Fig. 4.1
Tanner staging of puberty onset in boys from Palmer and Dunkel [4]. Copyright © (2012) Massachusetts Medical Society. Reprinted with permission
Table 4.1
Details of the Tanner stages of puberty
Tanner stage | Males | |
|---|---|---|
Genitalia | Pubic hair | |
1 | Testis 1–3 ml | None |
2 | Testis >3 ml Scrotal enlargement | Sparse, lightly pigmented |
3 | Testis continue to enlarge, penis lengthens | Increases in amounts, darkens, and starts to curl |
4 | Scrotum darkens, widening of glans penis | Resembles adult type but not spread to medial thighs |
5 | Adult size and morphology | Spreads to medial thighs, adult distribution |
Timing of Puberty
In the general population, there is a near-normal distribution of the timing of puberty, with the mean age of onset of G2 at 11.5 years in boys (Fig. 4.2). In healthy boys, the normal age limits for G2 development are between 9.5 and 14 years [4]. While a large variability in the timing of pubertal onset exists, clear age cutoffs for normal pubertal development have been established. However, the age limits for identifying children who need evaluation for precocious or delayed puberty may vary among different ethnic groups. Within this distribution, there has been in recent years an increasing degree of skew at both ends of the spectrum, as an increasing prevalence of an earlier age of pubertal onset (G2) has been documented in some populations as well as an increase in the number of children completing puberty at a later age [5].
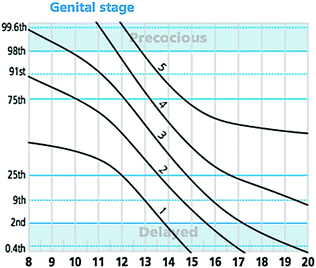
Fig. 4.2
Puberty normogram of the timing of genital Tanner staging in healthy boys. This staged line drawing gives age in years on the x-axis and centiles on the y-axis. Each curved line represents one genital tanner stage from 1–5 and illustrates the distribution of the timing of each stage in the normal population. These data have been incorporated into UK growth charts and are available at www.growthcharts.rcpch.ac.uk. Original concept and data from van Buuren [167]
Peak height velocity (PHV) and peak pubertal growth hormone production coincide approximately with the midpoint of pubertal development (Fig. 4.3). In boys, the PHV coincides with G3-4 at an average age of 13.5 years and achieves an incremental rate of 9.5 cm/yr [6]. Up to 25% of total adult height is achieved from growth during puberty, but the amplitude and peak velocity of the pubertal growth spurt are not fixed and vary with age at onset of puberty [7]. Early puberty is associated with a large pubertal growth spurt, while late maturers, who have a longer prepubertal period of growth, in turn experience a less pronounced pubertal growth spurt [8]. Therefore, although extremes of pubertal timing may lead to a small degree of final height reduction due to the reduced overall period of growth in precocious puberty, or poor PHV in delayed puberty [9], in general within wide limits, the timing of puberty does not influence adult height.
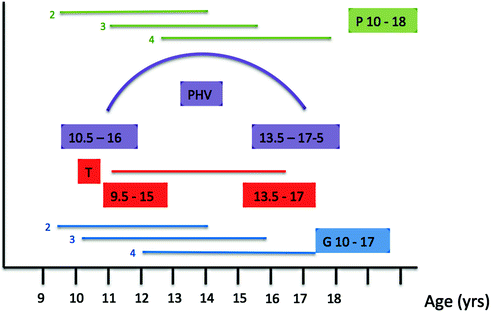
Fig. 4.3
Relationship between peak height velocity and pubertal development (male). P pubic hair stage; PHV peak height velocity; T testicular growth; G genital stage. Age ranges presented: in linear format—3rd–97th centiles for each Tanner stage, e.g., G2; in colored boxes—age range in years for each parameter
Regulators of the Timing of Puberty
Variability in the timing of puberty in healthy adolescents is governed by complex regulatory mechanisms involving genetic, environmental, and other factors [10]. Nutritional status, adoption, geographical migration, and emotional well-being have all been shown to affect pubertal timing [11–13]. While the timing of pubertal onset in girls in most countries in the developed world exhibited a rapid decrease in the first half of the twentieth century [14, 15], in boys, these trends are less clear. A small but significant change in the normal spectrum of timing of G2 development was documented in a European cohort [16] and in the USA [17] but remains controversial [18]. Much has been postulated about this observed secular trend toward an earlier age of pubertal onset in the developed world [19]. It also follows that it is difficult to understand the pathophysiology of delayed puberty before we can interpret this population level advancement in pubertal age.
