Obesity, metabolic syndrome, and disorders of energy balance
NEUROENDOCRINE REGULATION OF ENERGY BALANCE
The Hypothalamus and the Starvation Response
The Nucleus Accumbens and the Hedonic Pathway of Food Reward
The Amygdala and the Stress Response
METABOLIC IMPACT OF CHILDHOOD OBESITY
FACTORS ASSOCIATED WITH THE CURRENT EPIDEMIC OF OBESITY
“Classic” Endocrine Disorders with an Obesity Phenotype
Monogenetic Disorders of the Negative Feedback Pathway
Pleiotropic Obesity/Mental Retardation Disorders
EVALUATION AND TREATMENT OF THE OBESE CHILD
Introduction
Energy balance is the “final frontier” of endocrinology. Prior to 1994, with the discovery of leptin, disorders of energy balance were not even considered to be endocrine diseases. Today, obesity can account for up to 25% of pediatric endocrine practice referrals, and type 2 diabetes accounts for up to 33% of the new referrals for pediatric diabetes, the majority of whom are also obese. Since the discovery of leptin, the negative feedback pathway of energy balance has been elucidated, and endocrinologists have embraced disorders of energy balance as part of their management portfolio. Thus, the study of energy balance has become a matter of continuing education for pediatric endocrinologists. The whole field is a work in progress, which is problematic because our diagnostic armamentarium and our treatment options for the most part do not target the multiple hormonal feedback loops that govern energy balance. This chapter conveys a clear and up-to-date basic understanding of the energy balance pathway and provides a clinical rationale and formulation for evaluating and treating these patients.
Neuroendocrine regulation of energy balance
The negative feedback axis of energy balance and its function during homeostasis has been largely delineated through studies in animal models; human data are presented where available. The axis is composed of three arms (Figure 22-1). The first is the afferent arm, which conveys peripheral information on hunger and peripheral metabolism, in the form of hormonal and neural inputs, to the hypothalamus. The second is a central processing unit, consisting of various areas within the hypothalamus. The ventromedial hypothalamus (VMH; consisting of the ventromedial [VMN] and arcuate [ARC] nuclei), integrates the afferent peripheral signals, along with other central stimuli; the paraventricular nuclei (PVN) and lateral hypothalamic area (LHA) serve as a gated neurotransmitter system to alter neural signals for changes in feeding and energy expenditure. The third component is the efferent arm and consists of a complex network of autonomic effectors, which regulate energy intake, and energy expenditure versus storage.1,2 Anatomic disruptions or genetic or metabolic alterations of the afferent, central processing, or efferent arms can alter energy intake or expenditure in stereotyped ways, which can lead to either obesity or cachexia.
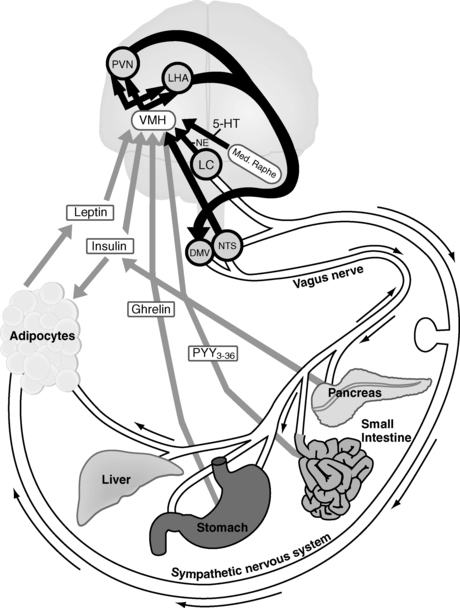
FIGURE 22-1  The homeostatic pathway of energy balance. Afferent (gray), central (black), and efferent (white) pathways are delineated. The hormones insulin, leptin, ghrelin, and peptide YY(3-36) (PYY3–36) provide afferent information to the ventromedial hypothalamus regarding short-term energy metabolism and energy sufficiency. From there, the ventromedial hypothalamus elicits anorexigenic (α-melanocyte stimulating hormone, cocaine-amphetamine regulated transcript) and orexigenic (neuropeptide Y, agouti-related protein) signals to the melanocortin-4 receptor in the paraventricular nucleus and lateral hypothalamic area. These lead to efferent output via the locus coeruleus, via the nucleus tractus solitarius, which activates the sympathetic nervous system, causing the adipocyte to undergo lipolysis, or via the dorsal motor nucleus of the vagus, which activates the vagus nerve to store energy, both by increasing pancreatic insulin secretion, and (in rodents) by increasing adipose tissue sensitivity to insulin. 5-HT, serotonin (5-hydroxytryptamine); DMV, dorsal motor nucleus of the vagus; LC, locus coeruleus; LHA, lateral hypothalamic area; NE, norepinephrine; NTS, nucleus tractus solitarius; PVN, paraventricular nucleus; VMH, ventromedial hypothalamus. (From Lustig, R. H. (2006). Childhood obesity: behavioral aberration or biochemical drive? Reinterpreting the First Law of Thermodynamics. Nature Clin Pract Endo Metab, 2, 447–458. Courtesy of Nature Publishing Group, with permission.)
The homeostatic pathway of energy balance. Afferent (gray), central (black), and efferent (white) pathways are delineated. The hormones insulin, leptin, ghrelin, and peptide YY(3-36) (PYY3–36) provide afferent information to the ventromedial hypothalamus regarding short-term energy metabolism and energy sufficiency. From there, the ventromedial hypothalamus elicits anorexigenic (α-melanocyte stimulating hormone, cocaine-amphetamine regulated transcript) and orexigenic (neuropeptide Y, agouti-related protein) signals to the melanocortin-4 receptor in the paraventricular nucleus and lateral hypothalamic area. These lead to efferent output via the locus coeruleus, via the nucleus tractus solitarius, which activates the sympathetic nervous system, causing the adipocyte to undergo lipolysis, or via the dorsal motor nucleus of the vagus, which activates the vagus nerve to store energy, both by increasing pancreatic insulin secretion, and (in rodents) by increasing adipose tissue sensitivity to insulin. 5-HT, serotonin (5-hydroxytryptamine); DMV, dorsal motor nucleus of the vagus; LC, locus coeruleus; LHA, lateral hypothalamic area; NE, norepinephrine; NTS, nucleus tractus solitarius; PVN, paraventricular nucleus; VMH, ventromedial hypothalamus. (From Lustig, R. H. (2006). Childhood obesity: behavioral aberration or biochemical drive? Reinterpreting the First Law of Thermodynamics. Nature Clin Pract Endo Metab, 2, 447–458. Courtesy of Nature Publishing Group, with permission.)
The afferent system
Alimentary afferents
Hunger
The afferent vagus.
