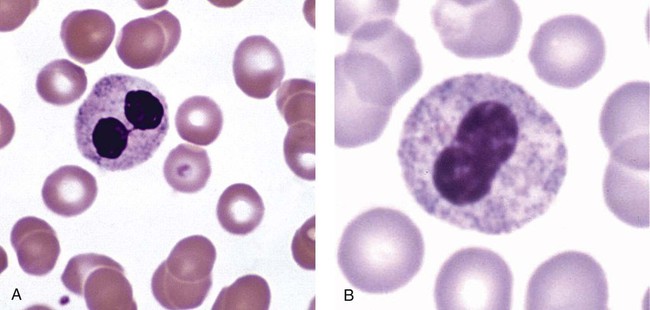
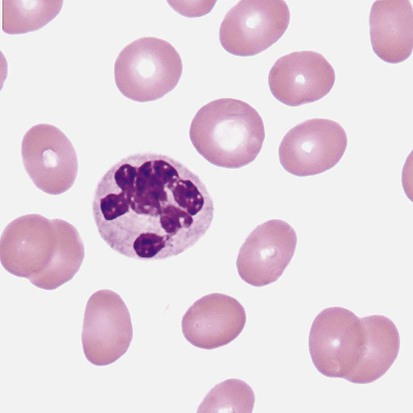
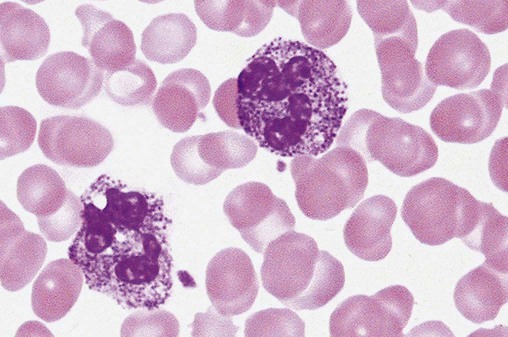
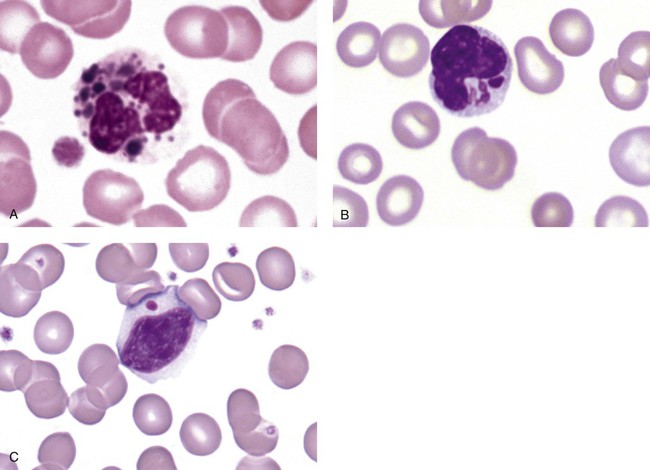
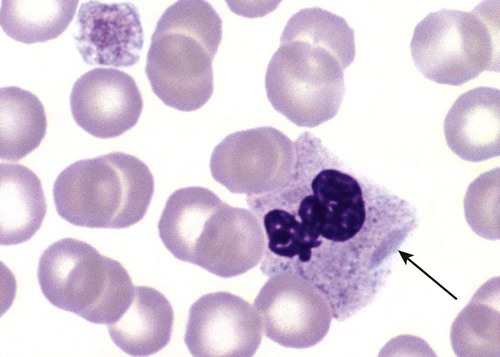
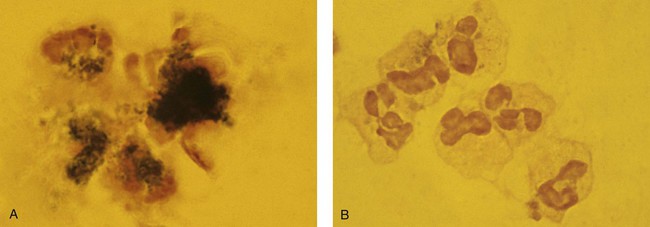
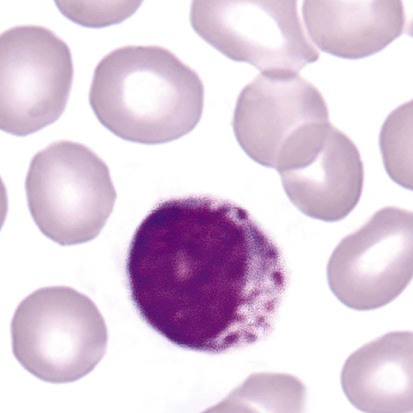
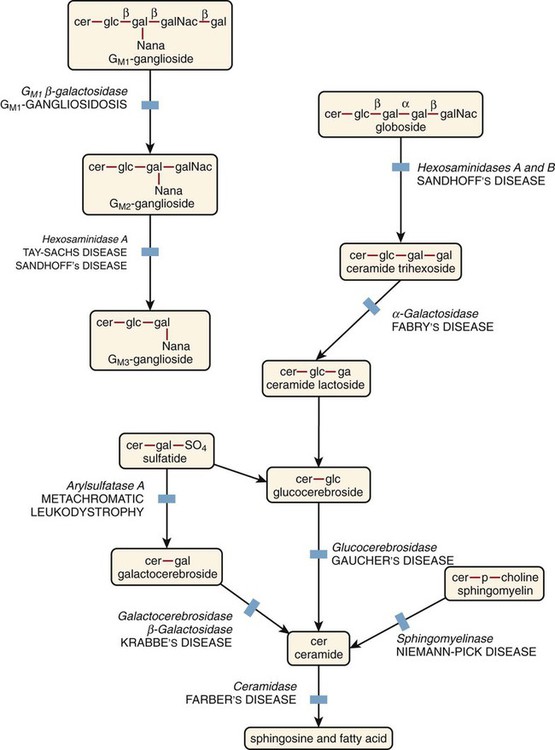
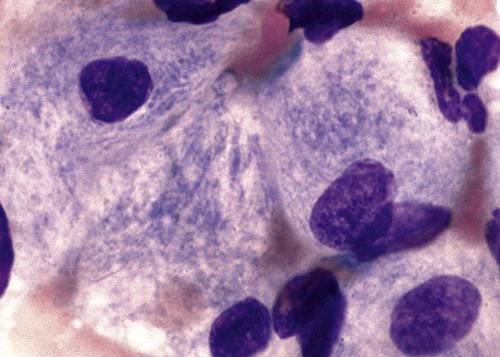
After the completion of this chapter, the reader will be able to: 1. Describe the basic genetic defect and the morphologic consequences in Pelger-Huët anomaly. 2. Indicate with what cell type Pelger-Huët cells might be confused and discuss what is meant by “pseudo-Pelger Huët.” 3. Compare and contrast two genetic causes of neutrophilic hypersegmentation and indicate what other disorder must be ruled out. 4. Describe the basic genetic defect and the morphologic consequences in Alder-Reilly anomaly, Chédiak-Higashi syndrome, and May-Hegglin anomaly. 5. Indicate how inclusions in Alder-Reilly anomaly and May-Hegglin anomaly might be confused with reactive morphology. 6. Discuss the defect in and functional consequences of chronic granulomatous disease. 7. Discuss the basic defects in and functional consequences of leukocyte adhesion disorders. 8. Discuss at least three other miscellaneous granulocyte disorders. 9. Describe the characteristic macrophage morphology associated with the mucopolysaccharidoses, Gaucher disease, and Niemann-Pick disease. 10. Describe the basic defect in genetic disorders leading to decreased T-lymphocyte production, decreased B-lymphocyte production, and the combined decrease of T, B, and natural killer lymphocytes. 11. Define what is meant by neutrophilia, neutropenia, lymphocytosis, lymphocytopenia, monocytosis, monocytopenia, eosinophilia, eosinopenia, and basophilia; give some examples of conditions in which each occurs. 12. Describe the nuclear and cytoplasmic alterations in granulocytes, monocytes, and lymphocytes that indicate a reaction to infection, inflammation, or stress. 1. What is the most likely cause of this child’s recurring infections? 2. Biochemically speaking, what is the specific nature of the problem? 3. Do all microorganisms produce equally severe disease in individuals affected with this disorder? 4. How is this disorder transmitted genetically in the majority of cases? 1. Based on the differential results, what cells should the medical laboratory scientist look for on the patient’s blood film? 2. Where on the blood film should the examination be made and why? 3. What is the significance if the suspected cells are found? 