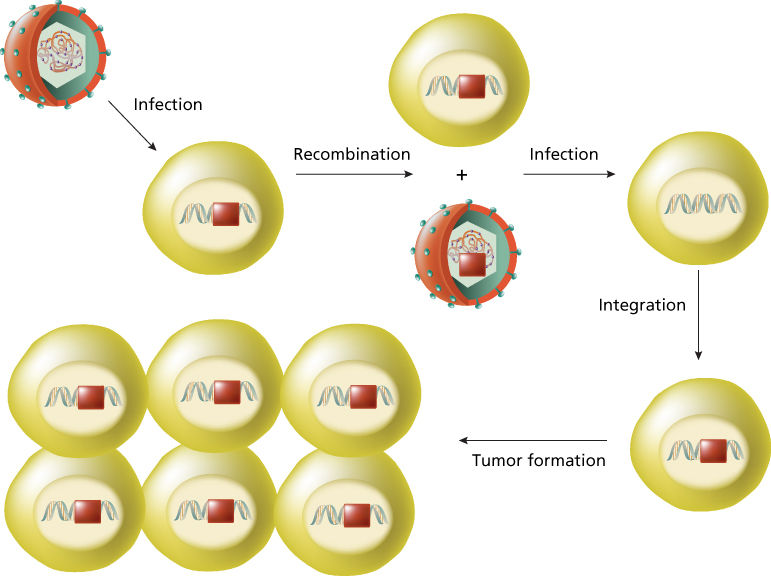
Figure 1 Retroviral transduction. An RNA tumor virus infects a human cell carrying an activated src gene (red box). After the process of recombination between retroviral genome and host DNA, the oncogene c-src* is incorporated into the retroviral genome and is renamed v-src. When the retrovirus carrying v-src infects a human cell, the viral oncogeneis rapidly transcribes and is responsible for the rapid tumor formation.
Table 1 Oncogenes
| Oncogene | Chromosome | Identification | Method neoplasm mechanism of activation | Protein function | |
| Growth factors | |||||
| V-SIS | 22q12.3–13.1 | Sequence homology | Glioma/fibrosarcoma | Constitutive production | B-chain PDGF |
| INT2 | 11q13 | Proviral insertion | Mammary carcinoma | Constitutive production | Member of FGF family |
| KS3 | 11q13.3 | DNA transfection | Kaposi sarcoma | Constitutive production | Member of FGF family |
| HST | 11q13.3 | DNA transfection | Stomach carcinoma | Constitutive production | Member of FGF family |
| Growth factor receptors | |||||
| Tyrosine kinases: integral membrane proteins | |||||
| EGFR | 7p1.1–1.3 | DNA amplification/DNA sequencing | Squamous cell carcinoma | Gene amplification/protein/point | EGF receptor |
| v-FMS | 5q33–34 (FMS) | Viral homolog | Sarcoma | Constitutive activation | CSF1 receptor |
| v-KIT | 4q11–21 (KIT) | Viral homolog/DNA sequencing | Sarcoma/GIST | Constitutive activation/point mutation | Stem cell factor receptor |
| v-ROS | 6q22(ROS) | Viral homolog | Sarcoma | Constitutive activation | Chimeric protein |
| MET | 7p31 | DNA transfection | MNNG-treated human osteocarcinoma cell line | DNA rearrangement/ligand-independent constitutive activation (fusion proteins) | HGF/SF receptor |
| TRK | 1q32–41 | DNA transfection | Colon/thyroid carcinomas | DNA rearrangement/ligand-independent constitutive activation (fusion proteins) | NGF receptor |
| NEU | 17q11.2–12 | Point mutation/DNA amplification | Neuroblastoma/breast carcinoma/NSCLC | Gene amplification/point mutation | Unknown ligand |
| RET | 10q11.2 | DNA transfection | Carcinomas of thyroid Men 2A/Men 2B | DNA rearrangement/point mutation (ligand-independent constitutive activation/fusion proteins) | GDNF/NTT/ART/PSP receptor |
| Receptors lacking protein kinase activity | |||||
| MAS | 6q24–27 | DNA transfection | Epidermoid carcinoma | Rearrangement of 5′ noncoding region | Angiotensin receptor |
| Signal transducers | |||||
| Cytoplasmic tyrosine kinases | |||||
| SRC | 20p12–13 | Viral homolog | Colon carcinoma | Constitutive activation | Protein tyrosine kinase |
| v-YES | 18q21–23 (YES) | Viral homolog | Sarcoma | Constitutive activation | Protein tyrosine kinase |
| v-FGR | 1p36.1–36.2 (FGR) | Viral homolog | Sarcoma | Constitutive activation | Protein tyrosine kinase |
| v-FES | 15q25–26 (FES) | Viral homolog | Sarcoma | Constitutive activation | Protein tyrosine kinase |
| ABL | 9q34.1 | Chromosome translocation | CML | DNA rearrangement (constitutive activation/fusion proteins) | Protein tyrosine kinase |
| Membrane-associated G proteins | |||||
| HRAS | 11p15.5 | Viral homolog/DNA transfection | Colon, lung, pancreas carcinomas | Point mutation | GTPase |
| KRAS | 12p11.1–12.1 | Viral homolog/DNA transfection | AML, thyroid carcinoma, melanoma/colon/lung | Point mutation | GTPase |
| NRAS | 1p11–13 | DNA transfection | Carcinoma, melanoma | Point mutation | GTPase |
| BRAF | 6 | DNA sequencing | Melanoma, thyroid, colon, ovary | Point mutation | Ser/Thr kinase |
| GSP | 20 | DNA sequencing | Adenomas of thyroid | Point mutation | Gs alpha |
| GIP | 3 | DNA sequencing | Ovary, adrenal carcinoma | Point mutation | Gi alpha |
| GTPase exchange factor (GEF) | |||||
| DBL | Xq27 | DNA transfection | Diffuse B-cell lymphoma | DNA rearrangement | GEF for Rho and Cdc42Hs |
| VAV | 19p13.2 | DNA transfection | Hematopoietic cells | DNA rearrangement | GEF for Ras? |
| Serine/threonine kinases: cytoplasmic | |||||