Nutritional changes clearly have an important role, as shown by the positive correlation between age at puberty onset and childhood body size [20], most markedly in girls [11]. He and Karlberg demonstrated in a large dataset (n = 3650) that one BMI unit increase between the ages of 2 and 8 years is associated with a 0.11 year advancement in the timing of puberty as measured by peak height velocity in both genders [21]. In boys, however, the data are less consistent, with some studies documenting an earlier onset of puberty with greater adiposity, and some a later onset. In particular, more European studies have noted the former trend, while US studies have more often shown the latter [22]. A recent study from the USA reported a far more complex relationship between fat mass and pubertal timing, with overweight status being associated with earlier pubertal onset, while obesity was associated with later onset. These effects also vary between ethnic groups [23]. Thus, one hypothesis in boys is that greater BMI leads to earlier pubertal timing up to the threshold at which obesity occurs. Obesity may delay pubertal timing in boys due to the suppression of the HPG axis with adiposity leading to excess aromatase activity and thus increased estrogen production.
Additional data point to an earlier trend in the age of puberty onset that is independent of BMI [24]. As detailed above, some studies suggest that over the last decade, the age of completion of puberty in males in some populations has become skewed toward later ages [5]. The effect of possible endocrine-disrupting chemicals (EDCs) on the timing of puberty is also an ongoing concern [5, 25]. Polybrominated biphenyls, bisphenol A, atrazine (herbicides), and phthalates, among others, have been suggested as possible EDCs responsible for contributing to this observed trend [26]. For example, children migrating for international adoption, and previously exposed to the estrogenic insecticide DTT in their country of origin, displayed early or precocious pubertal timing [27]. However, a mechanism of action for EDCs through the early initiation of the pulsatility of GnRH has not been conclusively demonstrated. Studies are complicated by the likely differing and possibly divergent influence of variable doses and mixtures of EDCs, and differing effects depending on age and length of exposure [5, 27, 28].
Epigenetic regulators are potential mediators of the effects of the environment on the hypothalamic control of puberty. However, while experimental data from rats provide evidence for changes in histone acetylation and gene methylation leading to altered gene expression during sexual development, the link between environmental factors and the epigenetic control of puberty has not been established. Although the window of opportunity for the effects of EDC exposure was historically considered to occur in the late prepubertal period, evidence of a fetal and neonatal origin for changes in pubertal timing counters this dogma. A multicenter US study found that prenatal exposure of boys to EDCs such as phthalates, based on the concentrations of four phthalate metabolites in a sample of urine, was associated with a short anogenital index, a marker for reduced masculinization of genital structures [29] (see Chap. 14). Epigenetic changes during fetal life are a potential mechanism for the effects of EDCs in utero [5]. Recent evidence suggests that effects of EDCs may persist in pregnant rats in not only their unborn fetus but into the next generation [30].
Despite the demonstrated importance of environmental factors, the genetic influence on pubertal timing is clearly fundamental. While the timing of pubertal onset varies within and between different populations, it is a highly heritable trait. The timing of sexual maturation is highly correlated within families and in twin studies, suggesting strong genetic determinants [31]. Previous epidemiological studies and genetic approaches estimate that 60–80% of the variation in pubertal onset is under genetic regulation [14, 32, 33]. Despite this strong heritability, however, little is known about the genetic control of human puberty either in the normal population or in cases of disturbed pubertal timing [4]. A lack of clear understanding of the genetic factors that control and trigger the onset of puberty is an important barrier to deciphering the mystery of EDC and other environment/external cues to understand the secular trend toward earlier puberty [34].
Endocrine Changes During Puberty
Gonadotropin-Releasing Hormone and Gonadotropins
The hypothalamus–pituitary–gonadal (HPG) axis is already functional in the late fetal and neonatal period. The development of the HPG axis is exceptional in that GnRH is produced by hypothalamic neurons that develop outside of the central nervous system. The embryonic migration of GnRH neurons from the posterior wall of the nasal cavity to hypothalamus is key for the creation of the neuroendocrine pathways that allow normal pubertal development [35]. The whole process of migration involves no more than a few hundred neurons per hemisphere in the mouse (several thousand in primates or humans) [36]. The absolute number of GnRH neurons required for pubertal development is not known, but there appears to be a degree of redundancy in the system [37]. In addition, adult reeler mice have significantly fewer GnRH neurons in the hypothalamus and display a phenotype of delayed pubertal maturation and low fertility [38].