The vagus nerve is the primary neural connection between the brain and the gut. The afferent vagus nerve conveys sensory information regarding the mechanical stretch of the stomach and duodenum and sensations of gastric fullness to the nucleus tractus solitarius (NTS).3 Of note is that each of the following alimentary neuropeptide effects on hunger and satiety is obviated by concomitant vagotomy, implicating the afferent vagus as the primary mediator of alimentary energy balance signals.4–6
Ghrelin.
Ghrelin, an octanoylated 28-amino-acid peptide, was discovered serendipitously while looking for the endogenous ligand of the growth hormone secretagogue receptor (GHS-R).7 Ghrelin induces the release of growth hormone (GH) through stimulation of the pituitary GHS-R. The endogenous secretion of ghrelin from the fasting stomach is high, but the administration of nutrients decreases it; volumetric stretching of the stomach wall has no effect. However, ghrelin also binds to the GHS-R in the VMH, which increases hunger, food intake, and fat deposition.8,9 Ghrelin also increases the respiratory quotient (RQ) in rats, suggesting a reduction of fat oxidation and promotion of fat storage. Ghrelin appears to tie the lipolytic effect of GH with the hunger signal and is probably important in the acute response to fasting. In humans, ghrelin levels rise with increasing subjective hunger and peak at the time of voluntary food consumption,10 suggesting that ghrelin acts on the VMH to trigger meal initiation. Ghrelin infusion increases food intake in humans.11 However, plasma ghrelin levels are low in obese individuals and increase with fasting,12 suggesting that ghrelin is a response to, rather than a cause of, obesity.
Satiety
Peptide YY 3-36 (PYY3-36).
A hormonal signal to control meal volume is PYY3-36.13 This peptide fragment is secreted by intestinal L cells in response to the exposure to nutrient, crosses the blood-brain barrier, and binds to the Y2 receptor in the VMH. Activation of this receptor causes a decrease in neuropeptide Y (NPY) mRNA in neurons of the orexigenic arm of the central processing system (discussed later). In nonobese humans, infusion of PYY3-36 during a 12-hour period decreased the total amount of food ingested from 2200 to 1500 kcal, but without an effect on food ingested during the next 12-hour interval.13 Although the pharmacology of this peptide is being elucidated, its specific role in obesity is not yet known.
Glucagon-like peptide-1 (GLP-1).
Those same intestinal L cells produce GLP-1 through the posttranslational processing of preproglucagon. Two equipotent forms of GLP-1 are generated: a glycine-extended form GLP-1 (7–37) and the amidated peptide GLP-1(7–36)amide.14 GLP-1 acts on the stomach to inhibit gastric emptying; this prolongs the time it takes to absorb a meal. GLP-1 also activates its receptor on pancreatic β cells to stimulate cAMP production, protein kinase A activation, and insulin secretion (Figure 22-2), thereby improving glucose tolerance, a mechanism of the “incretin” effect. GLP-1 also acts on rodent β cells to stimulate neogenesis, thereby increasing β-cell mass.15 Lastly, GLP-1 also exerts potent effects on reduction of appetite, both through reduction in gastric emptying and through direct decreases of corticotropin-releasing hormone (CRH) signaling in the PVN and increasing leptin signaling in the VMH.16
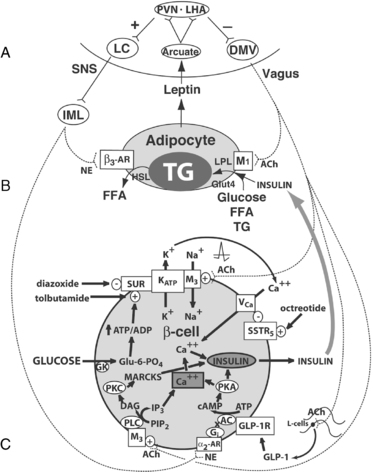
FIGURE 22-2  Central regulation of leptin signaling, autonomic innervation of the adipocyte and β cell, and the starvation response. A, The arcuate nucleus transduces the peripheral leptin signal as one of sufficiency or deficiency. In leptin sufficiency, efferents from the hypothalamus synapse in the locus coeruleus, which stimulates the sympathetic nervous system. In leptin deficiency or resistance, efferents from the hypothalamus stimulate the dorsal motor nucleus of the vagus. B, Autonomic innervation and hormonal stimulation of white adipose tissue. In leptin sufficiency, norepinephrine binds to the β3-adrenergic receptor, which stimulates hormone-sensitive lipase, promoting lipolysis of stored triglyceride into free fatty acids. In leptin deficiency or resistance, vagal acetylcholine increases adipose tissue insulin sensitivity (documented only in rats to date), promotes uptake of glucose and free fatty acids for lipogenesis, and promotes triglyceride uptake through activation of lipoprotein lipase. C, Autonomic innervation and hormonal stimulation of the β cell. Glucose entering the cell is converted to glucose-6-phosphate by the enzyme glucokinase, generating ATP, which closes an ATP-dependent potassium channel, resulting in cell depolarization. A voltage-gated calcium channel opens, allowing for intracellular calcium influx, which activates neurosecretory mechanisms leading to insulin vesicular exocytosis. In leptin sufficiency, norepinephrine binds to α2-adrenoceptors on the β-cell membrane to stimulate inhibitory G proteins, decrease adenyl cyclase and its product cAMP, and thereby reduce protein kinase A levels and insulin release. In leptin deficiency or resistance, the vagus stimulates insulin secretion through three mechanisms.99 First, acetylcholine binds to a M3 muscarinic receptor, opening a sodium channel, which augments the ATP-dependent cell depolarization, increasing the calcium influx, and insulin exocytosis. Second, acetylcholine activates a pathway that increases protein kinase C, which also promotes insulin secretion. Third, the vagus innervates L cells of the small intestine, which secrete glucagon-like peptide-1, which activates protein kinase A, contributing to insulin exocytosis. Octreotide binds to a somatostatin receptor on the β cell, which is coupled to the voltage-gated calcium channel, limiting calcium influx and the amount of insulin released in response to glucose (reprinted with kind permission of Springer Science and Business media). α2-AR, α2-adrenergic receptor; β3-AR, β3-adrenergic receptor; AC, adenyl cyclase; ACh, acetylcholine; DAG, diacylglycerol; DMV, dorsal motor nucleus of the vagus; FFA, free fatty acids; Gi, inhibitory G protein; GK, glucokinase; GLP-1, glucagon-like peptide-1; GLP-1R, GLP-1 receptor; Glu-6-PO4, glucose-6-phosphate; Glut4, glucose transporter-4; HSL, hormone-sensitive lipase; IML, intermediolateral cell column; IP3, inositol triphosphate; LC, locus coeruleus; LHA, lateral hypothalamic area; LPL, lipoprotein lipase; MARCKS, myristoylated alanine-rich protein kinase C substrate; NE, norepinephrine; PIP2, phosphatidylinositol pyrophosphate; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; PVN, paraventricular nucleus; SSTR5, somatostatin-5 receptor; TG, triglyceride; VCa, voltage-gated calcium channel; VMH, ventromedial hypothalamus; SUR, sulfonylurea receptor. (From Lustig, R. H. (2006). Childhood obesity: behavioral aberration or biochemical drive? Reinterpreting the first law of thermodynamics. Nature Clin Pract Endo Metab, 2, 447–458. Courtesy of Nature Publishing Group, with permission.)