4. What preparation other than a blood film might be helpful in this situation? Pelger-Huët anomaly (PHA) is an autosomal dominant disorder resulting in decreased nuclear segmentation that is most apparent in neutrophils. It is the most common genetic disorder of leukocytes and is caused by a mutation of the lamin B receptor. The lamin B receptor is an integral protein in the inner nuclear membrane,1 and the associated gene is located on chromosome band 1q41-43.2 PHA is one of a large number of so-called laminopathies ranging from a type of muscular dystrophy to premature aging.3 Heterozygous PHA is characterized by granulocytes whose nucleus is segmented only once, taking the form of “two very distinct segments connected by a very thin filament.”4 In addition, what appear to be band forms may be present; however, the chromatin shows a highly clumped pattern and stains densely. The heterozygous mutation does not affect the function of neutrophils.5 Homozygous PHA is relatively rare and is characterized by granulocytes with round or oval nuclei (no segmentation) as well as the band-form nuclei described earlier. The chromatin pattern is also very dense and clumped. The homozygous form may be accompanied by impaired cognitive development, skeletal abnormalities, and heart defects.5 Leukocyte differential counts performed on blood samples from patients with either heterozygous or homozygous PHA may mistakenly identify the abnormal cells as immature granulocytes if the highly clumped, dense chromatin pattern is not noticed (Figure 28-1). Medical laboratory scientists must distinguish inherited PHA from an acquired form of nuclear hyposegmentation known as pseudo–Pelger-Huët anomaly (pseudo-PHA) that may be seen in a variety of malignant myeloproliferative neoplasms.6 Two distinctions between inherited PHA and pseudo-PHA are the following: (1) the percentage of affected neutrophils is much higher in the inherited form (usually over 80%), whereas in the acquired form fewer than 50% are usually affected; and (2) pseudo-PHA alterations are usually accompanied by other morphologic indications of malignancy, such as the presence of blast forms. Hereditary neutrophil hypersegmentation designates a group of at least two disorders. The first is a benign autosomal dominant condition characterized by hypersegmented neutrophils (Figure 28-2) with no clinical signs or symptoms. The major concern is to distinguish this condition from the neutrophilic hypersegmentation found in megaloblastic anemia. In the hereditary form, a large majority of neutrophils are hypersegmented and there is no macrocytic anemia. In megaloblastic anemia, fewer than 50% of neutrophils are usually hypersegmented. The second disorder is myelokathexis, an inherited form of neutropenia characterized by hypersegmented neutrophils and bone marrow hyperplasia of myeloid cells. There is increased programmed cell death (apoptosis) of granulocyte precursors.7 In this case the neutrophil nuclei, in addition to being hypersegmented, are pyknotic (to be described later), and the