During gestation, there is an increase in GnRH content, which peaks at 34–38 weeks in the male fetus [39]. At mid-gestation, there is a striking rise in circulating gonadotropin levels in both male and female fetuses which fall to low levels in late gestation. This change in gonadotropin secretion is thought to result from the development of sex steroid negative feedback and from inhibiting influences from the CNS to GnRH neurons.
LH and FSH secretions rise during the first month after birth, probably because the negative feedback effect of placental estrogens is withdrawn. LH is secreted in pulses during this postnatal period indicating control by GnRH [40]. While the HPG axis is active during this ‘mini-puberty,’ it becomes dormant in young children between the age of 2 and 8–9 years [41]. Suppression of the axis is not absolute, however, as LH pulsatility is detectable during this stage using ultrasensitive assays, but pulses are infrequent, of low amplitude and occur mostly at night [42].
The clinical features of puberty are initiated by reactivation of the HPG axis after this relative quiescence during childhood. During this reactivation, there is a gradual development of a dynamic interplay between the central production of GnRH and gonadotropins, and gonadal sex steroid production, with progressive maturation of negative and positive feedback loops. The central suppressant drive from the CNS gradually abates, and intensifying positive feedback results from the increase in sex steroid production by the gonads. The gonadal inhibition of the hypothalamic–pituitary system occurs later, becoming operative only at mid-puberty, and eventually becomes dominant over the central inhibitory feedback drive (Fig. 4.4). Both mean LH and FSH levels increase through pubertal development, although LH rises to a greater extent, probably due to mechanistic differences in feedback by estradiol and inhibin for these two hormones [43]. These rises are due to both an increase in basal levels of LH and FSH, and to a greater number and amplitude of LH pulses.
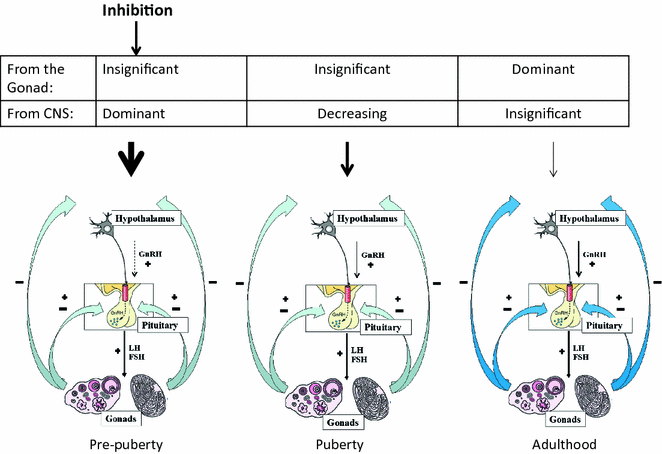
Fig. 4.4
A model showing the regulation of the hypothalamic–pituitary–gonadal axis during pubertal development
Sex Steroids and Inhibin-B
LH controls sex steroid production by upregulating expression of the steroidogenic enzymes and by controlling metabolic activity of the steroid-producing Leydig cells (see Chap. 2). LH may also stimulate Leydig cell proliferation and differentiation [44]. During puberty, plasma testosterone levels increase dramatically. Table 4.2 summarizes the levels of testosterone at various developmental stages with respective testis sizes. The pubertal increase in testis size results primarily from an increased number of proliferating and differentiating germ cells and to an increase in the number of Sertoli cells [45]. In early and mid-puberty, there is a pronounced diurnal rhythm with a morning peak in testosterone levels, but this is less pronounced in later puberty, and declines gradually with age [46], probably due to decreased day–night ratios of gonadotropins [47].