Central regulation of leptin signaling, autonomic innervation of the adipocyte and β cell, and the starvation response. A, The arcuate nucleus transduces the peripheral leptin signal as one of sufficiency or deficiency. In leptin sufficiency, efferents from the hypothalamus synapse in the locus coeruleus, which stimulates the sympathetic nervous system. In leptin deficiency or resistance, efferents from the hypothalamus stimulate the dorsal motor nucleus of the vagus. B, Autonomic innervation and hormonal stimulation of white adipose tissue. In leptin sufficiency, norepinephrine binds to the β3-adrenergic receptor, which stimulates hormone-sensitive lipase, promoting lipolysis of stored triglyceride into free fatty acids. In leptin deficiency or resistance, vagal acetylcholine increases adipose tissue insulin sensitivity (documented only in rats to date), promotes uptake of glucose and free fatty acids for lipogenesis, and promotes triglyceride uptake through activation of lipoprotein lipase. C, Autonomic innervation and hormonal stimulation of the β cell. Glucose entering the cell is converted to glucose-6-phosphate by the enzyme glucokinase, generating ATP, which closes an ATP-dependent potassium channel, resulting in cell depolarization. A voltage-gated calcium channel opens, allowing for intracellular calcium influx, which activates neurosecretory mechanisms leading to insulin vesicular exocytosis. In leptin sufficiency, norepinephrine binds to α2-adrenoceptors on the β-cell membrane to stimulate inhibitory G proteins, decrease adenyl cyclase and its product cAMP, and thereby reduce protein kinase A levels and insulin release. In leptin deficiency or resistance, the vagus stimulates insulin secretion through three mechanisms.99 First, acetylcholine binds to a M3 muscarinic receptor, opening a sodium channel, which augments the ATP-dependent cell depolarization, increasing the calcium influx, and insulin exocytosis. Second, acetylcholine activates a pathway that increases protein kinase C, which also promotes insulin secretion. Third, the vagus innervates L cells of the small intestine, which secrete glucagon-like peptide-1, which activates protein kinase A, contributing to insulin exocytosis. Octreotide binds to a somatostatin receptor on the β cell, which is coupled to the voltage-gated calcium channel, limiting calcium influx and the amount of insulin released in response to glucose (reprinted with kind permission of Springer Science and Business media). α2-AR, α2-adrenergic receptor; β3-AR, β3-adrenergic receptor; AC, adenyl cyclase; ACh, acetylcholine; DAG, diacylglycerol; DMV, dorsal motor nucleus of the vagus; FFA, free fatty acids; Gi, inhibitory G protein; GK, glucokinase; GLP-1, glucagon-like peptide-1; GLP-1R, GLP-1 receptor; Glu-6-PO4, glucose-6-phosphate; Glut4, glucose transporter-4; HSL, hormone-sensitive lipase; IML, intermediolateral cell column; IP3, inositol triphosphate; LC, locus coeruleus; LHA, lateral hypothalamic area; LPL, lipoprotein lipase; MARCKS, myristoylated alanine-rich protein kinase C substrate; NE, norepinephrine; PIP2, phosphatidylinositol pyrophosphate; PKA, protein kinase A; PKC, protein kinase C; PLC, phospholipase C; PVN, paraventricular nucleus; SSTR5, somatostatin-5 receptor; TG, triglyceride; VCa, voltage-gated calcium channel; VMH, ventromedial hypothalamus; SUR, sulfonylurea receptor. (From Lustig, R. H. (2006). Childhood obesity: behavioral aberration or biochemical drive? Reinterpreting the first law of thermodynamics. Nature Clin Pract Endo Metab, 2, 447–458. Courtesy of Nature Publishing Group, with permission.)
Metabolic afferents
Leptin.
Energy intake versus expenditure is normally regulated very tightly (within 0.15% per year) by the hormone leptin. Leptin is a 167-amino-acid hormone, produced by adipocytes, which transmits the primary long-term signal of energy depletion/repletion to the VMH.17,18 Leptin’s primary neuroendocrine role is to mediate information about the size of peripheral adipocyte energy stores to the VMH. Leptin is a prerequisite signal to the VMH for the initiation of high-energy processes, such as puberty and pregnancy.19,20 Leptin reduces food intake and increases the activity of the sympathetic nervous system (SNS).21 Conversely, low circulating levels of leptin infer diminished energy stores, which signal via the VMH to reduce energy expenditure, inhibit metabolic processes, and increase appetite. Serum leptin concentrations drop precipitously and in excess of body fat loss during periods of short-term fasting,22,23 and it seems likely that leptin functions primarily as a peripheral signal to the hypothalamus of inadequate caloric intake rather than specifically as a satiety signal.24
In the fed state, circulating levels of leptin correlate with the percentage of body fat.25,26 Leptin production by adipocytes is stimulated by insulin and glucocorticoids27,28 and is inhibited by β-adrenergic stimulation.24 Programming of relative leptin concentrations by early caloric intake may be one mechanism that links early overnutrition with later obesity.29
Leptin binds to its receptor (a member of the cytokine receptor superfamily) on target VMH neurons. Four receptor isoforms are formed by differential mRNA splicing: ObRa, an isoform with a shortened intracellular domain, which may function as a transporter; ObRb, the intact full-length receptor; ObRc, also with a short intracellular domain; and ObRe, without an intracellular domain, but which may function as a soluble receptor.30 As leptin binds to its VMH receptor, three neuronal signals are transduced. The first is the opening of an adenosine triphosphate (ATP)–sensitive potassium channel, which hyperpolarizes the neuron and decreases its firing rate.31 The second is the activation of a cytoplasmic janus kinase 2 (JAK2), which phosphorylates a tyrosine moiety on proteins of a family called signal transducers and activators of transcription (STAT-3).32 The phosphorylated STAT-3 translocates to the nucleus, where it promotes leptin-dependent gene transcription.33 However, leptin also activates the insulin receptor substrate 2/phosphatidyl inositol-3-kinase (IRS2/PI3K) second messenger system in VMH neurons, which increases neurotransmission of the central anorexigenic signaling pathway.34
Insulin.