| v-MOS | 8q11 (MOS) | Viral homolog | Sarcoma | Constitutive activation | Protein kinase (ser/thr) |
| v-RAF | 3p25 (RAF-1) | Viral homolog | Sarcoma | Constitutive activation | Protein kinase (ser/thr) |
| PIM-1 | 6p21 (PIM-) | Insertional mutagenesis | T-cell lymphoma | Constitutive activation | Protein kinase (ser/thr) |
| Cytoplasmic regulators | |||||
| v-CRK | 17p13 (CRK) | Viral homolog | — | Constitutive tyrosine phosphorylation of cellular substrates (e.g., paxillin) | SH-2/SH-3 adaptor |
| Transcription factors | |||||
| v-MYC | 8q24.1 (MYC) | Viral homolog | Carcinoma myelocytomatosis | Deregulated activity | Transcription factor |
| N-MYC | 2p24 | DNA amplification | Neuroblastoma: lung | Deregulated activity | Transcription factor |
| L-MYC | 1p32 | DNA amplification | Carcinoma of lung | Deregulated activity | Transcription factor |
| v-MYB | 6q22–24 | Viral homolog | Myeloblastosis | Deregulated activity | Transcription factor |
| v-FOS | 14q21–22 | Viral homolog | Osteosarcoma | Deregulated activity | Transcription factor API |
| v-JUN | p31–32 | Viral homolog | Sarcoma | Deregulated activity | Transcription factor API |
| v-SKI | 1q22–24 | Viral homolog | Carcinoma | Deregulated activity | Transcription factor |
| v-REL | 2p12–14 | Viral homolog | Lymphatic leukemia | Deregulated activity | Mutant NF-kappa B |
| v-ETS-1 | 11p23–q24 | Viral homolog | Erythroblastosis | Deregulated activity | Transcription factor |
| v-ETS-2 | 21q24.3 | Viral homolog | Erythroblastosis | Deregulated activity | Transcription factor |
| v-ERB A1 | 17p11–21 | Viral homolog | Erythroblastosis | Deregulated activity | T3 Transcription factor |
| v-ERB A2 | 3p22–24.1 | Viral homolog | Erythroblastosis | Deregulated activity | T3 Transcription factor |
| Others | |||||
| BCL2 | 18q21.3 | Chromosomal translocation | B-cell lymphomas | Constitutive activity | Antiapoptotic protein |
| MDM2 | 12q14 | DNA amplification | Sarcomas | Gene amplification/increased protein | Complexes with p53 |
Abbreviations: AML, acute myeloid leukemia; CML, chronic myelogenous leukemia; GTPase, guanosine triphosphatase; PDGF, platelet-derived growth factor.
In contrast, weakly oncogenic retroviruses do not carry viral oncogenes. These retroviruses, which include mouse mammary tumor virus (MMTV) and various animal leukemia viruses, induce tumors by a process called insertional mutagenesis (Figure 2).6 This process results from the integration of the provirus DNA into the host genome in infected cells and acquires relevance when (rarely) the provirus is inserted near a proto-oncogene, whose expression is then abnormally driven by the transcriptional regulatory elements contained within the long terminal repeats of the provirus.6 Therefore, proviral integration represents a mutagenic event that activates a proto-oncogene. The long latent period of tumor formation of weakly oncogenic retroviruses is therefore due to the rarity of the provirus insertional event that leads to tumor development from a single transformed cell. Insertional mutagenesis by weakly oncogenic retroviruses, first demonstrated in bursal lymphomas of chickens, frequently involves the same oncogenes (such as myc, myb, and erb B) that are carried by acutely transforming retroviruses.6, 14 In many cases, however, insertional mutagenesis has been used as a tool to identify new oncogenes, including int-1, int-2, pim-1, and lck.6
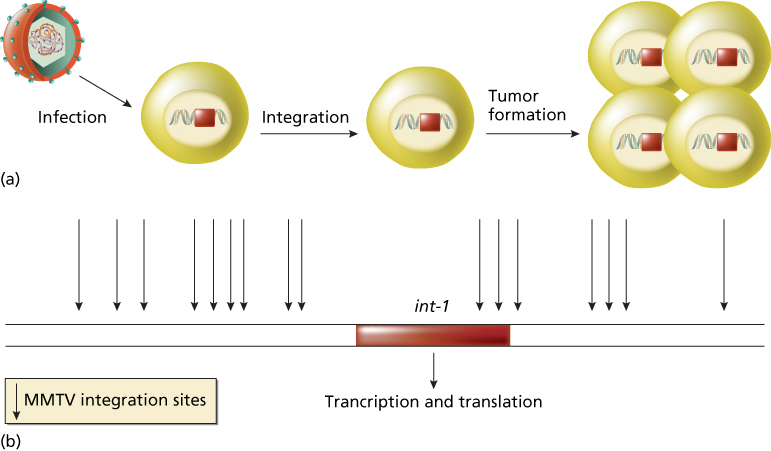
Figure 2 Insertional mutagenesis. (a) The process is independent from genes carried by the retrovirus. Retrovirus, for example, MMTV, infects a human cell. The proviral DNA is integrated into the host genome in infected cells. Rarely, the provirus inserts near a proto-oncogene (e.g., int-1) and activates the proto-oncogene. Activated proto-oncogene results in cell transformation and in tumor formation. (b) Sites of integration of MMTV retrovirus near the proto-oncogene int-1. All sites determine int-1 activation.