Table 4.2
Pubertal stages (according to Tanner) with respective testis volumes and plasma testosterone concentrations
Pubertal stage (Tanner) | Testicular volume (mL) | Plasma testosterone (ng/dL) | Plasma testosterone (nmol/L) |
|---|---|---|---|
1 | <4 | <10 | <0.3 |
2 | 4–8 | 12–69 | 0.4–2.4 |
3 | 8–10 | 60–275 | 2.1–9.5 |
4 | 10–20 | 142–515 | 4.9–17.9 |
5 | 20–25 | 319–775 | 11.1–26.9 |
Inhibin-B is a heterodimeric glycoprotein produced by Sertoli cells beginning in the fetus. Several studies show that serum inhibin-B levels in children change in concert with the secretion of gonadotropins [48–50]. During the ‘mini-puberty,’ serum inhibin-B levels increase to similar [51, 52] or even higher [49] levels than in adolescent boys and adult men. This early inhibin-B secretion is sustained until the age of 18–24 mo; thereafter, serum concentrations decline to lower but readily measurable levels [49]. Early in puberty, between Tanner stages G1 and G2, serum inhibin-B concentrations again increase to reach peak levels at the Tanner stage G2, but then the levels plateau [48, 50].
Central Control of Puberty via GnRH Upstream Pathways
GnRH pulsatility is coordinated by a balancing act between a number of inhibitory and excitatory neuronal and glial inputs (Fig. 4.5) [53]. Inhibitory inputs are primarily from GABAergic and opiatergic neurons, while glutamate and kisspeptin are the central excitatory neuronal signals. Glial cells additionally facilitate GnRH secretion via growth factor-derived signaling [54]. The onset of puberty is triggered by a decline in these inhibitory signals and an amplification of excitatory inputs, leading to increased frequency and amplitude of GnRH pulses [55]. Before the onset of puberty, GABA release in the preoptic area decreases in female rats [56], and in the rhesus monkey, GABA release into the median eminence decreases concomitant with the pubertal increase of GnRH secretion [57]. In female rats, glutamine synthase is downregulated, and glutamate dehydrogenase becomes more abundant in the hypothalamus at puberty, both leading to increased availability of glutamate [58]. Glutamate agonists are potent stimulators of GnRH secretion, and administration in prepubertal primates can stimulate LH and testosterone secretion [59].
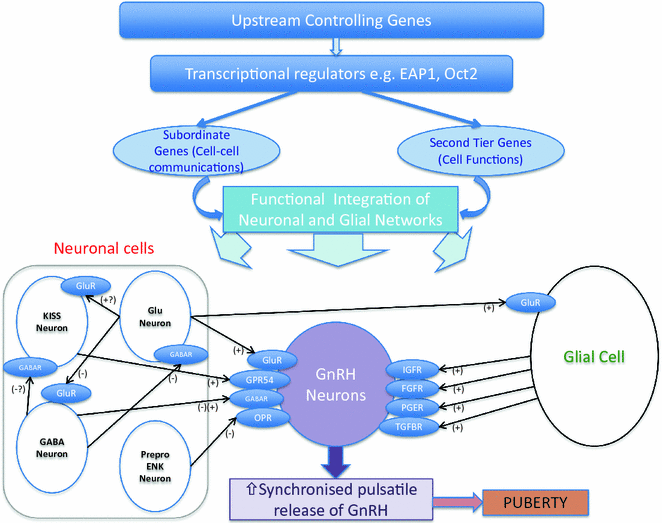
Fig. 4.5
Genetic regulators in the trans-synaptic and glial control of GnRH neurons during puberty. Adapted from Ojeda et al. [53]
Another vital piece in the puzzle of the central control of GnRH release came with the discovery of Kisspeptin. This excitatory neuropeptide was identified as a permissive factor in puberty onset by the discovery of patients with GnRH deficiency who had loss-of-function mutations in the KISS1 receptor, KISS1R (previously known as GPR54) [60, 61]. Mice with knockout of kiss1r were simultaneously discovered to be infertile despite anatomically normal GnRH neurons and normal hypothalamic GnRH content [61]. Their phenotype can be rescued by exogenous delivery of GnRH. Kiss1 knockout mice also have a phenotype consistent with normosmic GnRH deficiency. However, to date, only very rarely have mutations in KISS1 been found in patients with delayed or absent puberty [62], although two potential activating mutations were identified in patients with central precocious puberty (CPP) [63].