Insulin plays an extremely important role in energy balance,35 as it is part of both the afferent and efferent systems. On the afferent side, there is a significant insulin receptor density in a subpopulation of VMH neurons,36 and there is coordinated transport of insulin across the blood-brain barrier,37 suggesting a central role for this hormone. In animals, acute and chronic intracerebroventricular (ICV) infusions of insulin decrease feeding behavior and induce satiety.38–40 The data on acute and chronic peripheral insulin infusions are less clear. Studies of overinsulinized diabetic rats demonstrate increased caloric intake (to prevent subacute hypoglycemia) and the development of peripheral insulin resistance.41,42 Chronic experimental peripheral insulin infusions decrease hepatic and skeletal muscle glucose uptake by decreasing Glut4 expression, but they do not alter adipose tissue glucose uptake.43,44 One study on humans showed that injecting short-term insulin peripherally during meals did not have an effect on satiety.45
Insulin normally activates the insulin receptor substrate 2/phosphatidyl inositol-3-kinase (IRS2/PI3K) second messenger system in VMH neurons,46 which increases neurotransmission of the central anorexigenic signaling pathway (discussed later). The importance of central nervous system (CNS) insulin action was underscored by the development of a brain/neuron–specific insulin receptor knockout (NIRKO) mouse, which cannot transduce a CNS insulin signal.47 Such mice become hyperphagic, obese, and infertile, with high peripheral insulin levels. These data suggest that peripheral insulin mediates a satiety signal in the VMH to help control energy balance.48 Various knockouts of the insulin signal transduction pathway that reduce insulin signaling lead to an obese phenotype,49,50 whereas those that improve insulin signaling lead to a lean phenotype.51,52
Central processing
The peripheral afferent signals outlined earlier reach neurons in the VMH, where they are integrated by a gated neural circuit, designed to promote or diminish both energy intake and expenditure (see Figure 22-2). This circuit consists of two arms: the anorexigenic arm, which contains neurons expressing the colocalized peptides proopiomelanocortin (POMC) and cocaine/amphetamine-regulated transcript (CART); and the orexigenic arm, which contains neurons with the colocalized peptides neuropeptide Y (NPY) and agouti-related protein (AgRP). Ghrelin receptor-immunoreactivity colocalizes with NPY and AgRP neurons, whereas insulin and leptin receptors are located on both POMC/CART and NPY/AgRP neurons in the VMH,53 suggesting divergent regulation of each arm. These two arms compete for occupancy of melanocortin receptors (MCRs; either MC3R or MC4R) in the PVN and LHA.
Anorexigenesis, POMC/α-MSH, and CART
POMC is differentially cleaved in different tissues and neurons. The ligand α-melanocyte stimulating hormone (α-MSH) is the primary product involved in anorexigenesis. Both overfeeding and peripheral leptin infusion induce the synthesis of POMC and α-MSH within the ARC.54 α-MSH induces anorexia by binding to melanocortin receptors within the PVN or LHA. CART is a hypothalamic neuropeptide induced by leptin and reduced by fasting. Intrahypothalamic infusion blocks appetite, whereas antagonism of endogenous CART increases caloric intake.55
Orexigenesis, NPY, and AgRP
NPY and AgRP colocalize to a different set of neurons within the ARC, immediately adjacent to those expressing POMC/CART.56 NPY has numerous functions within the hypothalamus, including initiation of feeding, puberty, regulation of gonadotropin secretion, and adrenal responsiveness.57,58 NPY is the primary orexigenic peptide. ICV infusion of NPY in rats rapidly leads to hyperphagia, energy storage, and obesity,59,60 mediated through Y1 and Y5 receptors. Fasting and weight loss increase NPY expression in the ARC, accounting for increased hunger, whereas PYY3-36 (through Y2 receptors) and leptin decrease NPY mRNA13,61 AgRP is the human homolog of the protein agouti, which is present in abundance in the yellow (Ay-a) mouse.62 This protein is an endogenous competitive antagonist of all melanocortin receptors (MCRs), accounting for the yellow color in these mice. In the presence of large amounts of AgRP at the synaptic cleft in the PVN, α-MSH cannot bind to the MC4R to induce satiety.63
Other neuroendocrine modulators of energy balance
Norepinephrine (NE).
NE neurons in the locus coeruleus synapse on VMH neurons to regulate food intake.64 The actions of NE on food intake seem paradoxical, as intrahypothalamic NE infusions stimulate food intake through effects on central α2– and β-adrenergic receptors,65 whereas central infusion of α1-agonists markedly reduces food intake.66
Serotonin (5-HT).
5-HT has been implicated in the perception of satiety based on many lines of evidence: (1) injection of 5-HT into the hypothalamus increases satiety, particularly with respect to carbohydrate67; (2) central administration of 5-HT2c receptor agonists increase satiety, whereas antagonists induce feeding68; (3) administration of selective 5-HT reuptake inhibitors induce early satiety69; (4) leptin increases 5-HT turnover70; and (5) the 5-HT2cR-KO mouse exhibits increased food intake and body weight.71 The role of 5-HT in the transduction of the satiety signal may have both central and peripheral components, as intestinal 5-HT is secreted into the bloodstream during a meal, where it may have an impact on gastrointestinal (GI) neuronal function and muscle tone, and may bind to 5-HT receptors in the NTS (discussed previously) to promote satiety.72
Melanin-concentrating hormone (MCH).
MCH is a 17-amino-acid peptide expressed in the zona incerta and LHA. MCH neurons synapse on neurons in the forebrain and the locus coeruleus. MCH appears to be important in conditions such as anxiety and aggression around food.73 Expression of this peptide is up-regulated in ob/ob mice. MCH-knockout mice are hypophagic and lean,74 whereas transgenic MCH-overexpressing mice develop obesity and insulin resistance.75 ICV administration of MCH stimulates food intake, similar to that seen with NPY administration.76
Orexins A and B.
These 33- and 28-amino-acid peptides, respectively, have been implicated in both energy balance and autonomic function in mice.77 Orexin knockout mice demonstrate narcolepsy, hypophagia, and obesity,78 suggesting that orexins bridge the gap between the afferent and efferent energy balance systems.79 Orexins in the LHA stimulate neuropeptide Y (NPY) release, which may account for their effects on orexigenesis, and also stimulate the corticotropin-releasing factor (CRF) and sympathetic nervous system (SNS) output to increase wakefulness and energy expenditure, learning and memory, and the hedonic reward system (discussed later).80 Conversely, orexin neurons in the perifornical and dorsomedial hypothalamus regulate arousal and response to stress.
Endocannabinoids (EC).
It has long been known that tetrahydrocannabinol stimulates food intake. This observation led to the identification of endogenous EC and their receptor, termed CB1.81 The CB1 receptor is expressed in corticotropin-releasing factor (CRH) neurons in the PVN, in CART neurons in the VMN, and in MCH- and orexin-positive neurons in the LHA and perifornical region. Fasting and feeding are associated with high and low levels of ECs in the hypothalamus, respectively. For example, CB1 receptor-knockout mice have increased CRH and reduced CART expression. In the ob/ob mouse, hypothalamic EC levels are increased, whereas leptin infused intravenously reduces these levels, indicating that a direct negative control is exerted by leptin on the EC system. Glucocorticoids increase food intake by stimulating EC synthesis and secretion, whereas leptin blocks this effect.82 Finally, the presence of CB1 receptors on afferent vagal neurons suggest that endocannabinoids may be involved in mediating satiety signals originating in the gut.