The demonstration of activated proto-oncogenes in human tumors was first shown by the DNA-mediated transformation technique.15, 16 This method, also called gene-transfer or transfection assay, verifies the ability of donor DNA from a tumor to transform a recipient strain of rodent cells called NIH 3T3, an immortalized mouse cell line (Figure 3).17 This sensitive assay, which can detect the presence of single-copy oncogenes in a tumor sample, also enables the isolation of the transforming oncogene by molecular cloning techniques.18 Overall, approximately 20% of individual human tumors have been shown to induce transformation of NIH 3T3 cells in gene-transfer assays. The value of transfection assay was reinforced by Weinberg’s laboratory, which showed that the ectopic expression of the telomerase catalytic subunit (hTERT), in combination with the simian virus 40 large T product and a mutated oncogenic H-ras protein, resulted in the direct tumorigenic conversion of normal human epithelial and fibroblast cells.19 Many of the oncogenes identified by gene-transfer studies are identical or closely related to oncogenes transduced by retroviruses.20 A number of new oncogenes (such as neu, met, and trk) have also been identified by the gene-transfer technique.21 In many cases, however, oncogenes identified by gene transfer were shown to be activated by rearrangement during the experimental procedure and are not activated in the human tumors that served as the source of the donor DNA, as in the case of ret that was subsequently found genuinely rearranged and activated in papillary thyroid carcinomas.22
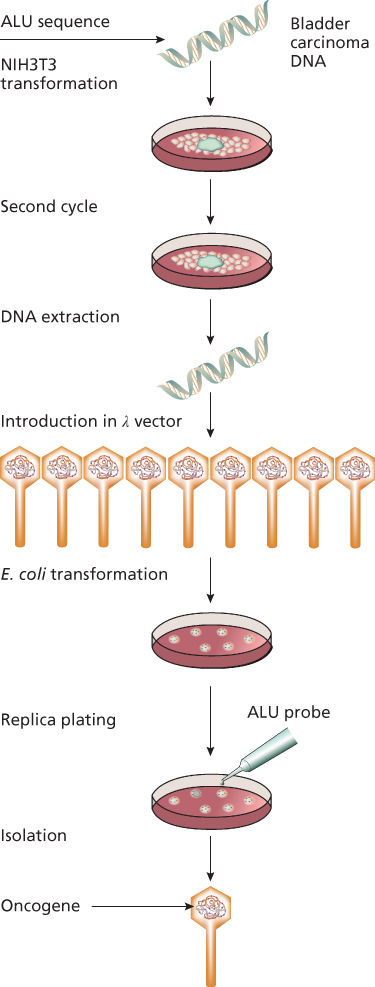
Figure 3 Transfection assay. DNA from a tumor (e.g., bladder carcinoma) is used to transform a rodent immortalized cell line (NIH3T3). After serial cycles, DNA from transformed cells was extracted and then inserted into p vector that was subsequently used to transform an appropriate Escherichia coli strain. Using a specific probe (Alu in the figure), it was possible to isolate and then characterize the involved human oncogene.
Chromosomal translocations have served as guideposts for the discovery of new oncogenes, especially in many hematological and solid tumors.23, 24 These abnormalities include chromosomal rearrangements as well as the gain or loss of whole chromosomes or chromosome segments. The first consistent karyotypic abnormality identified in a human neoplasm was a characteristic small chromosome in the cells of patients with chronic myelogenous leukemia (CML).23 Later identified as a derivative of chromosome 22, this abnormality was designated the Philadelphia chromosome, after its city of discovery. The application of chromosome banding techniques in the early 1970s enabled the precise cytogenetic characterization of many chromosomal translocations in human leukemia, lymphoma, and solid tumors.25 The subsequent development of molecular cloning techniques then enabled the identification of proto-oncogenes at or near chromosomal breakpoints in various neoplasms. Some of these proto-oncogenes, such as myc and abl, had been previously identified as retroviral oncogenes. In general, however, the cloning of chromosomal breakpoints has served as a rich source of discovery of new oncogenes involved in human cancer. More recently, the use of high-throughput sequencing technologies and bioinformatics from the Human Genome Project led to the discovery of new genes involved in cancer development, such as BRAF and PIK3CA.7, 10
Oncogenes, proto-oncogenes, and their functions
Proto-oncogenes encode proteins that are involved in the control of cell growth. Alteration of the structure and/or expression of proto-oncogenes can activate them to become oncogenes capable of inducing in susceptible cells the neoplastic phenotype. Oncogenes can be classified into five groups based on the functional and biochemical properties of protein products of their normal counterparts (proto-oncogenes). These groups are (1) growth factors, (2) growth factor receptors, (3) signal transducers, (4) transcription factors, and (5) others, including programmed cell death regulators. Table 1 lists examples of oncogenes according to their functional categories.