intrasegmental filaments are longer than normal. Alder-Reilly anomaly is transmitted as a recessive trait and is characterized by granulocytes with large, darkly staining metachromatic granules that may resemble toxic granulation (described later) (Figure 28-3). The granules are composed of mucopolysaccharides and are sometimes referred to as Alder-Reilly bodies or Reilly bodies. In more severe forms, the granules may also be found in monocytes and lymphocytes. The basic defect is the incomplete degradation of mucopolysaccharides (protein-carbohydrate complexes), and there may be a structural abnormality in the myeloperoxidase gene.8 These granules are also discussed later in the section on storage diseases. Chédiak-Higashi syndrome (CHS) is a rare and fatal autosomal recessive disease characterized by cells with enlarged lysosomal vesicles. All cells with lysosomal organelles are affected, including the melanosomes of melanocytes in the skin, the dense granules of platelets, and leukocyte granules. Patients usually have a tendency to bleed, frequent bacterial infections, symptoms of immunodeficiency, variable albinism, and progressive neurologic dysfunction.9 CHS is associated with a mutation in the LYST gene that encodes for a type of vesicle trafficking regulatory protein.10 Patients who do not die of bacterial infection develop a lymphohistiocytic infiltration into the major organs of the body, resulting in organ failure.9 Hematologic findings in CHS include giant lysosomal granules in granulocytes, monocytes, and lymphocytes (Figure 28-4). The platelet abnormality usually results in a prolonged bleeding time, and granule release by thrombin is impaired.11 May-Hegglin anomaly is one of a group of at least four overlapping autosomal dominant platelet disorders: May-Hegglin anomaly (MHA), Sebastian syndrome (SBS), Fechtner syndrome (FS), and Epstein syndrome (EPS). All are caused by mutations of the nonmuscle myosin heavy-chain type IIA gene referred to as MYH9.12 MHA, SBS, and FS are characterized by large basophilic inclusions in leukocytes, thrombocytopenia, and giant platelets (Figure 28-5). FS and EPS are also associated with nephritis and deafness. Patients with MHA and SBS generally do not have renal disease or deafness; however, there have been a few reports of renal disease in individuals with MHA.13 The leukocyte inclusions in MHA have been described as “Döhle body–like” (Döhle bodies are discussed later in this chapter); however, there are striking ultrastructural differences. Döhle bodies are made up of lamellar rows of rough endoplasmic reticulum, whereas the MHA inclusions consist of randomly placed rods in an amorphous background. In addition, Döhle bodies are found only in neutrophils, whereas MHA inclusions are seen in neutrophils, eosinophils, basophils, and monocytes.14 