Melanocortin receptors (MCR) and central neural integration
The human MC4R localizes to chromosome 2, and is a 7-transmembrane G-coupled receptor, encoded by an intron-less 1 kB gene. The binding of hypothalamic α-MSH to the MC4R in the PVN and LHA results in a state of satiety, whereas ICV administration of MC4R antagonists stimulates feeding, suggesting that MC4R transduces satiety information on caloric sufficiency. A different phenotype is observed in the MC3R knockout mouse. These animals are obese, but they are instead hypophagic and have increased body fat for their lean mass. They gain weight on either low- or high-fat chow and do not change substrate oxidation in response to changes in dietary fat content, suggesting a defect in energy expenditure.83 Thus, these two hypothalamic MCRs appear to modulate different aspects of energy metabolism. One hypothesis is that the MC4R modulates energy intake, whereas the MC3R modulates energy expenditure.84
The efferent system
The MCRs in the PVN and LHA transduce the anorexigenic and orexigenic information coming from the VMH, in order to modulate activity of the sympathetic nervous system (SNS), which promotes energy expenditure, and the efferent vagus, which promotes energy storage (see Figure 22-2). In this way, peripheral energy balance can be modulated acutely to provide requisite energy for metabolic needs, and store the rest.
The sympathetic nervous system (SNS) and energy expenditure
Anorexigenic pressure increases energy expenditure through activation of the SNS.85 For instance, leptin administration to ob/ob mice promotes increased brown adipose tissue lipolysis, thermogenesis, renovascular activity, and increased movement, all associated with increased energy expenditure, which assists in weight loss.86 Similarly, insulin administration acutely increases SNS activity in normal rats and in humans.87,88 The magnitude of energy expenditure also has a salutary effect on quality of life; factors that reduce resting energy expenditure (REE) (e.g., hypothyroidism) reduce quality of life, whereas factors that increase REE (e.g., caffeine) increase quality of life (at least acutely).
Activation of the SNS increases energy expenditure by the skeletal muscle, by activating β2-adrenergic receptors,89 which in turn increase the expression of numerous genes in skeletal muscle,90 especially those involved in carbohydrate metabolism. SNS activation stimulates glycogenolysis, incites myocardial energy expenditure, increases in glucose and fatty acid oxidation, and increases protein synthesis.91
Activation of the SNS in rodents stimulates the β3-adrenergic receptor of brown adipose tissue to promote lipolysis.92 In humans, activation of the β3-adrenergic receptor increases cAMP, which activates protein kinase A. PKA acts in two separate molecular pathways to increase energy expenditure. First, PKA phosphorylates the cyclic AMP response element binding protein (CREB), which induces expression of PPARγ-coactivator-1α (PGC-1α). PGC-1α then binds to enhancer elements on the uncoupling protein-1 (UCP1) gene, which increases the expression and activity of uncoupling proteins (UCPs) 1 and 2.93,94 Uncoupling proteins reduce the proton gradient across the inner membranes of mitochondria, thereby diverting protons from storage in the form of ATP to heat production. Originally, uncoupling proteins were discovered in brown adipose tissue and were found to be responsible for thermogenesis. UCP1 is an inner membrane mitochondrial protein that uncouples proton entry from ATP synthesis95; therefore, UCP1 expression dissipates energy as heat, thus reducing the energy efficiency of the adipose tissue. However, UCP2 has been found in most tissues and UCP3 in skeletal muscle. Second, PKA activation activates the enzyme hormone sensitive lipase (HSL), which is responsible for lipolysis of intracellular triglyceride to its component free fatty acids (FFAs). The FFAs also induce UCP1, further increasing energy expenditure. The FFAs released from the adipocyte also travel to the liver where they are utilized for energy by metabolizing into two-carbon fragments. Lipolysis reduces leptin expression; thus, a negative feedback loop is achieved between leptin and the SNS (see Figure 22-2).
The efferent vagus and energy storage
In response to declining levels of leptin or persistent orexigenic pressure, the LHA and PVN send efferent projections residing in the medial longitudinal fasciculus to the dorsal motor nucleus of the vagus nerve (DMV), activating the efferent vagus.96 The efferent vagus opposes the SNS by promoting energy storage in four ways: (1) by slowing the heart rate, myocardial oxygen consumption is reduced; (2) the vagus nerve promotes alimentary peristalsis, pyloric opening, and energy substrate absorption; (3) through direct effects on the adipocyte, the vagus nerve promotes insulin sensitivity to increase the clearance of energy substrate into adipose tissue; and (4) through effects on the β cells, the vagus increases postprandial insulin secretion, which promotes energy deposition into adipose tissue.97–100
Retrograde tracing of white adipose tissue reveals a wealth of efferents originating at the DMV.100 These efferents synapse on the M1 muscarinic receptor on the adipocyte, which increases insulin sensitivity of the adipocyte. Denervation of white adipose tissue results in a reduction of glucose and FFA uptake, and an induction of HSL, which promotes lipolysis—both of which reduce the efficiency of insulin-induced energy storage. Thus, vagal modulation of the adipocyte augments storage of both glucose and FFAs by improving adipose insulin sensitivity 101 (see Figure 22-2).
The DMV also sends efferent projections to the β cells of the pancreas.102 This pathway is responsible for the “cephalic” or preabsorptive phase of insulin secretion, which is glucose independent and can be blocked by atropine.103 Overactive vagal neurotransmission increases insulin secretion from β cells in response to an oral glucose load through three distinct but overlapping mechanisms104 (see Figure 22-2):
1. Vagal firing increases acetylcholine availability and binding to the M3 muscarinic receptor on the β cell, which is coupled to a sodium channel within the pancreatic β-cell membrane.105 As glucose enters the β cell after ingestion of a meal, the enzyme glucokinase phosphorylates glucose to form glucose-6-phosphate, increasing intracellular ATP, which induces closure of the ATP-dependent potassium channel. Upon channel closure, the β cell experiences an ATP concentration-dependent β cell depolarization106,107 and the opening of a separate voltage-gated calcium channel within the membrane. Intracellular calcium influx increases acutely, which results in rapid insulin vesicular exocytosis. Concomitant opening of the sodium channel by vagally mediated acetylcholine augments β-cell depolarization, which in turn augments the intracellular calcium influx and results in insulin hypersecretion.108–110
2. Vagally mediated acetylcholine increases phospholipases A2, C, and D within the β cell, which hydrolyze intracellular phosphatidylinositol to diacylglycerol (DAG) and inositol triphosphate (IP3).104 DAG is a potent stimulator of protein kinase C (PKC),111 which phosphorylates myristoylated alanine-rich protein kinase C substrate (MARCKS), which then binds actin and calcium-calmodulin and induces insulin vesicular exocytosis.112 IP3 potentiates the release of calcium within β cells from intracellular stores, which also promotes insulin secretion.113
3. The vagus also stimulates the release of GLP-1 from intestinal L cells, which circulates and binds to a GLP-1 receptor within the β-cell membrane. Activation of this receptor induces a calcium-calmodulin-sensitive adenyl cyclase, with conversion of intracellular ATP to cAMP, which then activates protein kinase A. PKA causes both the release of intracellular calcium stores and the phosphorylation of vesicular proteins, each contributing to an increase in insulin exocytosis.14,114
In the efferent pathway, insulin is responsible for shunting blood-borne nutrients into adipose for storage. Indeed, the primary hormonal signal for adipogenesis is insulin.115 Within the adipocyte, insulin increases (1) glut4 expression, (2) acetyl-CoA carboxylase, (3) fatty acid synthase, and (4) lipoprotein lipase.116 Thus, the net effect of insulin on the adipocyte is the rapid clearance and storage of circulating glucose and lipid. Thus, insulin promotes energy storage.