Kisspeptin signals directly to GnRH neurones to control pulsatile GnRH release. Kisspeptin is upregulated in both primates and mice in the peri-pubertal period, and its administration in prepubertal rodents advances the onset of puberty [64]. Kisspeptin also appears to be downregulated in functional hypogonadism, suggesting its role as a mediator of the action of environmental factors such as nutritional status and emotional well-being on puberty and reproductive capacity. Kisspeptin signaling is an important element of both positive and negative feedback loops in the HPG axis. While kisspeptin has been identified as a pivotal upstream regulator of GnRH neurons, whether kisspeptin is the key factor that triggers the onset of puberty remains unclear.
An additional excitatory neuropeptide, neurokinin B, has been implicated in the upstream control of GnRH secretion. Identification of this pathway was via the discovery of loss-of-function mutations in TAC3, encoding neurokinin B, and its receptor TACR3, in patients with normosmic GnRH deficiency and pubertal failure [65, 66]. Both KISS1 and TAC3 are expressed by neurons in the arcuate nucleus of the hypothalamus that project to GnRH neurons, and their expression is downregulated by estrogen [67]. However, studies of the effects of neurokinin B administration have provided conflicting results. While central administration of neurokinin B agonists failed to stimulate GnRH release in rodents, and Tacr3 knockout mice have grossly normal fertility [68, 69], primate studies showed that neurokinin B can act to stimulate GnRH release via kisspeptin signaling [70]. Additionally, neurokinin B is expressed more widely in the central nervous system than in kisspeptin, suggesting some differences in the roles of these two neuropeptides in the control of pubertal onset.
Dynorphin, an opioid peptide that is coexpressed with kisspeptin and neurokinin B in so-called KNDy neurons, inhibits the release of GnRH, and together, these peptides are currently believed to play a fundamental role in the GnRH pulse generator. Other neurons regulate GnRH including GABAergic signaling pathways which function in the stress-induced suppression of LH, and RFamide–related peptide gene (RFRP), the mammalian ortholog of the avian peptide gonadotrophin–inhibiting hormone (GnIH) [71].
Glial inputs appear to be predominantly facilitory during puberty, acting via growth factors and small diffusible molecules, including TGFβ1, IGF-1, and neuregulins that directly or indirectly stimulate GnRH secretion [72]. Glial cells in the median eminence regulate GnRH secretion through the production of growth factors that activate receptors with tyrosine kinase activity. FGF signaling is required for GnRH neurons to reach their final destination in the hypothalamus [73], as well as for GnRH neuronal differentiation and survival [74]. Additionally, GnRH neuron secretory activity is facilitated by IGF-1 and by members of the epidermal growth factor family such as neuregulin 1β [54, 75]. Moreover, plastic rearrangements of glia–GnRH neuron adhesiveness, mediated by soluble molecules such as neuronal cell adhesion molecule (NCAM) and synaptic cell adhesion molecule (SynCAM), coordinate the controlled delivery of GnRH to the portal vasculature [72], a process that is subject to sex steroid regulation [76].
The interplay between puberty and metabolism has been the focus of intense study. The role of fat mass in pubertal timing is thought to be mediated, at least in part, through the permissive actions of the metabolic hormone leptin, a key regulator of body mass, secreted from white adipose tissue (WAT) [77]. Absence of leptin signaling results in obesity and infertility, whereas leptin treatment decreases food intake and restores reproductive functions. During fasting, leptin levels decrease and gonadotropin secretion is suppressed. These findings imply that nutritional status, especially fat tissue and leptin, could contribute to pubertal development. Clinical observations have further supported a role for leptin signaling in the onset of puberty, e.g., humans and mice lacking leptin (Lep ob/ob) or the leptin receptor (LepR db/db) fail to complete puberty and are infertile [78], and in a 12-yr-old girl with congenital leptin deficiency, treatment with recombinant leptin was followed by a pubertal pattern of LH release [79].
GnRH neurons do not express the LepR. Instead, leptin appears to indirectly regulate GnRH via actions on cells that are afferent to GnRH neurons such as LEPR-expressing GABA neurons from the arcuate nucleus [80], or via cells that interact morphologically with GnRH neurons, partly through nitric oxide signaling, which is required for leptin action [81], and via kisspeptin/neuropeptide Y neurons [82, 83]. Cross-sectional studies showed that serum leptin levels rise in girls in early puberty [84] but not in boys. In careful longitudinal studies in male rhesus monkeys, leptin levels decreased during the juvenile period and were unchanged during puberty [85, 86], and leptin gene expression in the hypothalamus is unchanged during the development in the rat [87, 88]. Therefore, leptin is probably not the requisite metabolic trigger for the onset of puberty, but rather is a permissive factor signaling healthy energy balance.