Growth factors
Growth factors are secreted polypeptides that function as extracellular signals to stimulate the proliferation of target cells that possess a specific receptor in order to respond to a specific type of growth factor.2 A well-characterized example is platelet-derived growth factor (PDGF), an approximately 30-kDa protein consisting of two polypeptide chains.26 PDGF, released from platelets during the process of blood coagulation, stimulates the proliferation of fibroblasts, a cell growth process that plays an important role in wound healing. Other well-characterized examples of growth factors include nerve growth factor (NGF), epidermal growth factor, and fibroblast growth factor.
The link between growth factors and retroviral oncogenes was revealed by study of the sis oncogene of simian sarcoma virus, a retrovirus first isolated from a monkey fibrosarcoma. Sequence analysis showed that sis encodes the beta chain of PDGF.26 This discovery established the principle that inappropriately expressed growth factors may constitutively activate their receptor, resulting in self-sustained aberrant cell proliferation and therefore functioning as oncogenes. The mechanism behind is called autocrine stimulation (Figure 4).26 This model, derived from experimental animal systems, has been demonstrated in a human tumor, dermatofibrosarcoma protuberans (DP), an infiltrative skin tumor that was demonstrated to present specific cytogenetic features: reciprocal translocation and supernumerary ring chromosomes, involving chromosomes 17 and 22.26 Molecular cloning of the breakpoints revealed a fusion between the collagen type Ia1 (COL1A1) gene and PDGF-B gene. The fusion gene resulted in a deletion of PDGF-B exon 1 and a constitutive release of this growth factor. Subsequent experiments of gene transfer of DP’s genomic DNA into NIH 3T3 cells directly demonstrated the occurrence of an autocrine mechanism by the human rearranged PDGF-B gene involving the activation of the endogenous PDGF receptor.26 Another example of a growth factor that can function as an oncogene is int-2, a member of the fibroblast growth factor family. Int-2 is sometimes activated in mouse mammary carcinomas by MMTV insertional mutagenesis.27
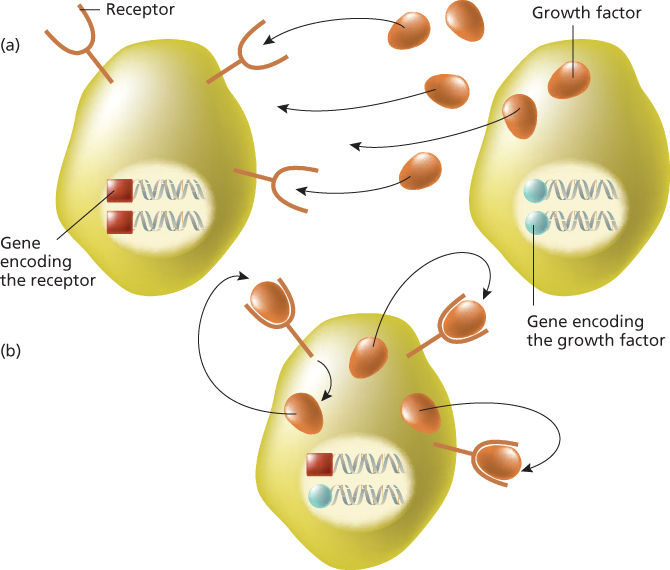
Figure 4 Paracrine and autocrine stimulation. (a) A growth factor produced by the cell on the right stimulates another cell carrying the appropriate receptor (left) on cell membrane. This process is named paracrine stimulation. (b) A growth factor is produced by the same cell expressing the cognate receptor. This process is designated autocrine stimulation.