CNS modulation of food intake
The hypothalamus and the starvation response
The regulation of the various components of the energy balance system is manifest during the starvation response. Everyone has a “personal leptin threshold,” probably genetically set, above which the brain interprets a state of energy sufficiency.117 Thus, the leptin-replete state is characterized by increased physical activity, decreased appetite, and increased feelings of well-being. However, in response to caloric restriction, leptin levels decline even before weight loss is manifest,22,23 which the VMH interprets as starvation. Gastric secretion of ghrelin increases, which increases pituitary GH release, in order to stimulate lipolysis to provide energy substrate for catabolism. Ghrelin stimulates NPY/AgRP to antagonize α-MSH/CART. Decline of leptin reduces α-MSH/CART as well. This leads to decreased MC4R occupancy so that the reduced anorexigenic pressure on the MC4R increases feeding behavior and energy efficiency (with reduced fat oxidation) in order to store energy as fat. In response, the efferent pathway of energy balance coordinates efforts at improving energy efficiency and increasing energy storage. Total and resting energy expenditures decline in an attempt to conserve energy.118 Specifically, UCP1 levels within adipose tissue decline119 as a result of decreased SNS activity in response to starvation.120 In spite of decreased SNS tone at the adipocyte, there is clearly an obligate lipolysis (due to insulin suppression and up-regulation of hormone-sensitive lipase), which is necessary to maintain energy delivery to the musculature and brain in the form of liver-derived ketone bodies. Additionally, in the starved state, vagal tone is increased in order to slow the heart rate and myocardial oxygen consumption, increase β-cell insulin secretion in response to glucose, and increase adipose insulin sensitivity—all directed to increase energy storage.120 These revert back to baseline once caloric sufficiency is reestablished, and leptin levels rise.
The nucleus accumbens and the hedonic pathway of food reward
The negative feedback pathway delineated beforehand is not the only site of central regulation of food intake. Complementary to insulin and leptin’s ability to alter energy balance, these hormones also modify the “hedonic pathway,” or the pleasurable and motivating responses to food. This is the same pathway that responds to drugs of abuse, such as nicotine and morphine. The hedonic pathway comprises the ventral tegmental area (VTA) and the nucleus accumbens (NA), with inputs from various components of the limbic system, including the striatum, amygdala, hypothalamus, and hippocampus. Food intake is a readout of the hedonic pathway; administration of morphine to the NA increases food intake in a dose-dependent fashion.121 When functional, the hedonic pathway helps curtail food intake in situations where energy stores are replete; however, when dysfunctional, this pathway can increase food intake leading to obesity.
The VTA appears to mediate feeding on the basis of palatability rather than energy need. The dopaminergic projection from the VTA to the NA mediates the motivating, rewarding, and reinforcing properties of various stimuli, such as food and addictive drugs. Leptin and insulin receptors are expressed in the VTA, and both hormones have been implicated in modulating rewarding responses to food and other pleasurable stimuli.122 For instance, fasting and food restriction (where insulin and leptin levels are low) increase the addictive properties of drugs of abuse, whereas ICV leptin can reverse these effects.123 In rodent models of addiction, increased addictive behavior (and pleasurable response from a food reward), as measured by dopamine release and dopamine receptor signaling, is greater after food deprivation.124 In humans with leptin deficiency, alterations in activity in the nucleus accumbens can be seen using functional MRI scanning, and these changes subside with administration of exogenous leptin.125 Acutely, insulin increases expression and activity of the dopamine transporter, which clears and removes dopamine from the synapse; thus, acute insulin exposure blunts the reward of food.126 Furthermore, insulin appears to inhibit the ability of VTA agonists (e.g., opioids) to increase intake of sucrose.127 Finally, insulin blocks the ability of rats to form a conditioned place preference association to a palatable food.128 However, insulin resistance of this pathway may lead to increased reward of food.
One question that has garnered increasing interest is whether any macronutrient has addictive properties. In animal studies, sugar has been shown to induce the four criteria for addiction: (1) bingeing, (2) withdrawal, (3) craving, and (4) cross-sensitization with other drugs of abuse.129 Within fast food, sugar and caffeine satisfy the criteria presented in the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-V) for dependence in humans.130 However, the question of whether food addiction exists, and whether it can explain patients with obesity, remains hotly contested.131
The amygdala and the stress response
The VMH and VTA-NA mediate satiety when energy stores are replete, but they appear to be easily overridden by amygdala activation and resultant stress, a state of physiologic insulin resistance. Numerous lines of evidence suggest that the stress glucocorticoid corticosterone (in the rat) or cortisol (in the human) is essential for the full expression of obesity, which helps to explain the disruptive role of stress in weight regulation.132
Stress and glucocorticoids are integral in promoting adiposity and the metabolic syndrome. Adrenalectomized rats maintained pharmacologically with high levels of corticosterone demonstrate that exogenous fat intake is directly proportional to circulating corticosterone concentrations,133 whereas amygdala activation by stress is dampened by the ingestion of energy-dense food.134 In intact rats, corticosterone stimulates eating, particularly of high-fat food, and in humans, cortisol administration increases food intake.135 Human research shows increased caloric intake of “comfort foods” (i.e., those with high energy density) after acute stress,136 and that the stress response contributes to leptin resistance (discussed later).137 Several studies in children have observed relationships between stress and unhealthy dietary practices, including increased snacking, and an elevated risk for problems with weight during adolescence and adulthood.138 In a controlled study of 9-year-olds, children who scored high on dietary restraint and who felt more stressed by lab challenges tended to eat more comfort food.139
Leptin resistance
Most obese children have high leptin levels but do not have receptor mutations, manifesting what is commonly referred to as “functional leptin resistance.” Leptin resistance prevents exogenous leptin administration from promoting weight loss.140 The response to most weight-loss regimens plateaus rapidly due to the rapid fall of peripheral leptin levels below a personal “leptin threshold,”141 which is likely genetically determined. Leptin decline causes the VMH to sense a reduction in peripheral energy stores, which modulates a decrease in REE to conserve energy, analogous to the starvation response (discussed earlier),118 but occurring at elevated leptin levels.