Neuropeptide Y (NPY) is involved in many CNS functions, including appetite control and reproduction. Evidence from primate studies suggests that NPY may also have a contributory role in the break restraining the onset of puberty in primates [68, 89]. Ghrelin and other gut-derived peptides may also form part of the mechanism by which energy homeostasis regulates reproductive development [90]. Both low birthweight and prematurity are associated with earlier onset of puberty [91, 92], particularly in those children with rapid increase in length or weight in the first two years of life [93]. It remains unclear, however, whether childhood obesity, insulin resistance, excess androgens, or other factors may explain this association [94, 95].
While GnRH pulsatility is clearly the central driver of pubertal onset, and aberrant GnRH neuronal development and function lead to hypogonadotropic hypogonadism (HH), our collective understanding of the upstream neurocircuitry regulating GnRH neurons remains incomplete. Data pointing to hypothalamic regulation via a hierarchical network of genes (Fig. 4.6) have mainly come from a systems biology approach and from animal models, with little data from human subjects [96]. Candidate transcriptional regulators that have been identified via these approaches include Oct–2, TTF–1, and EAP1 [97–99].
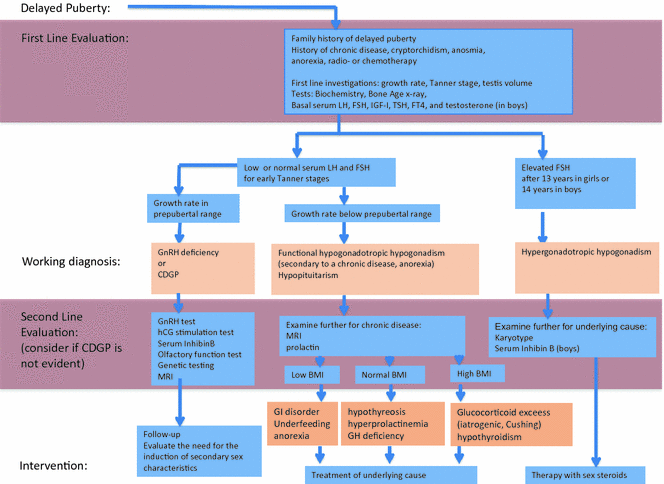
Fig. 4.6
Algorithm for the evaluation of boys with delayed puberty. CDGP constitutional delay of growth and puberty, GI gastrointestinal, GH growth hormone, GHD GH deficiency, PRL prolactin, and IGF-1 insulin-like growth factor 1. Adapted from Palmer and Dunkel [4]. Copyright © (2012) Massachusetts Medical Society. Reprinted with permission
Delayed Puberty
Presentation
Disturbances of puberty encompass an important group of pathologies within the field of pediatric endocrinology. First, they are common, affecting over 4% of adolescents. In addition, abnormal timing of pubertal development is associated with adverse health and psychosocial outcomes [100–102]. This has importance for the individual, but also has a potential major impact on public health, especially in view of the secular trend toward an earlier age of puberty onset [25]. Early puberty, in particular, is associated with adverse health outcomes, including breast [103] and endometrial cancer [104], obesity [105], type 2 diabetes [102], cardiovascular disease [106, 107], short stature [108], and even increased mortality [106]. However, persons with delayed puberty are also at risk of short adult height [109–112], decreased bone mineral density [113, 114], and psychological sequelae [115, 116].
Self-limited Delayed Puberty
Self-limited delayed puberty (DP), also known as constitutional delay of growth and puberty (CDGP), represents the most common cause of delayed puberty in both sexes. Up to 63% of boys with delayed puberty have self-limited DP [117]. Self-limited DP segregates within families, with the majority of families displaying an autosomal dominant pattern of inheritance (with or without complete penetrance) [31]. Fifty to 75% of subjects with self-limited DP have a family history of delayed puberty onset [118].
Patients with self-limited DP represent the extreme tail of normal puberty, defined as the markers of the onset of puberty occurring two or more standard deviation (SD) later than the population mean age for gender. In addition, self-limited DP may encompass older children with delayed pubertal progression, a diagnosis that is aided by the use of puberty nomograms (Fig. 4.2) [119]. The absence of a pathological medical history, signs and symptoms, and a positive family history of delayed puberty in one or both of the parents suggests a diagnosis of self-limited DP; however, before making that diagnosis, pathological conditions must be excluded.