Growth factor receptors
Some viral oncogenes are altered versions of normal growth factor receptors that possess intrinsic tyrosine kinase (TK) activity.28 Receptor tyrosine kinases (RTKs), as these growth factor receptors are collectively known, have a characteristic protein structure consisting of three principal domains: (1) the extracellular ligand-binding domain, (2) the transmembrane domain, and (3) the intracellular TK catalytic domain (Figure 5). RTKs are molecular machines that transmit information in a unidirectional manner across the cell membrane. The binding of a growth factor to the extracellular ligand-binding domain of the receptor results in the activation of the intracellular TK catalytic domain, usually after dimerization, leading to the activation of downstream proteins that physically interact with RTK, mainly represented by the mitogen-activated protein kinases (MAPKs) pathway, by the PI3K/AKT axis and by STAT proteins. These pathways are activated differently, depending on the specific RTK, overall resulting in abnormal cell duplication and in escaping from programmed cell death (apoptosis).28
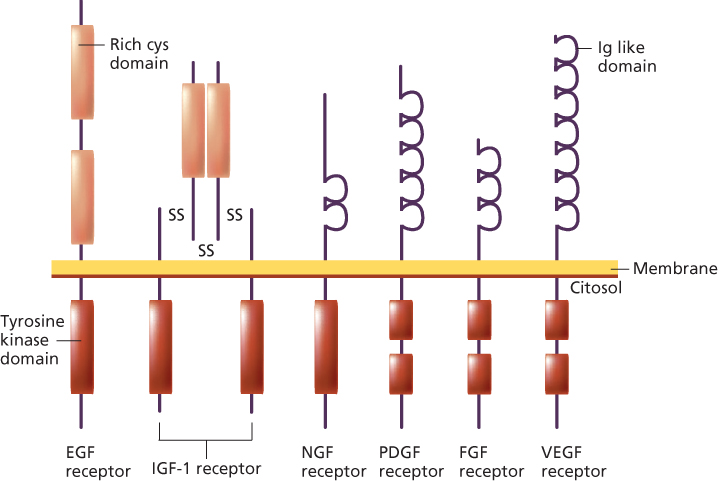
Figure 5 Representative examples of tyrosine kinase receptor families.
The list of RTK includes ERB B1, ERB B2, FMS, KIT, MET, RET, ROS, ALK, and TRK, which can be converted into oncogenes through different mechanisms, often depending on tumor type.28 ERB B1 (the epidermal growth factor receptor—EGFR) can be oncogenically activated by deletion of the ligand-binding domain, by point mutations in the TK domain in a subgroup of patients affected by non-small-cell lung cancer (NSCLC) (more commonly in Japan than in other western countries, suggesting that ethnic differences may have a significant impact on ERB B1 activation),29 or at germ-line level in patients showing multiple lung adenocarcinomas,30 by overexpression of ligands or by gene amplification in colorectal cancer (CRC) patients.31, 32
Another example is represented by ERB B2, whose growth factor is still unknown, which is altered by gene amplification in breast and gastric cancers and by point mutations in gastric, colorectal, and breast cancers. Interestingly, immunohistochemical staining for ERB B2 revealed no differences between tumors with or without ERB B2 mutations, indicating that overexpression probably does not accompany the mutation.33
Last example is KIT, whose activation in tumor cells can be due to three different mechanisms: (1) autocrine and/or paracrine stimulation of the receptor by its ligand, stem cell factor (SCF); (2) cross-activation by other kinases and/or loss of regulatory phosphatase activity; and (3) acquisition of activating mutations of several different exons of the KIT gene.34 KIT mutations (typically in exon 17) are most commonly found in mastocytosis/mast cell leukemia, acute myelogenous leukemia (AML), seminoma/dysgerminoma, and sinosal natural killer/T-cell lymphoma. In gastrointestinal stromal tumors (GISTs), more heterogeneous mutations are described: mutations most commonly occur in exon 11 (encoding the juxtamembrane region) (in about 65% of all GISTs), whereas about 10% of GISTs have kit exon 9 mutations and 2% exon 13 or exon 17 mutations. In GIST showing any alterations in the KIT gene, point mutations may occur in PDGFRA, a gene belonging to the same family as KIT. Approximately 5% of GIST have a constitutively activating mutations in PDGFRA, mostly (80%) found in exon 18 and the rest either in exon 12 (10–15%) or 14 (1–5%).34 KIT and PDGFRA mutations are mutually exclusive. Gastric GISTs with exon 11 deletions are more aggressive than those with substitutions. The less common KIT exon 9 codons 502–503 duplication occurs predominantly in small intestinal GISTs. PDGFRA mutations are associated with gastric GISTs, epithelioid morphology and a less malignant course of disease.34 Germ-line mutations in KIT gene have been found in patients manifesting multiple GIST arising at earlier age, urticaria pigmentosa, melanocytic nevi, melanomas, achalasia, or neuronal hyperplasia of the mesentheric plexus.
Signal transducers
Mitogenic signals are transmitted from RTK on the cell surface to the cell nucleus through a series of complex interlocking pathways collectively referred to as the signal transduction cascade.28 This relay of information is accomplished in part by the stepwise phosphorylation of interacting proteins in the cytosol. Signal transduction also involves guanine nucleotide-binding proteins and second messengers such as the adenylate cyclase system. The first retroviral oncogene discovered, SRC, was subsequently shown to be involved in signal transduction.13, 28
Many proto-oncogenes are members of signal transduction pathways.35 These consist of two main groups: nonreceptor protein kinases and guanosine triphosphate (GTP)-binding proteins. The nonreceptor protein kinases are subclassified into TKs (e.g., ABL, LCK, and SRC) and serine/threonine kinases (e.g., RAF-1, MOS, and PIM-1). GTP-binding proteins with intrinsic guanosine triphosphatase (GTPase) activity are subdivided into monomeric and heterotrimeric groups. Monomeric GTP-binding proteins are members of the important RAS family of proto-oncogenes that includes HRAS, KRAS, and NRAS.36 Heterotrimeric GTP-binding proteins (G proteins) implicated as proto-oncogenes currently include GSP and GIP. Signal transducers are often converted to oncogenes by mutations that lead to their unregulated activity, which in turn leads to uncontrolled cellular proliferation.36
Transcription factors
Transcription factors are nuclear proteins that regulate the expression of target genes or gene families.37 Transcriptional regulation is mediated by protein binding to specific DNA sequences or DNA structural motifs (such as zinc fingers), usually located upstream of the target gene. The mechanism of action of transcription factors also involves binding to other proteins, sometimes in heterodimeric complexes with specific partners. Transcription factors are the final link in the signal transduction pathway that converts extracellular signals into modulated changes in gene expression.