The cause of leptin resistance is unknown, but it may have several etiologies. Leptin crosses the blood-brain barrier via a saturable transporter, which limits the amount of leptin reaching its receptor in the VMH 142,143; this transporter operates more efficiently at lower levels of leptin, while preventing increased signaling at higher levels.144 Activation of the leptin receptor induces the intraneuronal expression of suppressor of cytokine signaling-3 (SOCS-3), which limits leptin signal transduction in an autoregulatory fashion.51
The standard method for producing insulin resistance and obesity in rodents is a high-fat diet. Dietary fat promotes leptin resistance through its effects on hypertriglyceridemia,145 which limits access of peripheral leptin to the VMH, and also by interfering with leptin signal transduction upstream of STAT-3, its primary second messenger.146 One likely modulator of this pathway is the enzyme phosphatidyl inositol-3-kinase (PI3K), which is the downstream effector of insulin action in POMC neurons147 and which appears to account for the effects of dietary fat on leptin resistance and obesity.148
Two clinical paradigms have been shown to improve leptin sensitivity. After weight loss through caloric restriction, exogenous administration of leptin can then increase REE back to baseline and permit further weight loss,149,150 suggesting that the weight loss itself improves leptin sensitivity. Second, suppression of insulin correlates with improvement in leptin sensitivity and promotes weight loss,151 suggesting that hyperinsulinemia promotes leptin resistance by interfering with leptin signal transduction in the VMH and VTA.152 Indeed, insulin reduction strategies can effectively promote weight loss in children with hyperinsulinemia by improving leptin sensitivity.153 These clinical substantiations of the PI3K data in animals has led to the hypothesis that chronic hyperinsulinemia blocks leptin signal transduction at the VMH and VTA, which turns a negative feedback cycle into a vicious feed-forward cycle.
Based on the concept that hyperinsulinemia promotes weight gain at the adipocyte, yet blocks leptin signaling in the hypothalamus and nucleus accumbens, the role of starvation, reward, and stress on weight gain and adiposity via their effects on insulin are a reasonable hypothesis, termed the “limbic triangle” (Figure 22-3).154 However, this hypothesis remains to be proven.
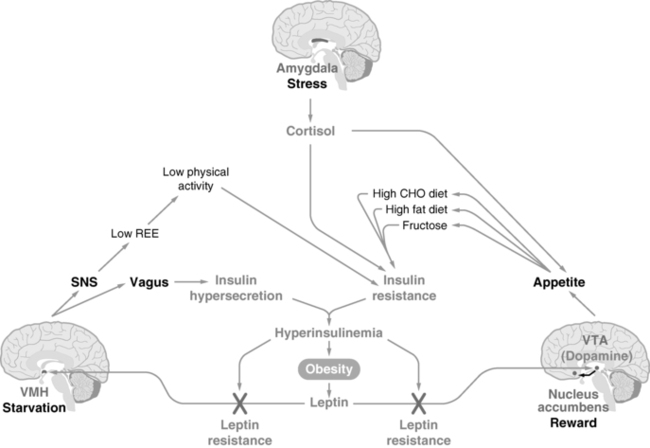
FIGURE 22-3  The “limbic triangle.” Three areas of the CNS conspire to drive food intake and reduce physical activity, resulting in persistent weight gain. The ventromedial hypothalamus (VMH) transduces the leptin signal from adipocytes to reduce energy intake and increase energy expenditure; however, hyperinsulinemia prevents leptin signaling, promoting the “starvation response.” The ventral tegmental area (VTA) transduces the leptin signal to reduce dopamine neurotransmission to the nucleus accumbens (NA), reducing food intake; however, hyperinsulinemia prevents leptin signaling here as well, increasing dopamine and promoting the “reward” of food. The amygdala transduces fear and stress, which results in increased cortisol release from the adrenal cortex. The elevated cortisol also drives energy-rich food intake and promotes insulin resistance, further interfering with leptin signaling at the other two CNS sites. Thus, activation of any aspect of the limbic triangle turns on a positive feedback loop, promoting continued weight gain and obesity. (From Mietus-Snyder, M. L., & Lustig, R. H. (2008). Childhood obesity: adrift in the “limbic triangle.” Ann Rev Med, 59, 119–134.)
The “limbic triangle.” Three areas of the CNS conspire to drive food intake and reduce physical activity, resulting in persistent weight gain. The ventromedial hypothalamus (VMH) transduces the leptin signal from adipocytes to reduce energy intake and increase energy expenditure; however, hyperinsulinemia prevents leptin signaling, promoting the “starvation response.” The ventral tegmental area (VTA) transduces the leptin signal to reduce dopamine neurotransmission to the nucleus accumbens (NA), reducing food intake; however, hyperinsulinemia prevents leptin signaling here as well, increasing dopamine and promoting the “reward” of food. The amygdala transduces fear and stress, which results in increased cortisol release from the adrenal cortex. The elevated cortisol also drives energy-rich food intake and promotes insulin resistance, further interfering with leptin signaling at the other two CNS sites. Thus, activation of any aspect of the limbic triangle turns on a positive feedback loop, promoting continued weight gain and obesity. (From Mietus-Snyder, M. L., & Lustig, R. H. (2008). Childhood obesity: adrift in the “limbic triangle.” Ann Rev Med, 59, 119–134.)
Energy excess—obesity
The rise in the prevalence of obesity in children and adolescents is one of the most alarming public health issues facing the world today. Although the rise in the prevalence of obesity in children and adolescents seems to have leveled in some parts of the world,155 many others, especially developing countries, are still experiencing a steady increase. Obesity is associated with significant health problems in children, is an early risk factor for a great many cases of adult morbidity and mortality,156 and is an important factor in increasing health care expenditures.157 Childhood obesity tends to track to adulthood,158 and those who remain obese as adults have a significant risk for the development of type 2 diabetes, atherosclerosis, and dyslipidemia. In contrast, obese children who lost weight and became nonobese adults do not have an increased risk for such morbidity159; thus, early and effective intervention against obesity during childhood is crucial.
Definition
The theoretical definition of obesity is a degree of somatic overweight that affords detrimental health consequences.160 Based on morbidity and mortality statistics, and with a desire to prevent future risk of morbidity, we practically define obesity as a statistical magnitude of overweight for a population, keeping in mind that morbidity and mortality vary with degree of overweight in different racial, ethnic, and socioeconomic groups,161 and indeed for individuals as well. The World Health Organization162 categorizes adult overweight into four subgroups based on body mass index (BMI; weight [kg] ÷ height [m]2): BMI 25 to 30 (overweight); BMI 30 to 35, grade 1 (moderately obese); BMI 35 to 40, grade 2 (severely obese); and BMI > 40, grade 3 (morbidly obese). Some make a further delineation at BMI > 60, denoting this as “superobesity,” as even surgical therapies are less effective in this range.