MHA inclusions may stain a very pale color with Wright stain and may be missed in monocytes whose cytoplasm is also blue-grey. Chronic granulomatous disease (CGD) is a disorder caused by the inability of phagocytes to produce superoxide and reactive oxygen species. The basic defect is one or more mutations in any of four genes responsible for proteins that make up a complex known as NADPH oxidase (NADPH is the reduced form of nicotinamide adenine dinucleotide phosphate). The majority of cases (approximately 60% to 65%) are X-linked recessive and 35% to 40% are autosomal recessive, depending on which gene has been mutated. The X-linked forms tend to carry a higher mortality rate.15 In the nitroblue tetrazolium reduction test, normal neutrophils, when stimulated, reduce the yellow water-soluble nitroblue tetrazolium to a dark blue insoluble formazan. CGD neutrophils cannot perform this reduction (Figure 28-6). Flow cytometry uses a fluorescent probe, such as dihydrorhodamine-123, to measure intracellular production of reactive oxygen species.15 Leukocyte adhesion disorders (LADs) result in the inability of neutrophils and monocytes to adhere to endothelial cells and to transmigrate from the blood to the tissues. The consequence of this inability is increased and potentially lethal bacterial infections. The basic defect is a mutation in the genes responsible for the formation of cell adhesion molecules, including integrins and selectins (see Chapter 12). LADs have been subdivided into three subcategories. LAD-I is caused by a mutation in the gene(s) responsible for β2 integrin subunits (CD11/CD18), resulting in either decreased or defective β2 integrins, which are necessary for adhesion to endothelial cells, recognition of bacteria, and outside-in signaling.16 In addition to experiencing recurrent infections, patients with LAD-I frequently have neutrophilia. LAD-II is considerably rarer than LAD-I and presents in a similar manner (recurrent infection, neutrophilia, and impaired pus formation); however, the leukocytes have normal β2 integrins. In addition, patients with LAD-II may have mental retardation, growth defects, and/or deficiency in H blood group antigen.17 In this case, the basic defect is faulty ligands for selectin adhesive molecules. Patients with LAD-III present with Glanzmann thrombasthenia–like bleeding problems (see Chapter 44) as well as LAD-I symptoms despite normal integrin expression. A recent report indicates that the basic defect is a mutation in KINDLIN3, a gene responsible for a protein that binds to the β2 integrin subunit.18 Miscellaneous granulocyte disorders include several relatively rare diseases. Hyperimmunoglobulinemia E, also known as Job syndrome, is characterized by elevated levels of immunoglobulin E, skeletal abnormalities, and recurrent