Many proto-oncogenes are transcription factors that were discovered through their retroviral homologs.37 Examples include ERB A, ETS, FOS, JUN, MYB, and C-MYC. Together, fos and jun form the AP-1 transcription factor, which positively regulates a number of target genes whose expression leads to cell division.38 ERB A is the receptor for the T3 thyroid hormone, triiodothyronine.39 Proto-oncogenes that function as transcription factors are often activated by chromosomal translocations in hematologic and solid neoplasms. In certain types of sarcomas, chromosomal translocations cause the formation of fusion proteins involving the association of EWS gene with various partners and resulting in an aberrant tumor-associated transcriptional activity. Interestingly, a role of the adenovirus E1A gene in promoting the formation of fusion transcript fli1/ews in normal human fibroblasts was recently reported.40 An important example of a proto-oncogene with a transcriptional activity in human hematologic tumors is the C-MYC gene, which helps to control the expression of genes leading to cell proliferation.41 As will be discussed later in this chapter, the C-MYC gene (which is encoded for a nuclear DNA-binding protein belonging to the helix-loop-helix/leucine zipper superfamily, involved in transcriptional regulation) is frequently activated by chromosomal translocations in human leukemia and lymphoma.
Programed cell death regulation
Normal tissues exhibit a regulated balance between cell proliferation and cell death. Programmed cell death is an important component in the processes of normal embryogenesis and organ development. A distinctive type of programmed cell death, called apoptosis, has been described for mature tissues.42 Studies of cancer cells have shown that both uncontrolled cell proliferation and failure to undergo programmed cell death can contribute to neoplasia and insensitivity to anticancer treatments.
The first proto-oncogene shown to regulate programmed cell death is BCL-2, discovered in human lymphomas. Experimental studies show that BCL-2 activation inhibits (in a dominant mode) programmed cell death in lymphoid cell populations. The BCL-2 gene encodes a protein localized to the inner mitochondrial membrane, endoplasmic reticulum, and nuclear membrane. The mechanism of action of the bcl-2 protein has not been fully elucidated, but studies indicate that it functions in part as an antioxidant that inhibits lipid peroxidation of cell membranes,43 and in part through protein–protein interaction with homolog proteins. Site-directed mutagenesis of BH1 and BH2 domains showed that these two regions are important for binding of bcl-2 to bax, a member of the bcl-2-family that promotes cell death and whose interaction with bcl-2 is necessary to regulate the apoptotic pathway (Figure 6). Although translocation is the main mechanism of BCL-2 gene activation, BCL-2 point mutations (in high-grade B-cell lymphomas) and amplification (in about 30% of high-grade diffuse large cell lymphomas (DLCL) lacking bcl-2 translocation) have been reported.44 Clinical relevance of BCL-2 expression has been shown in solid tumors, such as breast, prostate, thyroid, and lung.45, 46
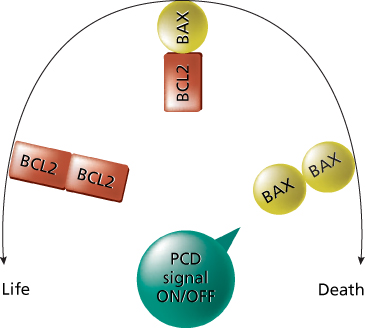
Figure 6 Effect of bcl-2 activity on the control of the cell life. In the presence of BAX only, the cell goes to apoptosis. bcl-2 regulates the cycle of the cell by the interaction with BAX. When bcl-2 is overexpressed, the cell cycle is deregulated and the apoptosis is prevented, eventually leading to tumor formation. This is an important cause for tumor formation. Abbreviations: PCD, program cell death or apoptosis.