In DP, probably because of a low estrogen concentration for chronological age (but not for bone age), growth hormone (GH) secretion is functionally and temporally reduced for age, and when this functional GH deficiency is prolonged, it may also impact adult height. After the onset of puberty or the initiation of appropriate treatment, growth velocity and GH secretion return to normal. Additionally, in about half of the subjects with constitutional delay, there is delayed maturation during early childhood, and consequently, they are shorter than their peers even before the mean age for the pubertal growth spurt [119].
It has been shown that those self-limited DP subjects who also have reduced growth in childhood may not fully exploit their genetic height potential, resulting in an adult height below their mid-parental target height [112, 120–122], with an average loss of 4.2 cm if untreated [122]. However, the majority of the patients seen at pediatric endocrinology clinics are short because their condition is compounded by two independent factors: short genetic height potential and DP. Other studies showed only a negligible difference in final height, even in DP subjects who have received no intervention [109, 123–128]. This may imply a pathophysiological mechanism in addition to lack of sex steroids contributing to the growth phenotype in some patients with DP, but not in others [128].
In self-limited DP, adrenarche may also occur later than usual [4]. Bone age in self-limited DP is retarded compared to chronological age, but the developmental milestones are achieved at a normal bone age, that is, onset of signs of pubertal development by a bone age of 13.5 years in boys. Gonadotropin and sex steroid concentrations increase in concert with the progression of the bone age. Thus, all stages of pubertal development occur at an age that is later than average.
There are three main differential diagnoses of self-limited DP [4, 117] (Table 4.3): hypergonadotropic hypogonadism, with primary gonadal failure leading to elevated gonadotropin levels (approximately 7% of males with delayed puberty); functional hypogonadotropic hypogonadism, where late pubertal development is due to maturational delay in the HPG axis secondary to a chronic disease [129] (found in 19–20%), malnourishment [130], excessive exercise [131, 132], or psychological or emotional stress [133]; and permanent hypogonadotropic hypogonadism (HH), characterized by low LH and FSH levels (9% of boys).
Table 4.3
Differential diagnoses of male self-limited delayed puberty
Hypergonadotropic hypogonadism | Hypogonadotropic hypogonadism | Functional hypogonadotropic hypogonadism | |
|---|---|---|---|
Common causes: | Klinefelter syndrome Gonadal dysgenesis Chemotherapy/radiation therapy | CNS tumors/infiltrative diseases Isolated hypogonadotropic hypogonadism Kallmann syndrome Combined pituitary hormone deficiency Chemotherapy/radiation therapy | Inflammatory bowel disease Celiac disease Anorexia nervosa Hypothyroidism Excessive exercise Cystic fibrosis |
Hypogonadotropic Hypogonadism
HH is often diagnosed during the second or third decades of life. Common presenting signs are delayed onset of puberty, poorly developed secondary sexual characteristics, eunuchoid body proportions, or infertility [134]. This condition can be due to a congenital hypothalamic or pituitary disorder or to an acquired central dysfunction secondary to irradiation, surgery, tumor, or a vascular lesion. Tumors causing delayed puberty most commonly interfere with GnRH synthesis or secretion, the most common being craniopharyngioma, germinoma, and Langerhans cell histiocytosis. A deficiency of other anterior pituitary hormones, diabetes insipidus, and visual disturbance is common. Congenital malformation of the pituitary, hypothalamic, and other midline structures may cause HH, often in conjunction with other pituitary hormone deficiencies.
A picture of ‘idiopathic’ hypogonadotropic hypogonadism (IHH) with no associated anatomical or functional defect in the hypothalamic–pituitary–gonadal axis occurs in 1–10 cases per 100,000 births. Because of different causes and incomplete penetrance, there is a wide spectrum of phenotypes, ranging from complete hypogonadotropic hypogonadism, with lack of pubertal development, to partial hypogonadism with an arrest of pubertal development, and even reversible HH in some patients post treatment [135]. Despite recent advances, with over twenty genes linked with this disorder having been identified, the pathophysiological basis of hypogonadotropic hypogonadism in the majority of individuals remains unclear [136] (see Chap. 5). The condition may be due to the failure of the development of gonadotropin-releasing hormone (GnRH) neurons, lack of activation of GnRH secretion, or disrupted GnRH signaling (including mutations in the GNRHR gene). Kallmann syndrome (hypogonadotropic hypogonadism associated with anosmia) is the most common form of isolated HH, accounting for 60% of cases.