severe bacterial infections.19 Neutrophils in these patients have poor directional motility. Evidence points to dysregulation in cytokine production by T cells, resulting in imbalances between interferon-γ and transforming growth factor β in relation to interleukin-4 (IL-4).20 Lazy leukocyte syndrome is extremely rare and is characterized by neutropenia and poor neutrophil response to chemotactic agents.21 Abnormalities in neutrophil cytoplasmic actin and myosin microfilaments are responsible for the dsyfunction.22 Myeloperoxidase (MPO) deficiency is probably the most common neutrophil abnormality; however, symptoms are relatively mild in most patients. The discovery of MPO deficiency is credited to the automated hematology analyzers that use MPO to identify cells. The MPO gene is located on chromosome 17, and up to 10 mutations have been described.23 Diabetes mellitus can be either inherited or acquired. It has been associated with poor neutrophil function, especially when glucose levels are very high.24 The most consistent abnormality is abnormal oxidative burst activity.25 Monocyte/macrophage lysosomal storage diseases can be subdivided into mucopolysaccharide (or glycosaminoglycan [GAG]) storage diseases and lipid storage diseases (Table 28-1). As a group, they represent inherited enzyme deficiencies or defects that result in flawed degradation of phagocytized material and buildup of the partially digested material within the phagocyte. All cells containing lysosomes can be affected, including T lymphocytes.26 TABLE 28-1 Variants of Monocyte/Macrophage Lysosomal Storage Disorders The mucopolysaccharidoses (MPSs) are a family of inherited disorders of GAG degradation. Each MPS is caused by deficient activity of an enzyme necessary for the degradation of dermatan sulfate, heparan sulfate, keratan sulfate, and/or chondroitin sulfate. The partially degraded GAG builds up in the lysosomes and eventually results in physical abnormality and sometimes mental retardation. The MPSs have been subdivided according to which enzyme is defective, which GAG is being stored, and whether the symptoms are severe or attenuated (see Table 28-1).27 The peripheral blood of a patient with MPS may appear relatively normal; however, metachromatic Reilly bodies may be seen in neutrophils, monocytes, and lymphocytes (Figure 28-7). Bone marrow may reveal macrophages with large amounts of metachromatic material. Diagnosis relies on assays for the specific enzymes involved. Treatment has consisted of enzyme replacement therapy or hematopoietic stem cell transplantation.27 Lipid storage diseases include a variety of disorders in which lipid catabolism is defective (Figure 28-8). Two of these disorders are characterized by macrophages with distinctive morphology and are discussed here. Gaucher disease is the