The second oncogene involved in apoptosis is caspase-9, which is activated by the following intrinsic pathway. Release of cytochrome c into the cytosol results in activation of the caspase adaptor Apaf-1 and procaspase-9, which form a holoenzyme complex named apoptosome.47 Caspase-9, in turn, activates downstream caspases, especially caspase-3 and also caspase-6, 7, and 8, leading to DNA fragmentation and apoptosis. It has been demonstrated that Akt may regulate apoptosome function by phosphorylation of caspase-9 at the Ser-196 level.48 This phosphorylation event has relevant functional consequences, leading to the suppression of apoptosis caspase-9-mediated. The Akt suppression is specific for caspase-9 and probably is due to the inactivation of the intrinsic catalytic activity. More recently, it has been demonstrated that bax is also involved, leading to caspase-9 stimulation (through Apaf-1) in response to mitochondrial membrane damage.49
Mechanisms of oncogene activation
The activation of oncogenes involves genetic changes to cellular proto-oncogenes. The consequence of these genetic alterations is to confer a growth advantage to the cell. Four genetic mechanisms activate oncogenes in human neoplasms: (1) mutation, (2) gene amplification, (3) chromosome rearrangements, and (4) overexpression. The first three mechanisms result in either an alteration of proto-oncogene structure or an increase in proto-oncogene expression (Figure 7). Because neoplasia is a multistep process, more than one of these mechanisms may occur, leading to a combination of proto-oncogene activation and tumor-suppressor gene loss or inactivation.
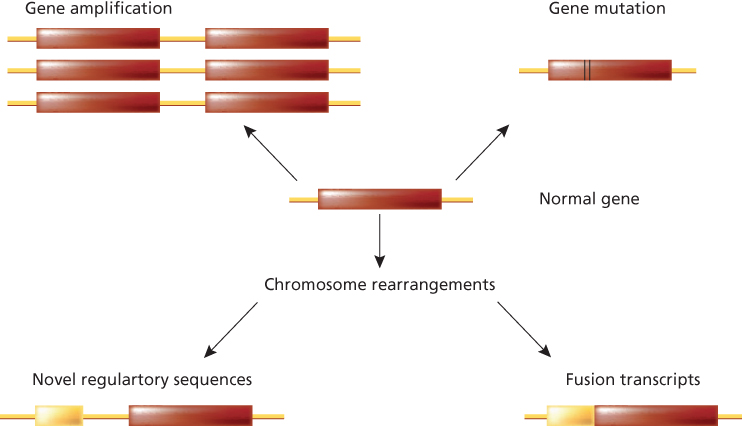
Figure 7 Schematic representation of the main mechanisms of oncogene activation (from proto-oncogenes to oncogenes). The normal gene (proto-oncogene) is depicted with its transcribed portion (rectangle). In the case of gene amplification, the latter can be duplicated 100-fold, resulting in an excess of normal protein. A similar situation can occur when following chromosome rearrangements such as translocation, the transcription of the gene is now regulated by novel regulatory sequences belonging to another gene. In the case of point mutation, single amino acid substitutions can alter the biochemical properties of the gene product, causing, in the example, its constitutive enzymatic activation. Chromosome rearrangements, such as translocation and inversion, can then generate fusion transcripts resulting in chimericoncogenic proteins.
Mutation
Mutations activate proto-oncogenes through structural alterations in their encoded proteins. These alterations, which usually involve critical protein regulatory regions or directly the catalytic domain, often lead to the uncontrolled, continuous activity of the mutated protein. Various types of mutations, such as base substitutions, deletions, and insertions, are capable of activating proto-oncogenes.50 Retroviral oncogenes, for example, often have deletions that contribute to their activation. Examples include deletions in the amino-terminal ligand-binding domains of the ERB B, KIT, ROS, MET, and TRK oncogenes. In human tumors, however, most characterized oncogene mutations are base substitutions (point mutations) that change a single amino acid within the protein.
Point mutations are frequently detected in the ras family of protooncogenes (KRAS, HRAS, and NRAS). The human ras genes encode for membrane-bound 21 kd proteins (189 amino acids) involved in signal transduction, with a guanine nucleotide-binding activity as well as an intrinsic GTPase activity. When activated, ras proteins transduce the signal by linking TKs to downstream serine/threonine kinases, such as raf, and MAP kinases (Figure 8).51 Stabilization of ras proteins in their active state causes a continuous flow of signal transduction, which results in malignant transformation. This status can be achieved after point mutation, mainly at codon 12 (in KRAS gene), 13 and 61 (mainly in NRAS gene) level. Rare mutations can be detected in codons 59, 117, and 146.52 Mutations of ras in human tumors have been linked to carcinogen exposure: for example, the occurrence of KRAS mutations in NSCLC seems to be due to smoking exposure, in particular to benzoapyrene.53 It has been estimated that as many as 15–20% of unselected human tumors may contain a RAS mutation. Mutations in KRAS predominate in tumors derived from endodermal tumors, including carcinomas of pancreas (90%), colorectal (40%) and lung.36, 51 This finding is due to the fact that KRAS, but not HRAS or NRAS, promotes