Downstream mutations in the GnRH signaling pathway can also present as delayed puberty. LH and FSH are encoded by a common α-subunit gene and a specific β-subunit gene. Mutations of the β-subunits genes of LH or FSH are extremely rare causes of HH [137, 138] (see Chap. 6).
Functional Hypogonadotropic Hypogonadism
Functional HH is seen in malnutrition and chronic diseases, as exemplified by patients with inflammatory bowel disease, celiac disease, chronic kidney disease, cystic fibrosis, and sickle cell anemia. Undernutrition in states of starvation such as anorexia nervosa, or an imbalance due to excess energy consumption in extreme athletic training, can also cause central GnRH suppression. The factors contributing to these states of functional HH include low fat mass, inflammation, and stress [139, 140].
Hypergonadotropic Hypogonadism
Hypergonadotropic states (chromosomal alterations, syndromes, genetic disorders, and radiotherapy/chemotherapy) are covered elsewhere in this volume (see Chaps. 9 and 10) and may present as pubertal delay. Testicular abnormalities are characterized by elevated gonadotropin and low inhibin-B concentrations, and patients with these abnormalities sometimes have specific physical features.
Delayed Puberty—Diagnosis
The cutoff age for boys who need an evaluation for delayed puberty may vary in different ethnic groups, but in most populations, early signs of secondary sexual development should be present by age 14 years. The evaluation of testicular volume is vital, as an accurate diagnosis of delayed puberty cannot be made without assessment of Tanner genital stage. A thorough medical history should note the symptoms and signs of anorexia nervosa, the intensity of athletic training, and the timing of puberty of both parents, as there is often a family history of DP in cases of self-limited DP (Fig. 4.6). A history of chronic illness, such as celiac disease or inflammatory bowel disease, suggests a temporary or secondary delay of puberty. Stature and height velocity should be evaluated using appropriate growth charts. Bone age (X-ray film of left hand and wrist read according to standards such as Greulich and Pyle) delay provides useful information in the growth analysis but contributes little to the differential diagnosis. It may be very difficult to distinguish clinically between the diagnosis of self-limited DP and congenital HH in the teenage years [141]. While gonadotropin levels are generally increased in primary testicular failure or in Klinefelter syndrome, single basal serum LH and FSH determinations are not useful in the differential diagnosis of self-limited delay vs hypogonadotropic hypogonadism.
In some cases, the diagnosis of permanent HH can be suspected before the age of pubertal onset, and importantly, if this suspicion arises in the first six months of life, it can be confirmed on the basis of low testosterone and gonadotropin levels, indicating an absence of the normal ‘mini-puberty.’ The presence or absence of ‘red flag’ features remains the strongest discriminator between self-limited DP and IHH. These red flags include cryptorchidism or micropenis, or the presence of the other components of the Kallmann syndrome which include anosmia or hyposmia due to hypoplasia of the olfactory bulbs, as well as cleft lip and palate, unilateral renal agenesis, short metacarpals, sensorineural hearing loss, synkinesia, and color blindness.
Investigation of the differential diagnosis of the two conditions may involve a number of physiological and dynamic tests including assessment of LH pulsatility by frequent blood sampling [42], the prolactin response to provocation [142, 143], the gonadotropin response to GnRH [144, 145] and analogs [146], the testosterone response to hCG [146–148], and first morning-voided urinary FSH and LH levels [149]. Boys with self-limited delay who are destined to undergo spontaneous pubertal development within 6 to 12 months may have a pubertal pattern of response to GnRH (post-GnRH maximum LH levels higher than maximum FSH levels). However, a low prepubertal LH response to GnRH is usually found in boys with self-limited DP who will develop later than that, as well as in boys with permanent HH. Most recently, a single measurement of inhibin-B <35 pg/mL has been shown to help discriminate prepubertal boys with IHH from DP with high sensitivity [150]; nevertheless, patient follow-up is necessary for a definitive diagnosis in the majority of cases. With the discovery in the last two decades of genes causing HH, genetic testing will no doubt modify the management of many of our male delayed puberty patients [151].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