Most obesity in adulthood has its origins in childhood,163,164 making obesity a pediatric concern and the prevention and treatment of obesity a pediatric goal. BMI is also the accepted marker in children.165 In childhood, a comparison of BMI to normal curves for age166 allows for categorization of BMI above the 85th percentile as overweight and that above the 95th percentile as obese (Figure 22-4A and B).
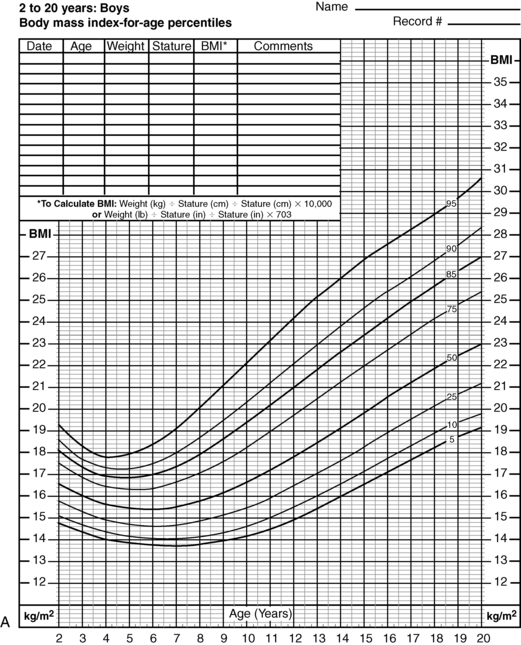
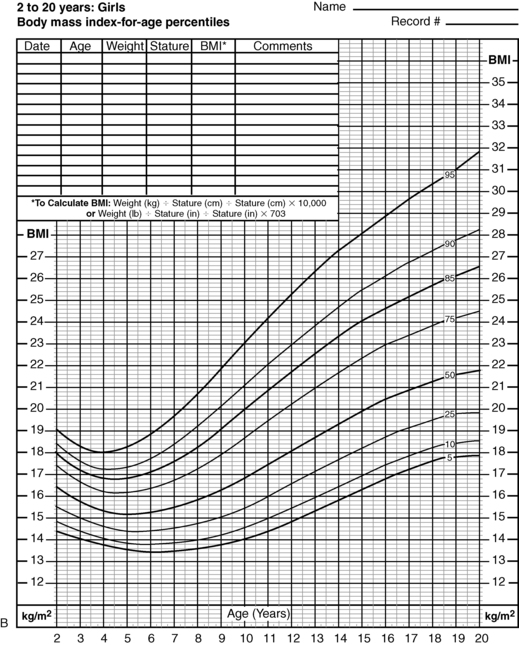
FIGURE 22-4  Body mass index (BMI)-for-age percentiles for U.S. boys (A) and U.S. girls (B), 2 to 20 years of age. Note that the 95th percentile signifies obesity. The adiposity rebound occurs at approximately 5 years of age in both sexes; the earlier the adiposity rebound occurs, the more likely an organic etiology for the weight gain can be inferred. (From www.cdc.gov/growthcharts [National Center of Health Statistics, 2000].)
Body mass index (BMI)-for-age percentiles for U.S. boys (A) and U.S. girls (B), 2 to 20 years of age. Note that the 95th percentile signifies obesity. The adiposity rebound occurs at approximately 5 years of age in both sexes; the earlier the adiposity rebound occurs, the more likely an organic etiology for the weight gain can be inferred. (From www.cdc.gov/growthcharts [National Center of Health Statistics, 2000].)
Although BMI is the standard for obesity for statistical purposes and within populations, it should be noted that BMI takes into account muscle and bone, as well as both subcutaneous and visceral fat. Furthermore, BMI in children is age and puberty dependent, thus the BMI z-score is a more accurate assessment of childhood adiposity. Lastly, waist circumference (an indirect measure of visceral fat) has emerged as a more accurate indicator of metabolic disturbance in children.167
Prevalence and epidemiology
The prevalence of childhood obesity in the United States has increased dramatically since the 1980s168,169 and continues to do so, although the comparison of longitudinal and cross-sectional data is difficult due to different definitions and measurement parameters between epidemiologic studies. Estimates of obesity prevalence in the United States based on data from the 2009-2010 National Health and Nutrition Examination Survey (NHANES IV)155 demonstrate that the epidemic of childhood obesity is occurring at earlier ages: In 2009 to 2010, 9.7% (95% CI, 7.6% to 12.3%) of infants and toddlers had a high weight-for-recumbent length and 16.9% (95% CI, 15.4% to 18.4%) of children and adolescents from 2 through 19 years of age were obese. Trend analyses over a 12-year period indicated a significant increase in obesity prevalence between 1999-2000 and 2009-2010 in males ages 2 through 19 years (odds ratio, 1.05; 95% CI, 1.01 to 1.10) but not in females (odds ratio, 1.02; 95% CI, 0.98 to 1.07) per 2-year survey cycle. There was a significant increase in BMI among adolescent males ages 12 through 19 years (P = .04) but not among any other age group or among females. Thus, in 2009-2010, the prevalence of obesity in children and adolescents was 16.9%; this was not changed compared with 2007-2008. Importantly, whereas the prevalence of obesity in Caucasian children declined slightly, the prevalence of African-American and Latino obesity continued to climb. Furthermore, the prevalence of severe obesity (BMI > the 97th percentile) is still on the rise.170 Lastly, projections argue that by 2030, 42% of American adults will be obese.157
Global prevalence
Obesity has overtaken AIDS and malnutrition as the number one public health problem in the world.171 The global prevalence of childhood obesity has been increasing worldwide at an alarming rate since the 1990s. Rates have increased 2.7-fold to 3.8-fold over 29 years in the United States,169 2-fold to 2.8-fold over 10 years in England, 3.4-fold to 4.6-fold over 10 years in Australia, and 3.4-fold to 3.6-fold over 23 years in Brazil. In Asia, the prevalence has increased 1.1-fold to 1.4-fold over 6 years in China and 2.3-fold to 2.5-fold over 26 years in Japan. In Africa, the prevalence has increased 3.9-fold over 18 years in Egypt, 3.8-fold over 6 years in Ghana, and 2.5-fold over 5 years in Morocco.172 Based on data from 2008, nearly 20% of Indian boys and 18% of Indian girls from 2 to 17 years of age are considered overweight. Even higher prevalence rates are reported from New Zealand and Taiwan, making the Far East and Oceania regions the current hub of the obesity epidemic.170

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