the expansion of an endodermal stem/progenitor cell and blocks its differentiation.54 NRAS mutations are preferentially found in hematologic malignancies, with up to a 25% incidence in AMLs and myelodysplastic syndromes, or in melanoma and in a subgroup of CRC.55 The majority of thyroid carcinomas have been found to have RAS mutationsdistributed among KRAS, HRAS, and NRAS, without preference for a single RAS family member, but showing an association with the follicular type of differentiated thyroid carcinomas. Moreover, again in CRC, the identification of KRAS mutations in stool samples may be used in early diagnosis,56 and in plasma samples for monitoring patients’ follow-up.57
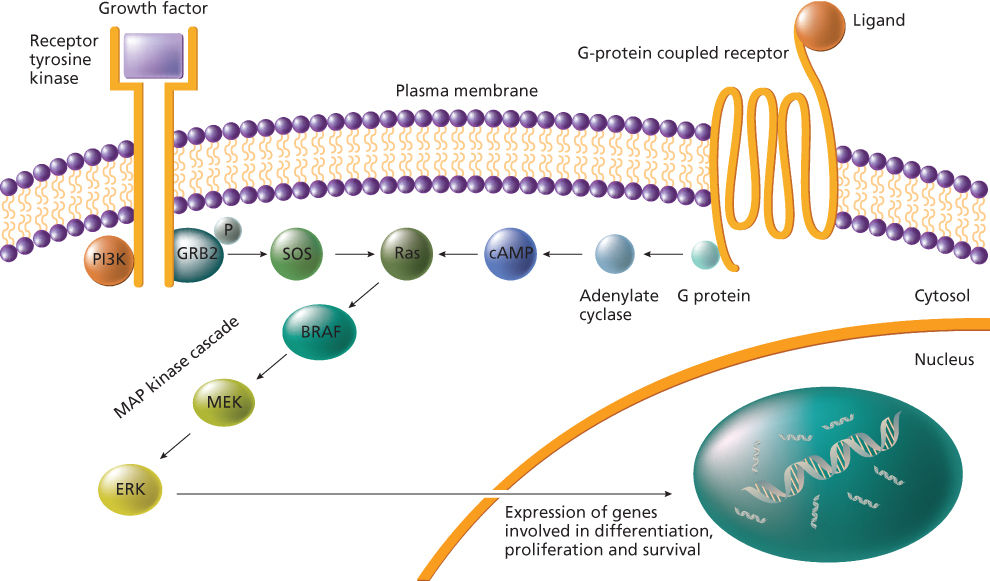
Figure 8 The ras–raf–MAPK signaling pathway.
In addition to cancer, RAS mutations are involved in other diseases. Specifically, NRAS mutations cause a human autoimmune lymphoproliferative syndrome,58 while HRAS and KRAS germ-line mutations underlie disorders of the Noonan syndrome spectrum.51 The last data, coupled with the evidence that cancers rarely occur in these individuals, is leading to a re-evaluation of the effective role played by RAS genes in carcinogenesis.
Another example of activating point mutations is represented by those occurring in BRAF gene, the first result of the human genome project in the screening of cancer genes using high-throughput genomic technologies.7 The BRAF gene product is recruited to the plasma membrane upon binding to ras-GTP and represents a key point in the signal transduction through the MAP kinase pathway (Figure 8). The most common oncogenic mutation of BRAF, occurring in more than 90% of cases, is the valine to glutamic acid change at codon 600, which mimics the phosphorylation of threonine 599 and serine 602, required for BRAF activation.7 In tissue specimens, BRAF mutations occur in melanomas (75%), thyroid (45%), colorectal (12%), and ovarian cancer (14%)59 and in acute lymphoblastic leukemia (ALL).60 Furthermore, it has been shown that BRAF mutations occur in CRC only when tumors do not carry mutations in the KRAS gene, and in papillary thyroid carcinoma when RET or TRK rearrangements are absent. These mutual exclusions have led to the assumption that BRAF and K-ras alterations, or BRAF, RET, and TRK alterations, could have the same functional effect in colorectal or in thyroid carcinogenesis, respectively.59, 61 As regards CRC, BRAF mutations are frequently present in sporadic cases with methylated hMLH1 promoter but not in HNPCC-related cancers, thus representing a possible strategy for exclusion criteria for HNPCC.62 Moreover, BRAF mutations are frequently found in hyperplastic polyps and serrated adenomas,63 suggesting that they represent an early and critical event in these types of lesions. BRAF mutations may have an influence on patients’ prognosis depending on the tumor type where they occur: in ovarian cancer, BRAF mutations are associated with type I tumors that are slow growing and generally confined to the ovary at diagnosis64 and with the aggressive papillary thyroid and colorectal carcinomas.59 BRAF mutations, especially the V600E change that is very easy to investigate, are now acquiring a role in early diagnosis or disease monitoring in patients’ follow-up: indeed, they are investigated to improve the diagnosis in fine-needle aspiration biopsy with cytological findings suspicious for papillary thyroid carcinomas.65 On the contrary, in melanoma, BRAF mutations could be only used to monitor in serum the follow-up of patients receiving biochemotherapy66 and not for early diagnosis because they even precede the neoplastic transformation, as all types of nevi besides Spitz and Blue nevi show BRAF alterations at a high frequency.67
Another significant example of activating point mutations is represented by those affecting the RET proto-oncogene in multiple endocrine neoplasia (MEN) type 2 (2A and 2B) syndrome and familial medullary thyroid carcinomas (FMTC). MEN 2A is associated most frequently with mutations of codon 634 (85%), particularly C634R. This germ-line point mutation affecting one of the cysteine residues located in the juxtamembrane domain of the ret receptor has been found to confer an oncogenic potential through the abnormal formation of intermolecular disulfide bonding, resulting in a ligand-independent activation of the TK activity of the receptor. Most MEN2B patients carry the M918T mutation in the kinase domain, which confers an aggressive phenotype to MEN2B subtype. Sporadic
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