most common of the lysosomal lipid storage diseases. It is an autosomal recessive disorder caused by a defect in the catabolic enzyme β-glucocerebrosidase. This results in accumulation of glucocerebroside in macrophages throughout the body, including osteoclasts in bone and microglia in the brain. Gaucher disease has been subdivided into three types based on clinical signs and symptoms (Table 28-2).28 Hematologic features may include anemia and thrombocytopenia reflecting the hypersplenism that is common in these patients. Bone marrow contains distinctive macrophages called Gaucher cells, occurring individually or in clusters, that have abundant fibrillar blue-grey cytoplasm with a striated or wrinkled appearance (sometimes described as onion skin–like) (Figure 28-9). TABLE 28-2 Clinical Subtypes of Gaucher Disease Note: Absence and severity of features are indicated by – to +++. Treatment of Gaucher disease includes the use of recombinant glucocerebrosidase, which is quite expensive.29
Nonmalignant Leukocyte Disorders
Case Studies
Case 1
Case 2
Genetic Leukocyte Abnormalities
Nuclear Abnormalities
Pelger-Huët Anomaly
Hereditary Neutrophil Hypersegmentation
Cytoplasmic Abnormalities
Alder-Reilly Anomaly
Chédiak-Higashi Syndrome
May-Hegglin Anomaly
Functional Abnormalities
Chronic Granulomatous Disease
Leukocyte Adhesion Disorders
Miscellaneous Granulocyte Disorders
Monocyte/Macrophage Lysosomal Storage Diseases
Type
Name
Deficient Enzyme
Substance Stored
Mucopolysaccharidosis
MPS I—severe
Hurler syndrome
α-l-iduronidase
Dermatan sulfate, heparan sulfate
MPS I—attenuated
Scheie syndrome
α-l-iduronidase
Dermatan sulfate, heparan sulfate
MPS II— severe
Hunter syndrome
Iduronate sulfatase
Dermatan sulfate, heparan sulfate
MPS III
Sanfilippo syndrome type A
Heparan N-sulfatase
Heparan sulfate
Sanfilippo syndrome type B
α-N-acetylglucosaminidase
Heparan sulfate
Sanfilippo syndrome type C
Acetyl–coenzyme A:α-glucosaminide N-acetyltransferase
Heparan sulfate
MPS IV
Morquio syndrome type A
Galactose-6-sulfatase
Keratan sulfate, chondroitin-6-sulfate
Morquio syndrome type B
β-Galactosidase
Keratan sulfate
Lipid storage diseases
Gaucher disease
β-glucocerebrosidase
Glucocerebroside
Niemann-Pick disease
Sphingomyelinase
Sphingomyelin
Fabry disease
α-Galactosidase
Ceramide trihexoside
Tay-Sachs disease, Sandhoff disease
Hexosaminidase A
GM2 ganglioside
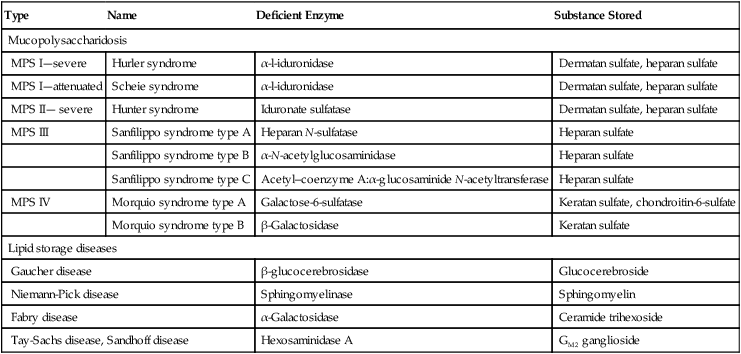
Clinical Features
Type I: Nonneuropathic
Type II: Acute Neuropathic
Type III: Subacute Neuropathic
Clinical onset
Childhood/adulthood
Infancy
Childhood
Hepatosplenomegaly
+
+
+
Skeletal abnormality
+
–
+
Neurodegeneration
–
+++
++
Death
Variable
By 2 years
2nd-4th decade
Ethnic predilection
Ashkenazi Jews
Panethnic
Swedes
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Nonmalignant Leukocyte Disorders