Non-Hodgkin’s lymphoma
Arnold S. Freedman, MD  Ann S. LaCasce, MD
Ann S. LaCasce, MD
Overview
The malignant lymphomas are neoplastic transformations of cells that reside predominantly within lymphoid tissues. Although Hodgkin lymphomas (HLs) and non-Hodgkin lymphomas (NHLs) infiltrate lymphohematopoietic tissues, their biologic and clinical behaviors are distinct. They differ with neoplastic cells of origin, sites of disease, presence of specific symptoms, and response to treatment. Although both are among the most sensitive malignancies to radiation and cytotoxic therapy, their cure rates markedly differ. HLs are cured in nearly 80% of all patients employing both conventional and salvage treatment strategies whereas NHLs are cured in fewer than 50% of patients.
Epidemiology and etiology
Incidence and mortality
In 2014, 70,890 new cases of NHL (non-Hodgkin lymphoma) were diagnosed in the United States1 with 18,990 deaths predicted. Cases increase steadily with age. There is a slight male predominance and the incidence is higher in Caucasians than in African Americans. Although the rate of increase has slowed since the mid-1990s, the incidence continues to rise by 1.5–2% each year.
The incidence of NHL subtype varies significantly by age. In children, Burkitt’s lymphoma (BL), lymphoblastic lymphoma (LBL), and diffuse large B-cell lymphoma (DLBCL) predominate. With increasing age, rates of follicular lymphomas (FLs) and other aggressive lymphomas continue to increase. Small lymphocytic and FLs are most commonly diagnosed in patients over age 60.
Exposures and diseases associated with increased risk of developing NHL
Infectious agents are involved in the pathogenesis of some NHLs (Table 1). Epstein–Barr virus (EBV) has a strong association with development of BL, natural killer cell lymphoma, and human immunodeficiency virus-related (HIV-1) lymphoma.2, 3 About 45–70% of HIV-associated NHLs are EBV related, as are nearly 100% of the primary central nervous system (CNS) lymphomas in HIV-1 positive individuals. Human T-cell lymphotropic virus (HTLV)-1 is responsible for adult T-cell leukemia/lymphoma, endemic to the Caribbean and southern Japan.4 Gastric marginal zone lymphoma (MZL) is associated with Helicobacter pylori infection.5 Splenic MZL is associated with hepatitis C infection.6 Chronic hepatitis B infection carries an increased risk of NHL.7 In Europe, ocular adnexal MZL is linked with Chlamydia psittaci infection8 and MZL involving the skin with Borrelia burgdorferi. Immunoproliferative small intestinal disease (Mediterranean lymphoma, alpha heavy chain disease) has been associated with Campylobacter jejuni.9 Kaposi sarcoma-associated herpes virus, also known as HHV-8 (human herpes virus-8), has been isolated from the neoplastic cells in patients with primary effusion lymphomas.10, 11
Table 1 Risk factors for the development of lymphoma
| Inherited immunodeficiency states | Acquired immunodeficiency states | Autoimmune and inflammatory disorders | Infectious agents (other than HIV) | Chemicals and drugs |
| Autoimmune lymphoproliferative disease | HIV-1 infection | Rheumatoid arthritis | Epstein–Barr virus | Herbicides, pesticides, organic solvents |
| Ataxia telangiectasia | Iatrogenic | Systemic Lupus Erythematosus | HTLV-1 | Ionizing radiation |
| Chediak–Higashi syndrome | Tumor necrosis factor agonists | Sjögren’s syndrome | HHV-8 | Chemotherapy, radiation therapy |
| Common variable immunodeficiency | Celiac disease | Helicobacter pylori | ||
| Wiskott–Aldrich syndrome | Hashimoto’s thyroiditis | Campylobacter jejuni | ||
| X-linked lymphoproliferative disease | Inflammatory bowel disease | Chlamydia psittaci | ||
| Borrelia burgdorferi | ||||
| HCV |
HIV-1, human immunodeficiency virus-1; HTLV-1, human T-cell lymphotropic virus-1; HHV-8, human herpes virus-8; HCV, hepatitis C virus.
An increased risk of NHL is associated with a number of exposures and/or disease states. Controversial evidence suggests certain chemical exposures, including the herbicide phenoxyacetic acid, arsenic, pesticides, fungicides, chlorophenols, organic solvents, halomethane, lead, vinyl chloride, or asbestos, increase the risk of NHL.12–14 Occupational exposures associated with an increased risk include agricultural work, welding, and work in the lumber industry.15, 16
Diseases of inherited and acquired immunodeficiency and autoimmune diseases are associated with an increased incidence of lymphoma.17, 18 The association between immunosuppression and NHLs is compelling given a percentage of lymphomas will regress with the withdrawal of immunosuppression.19 Patients undergoing organ transplantation necessitating chronic immunosuppression have a nearly 100-fold risk of NHL, which is greatest in the first-year posttransplant. DLBCL is most common NHL in this setting and is frequently associated with EBV.20 The rare inherited immunodeficiency diseases such as X-linked lymphoproliferative syndrome, Wiskott–Aldrich syndrome, Chédiak–Higashi syndrome, ataxia telangiectasia, and common variable immunodeficiency syndrome are complicated by highly aggressive lymphomas. An increased risk of NHL has been observed in first-degree relatives with NHL, CLL, and Hodgkin lymphoma (HL).21
Pathology, immunobiology, and natural history of NHL
The World Health Organization (WHO) published a new classification of tumors of the hematopoietic and lymphoid tissues (Table 2)22 that integrate morphology, immunotyping, genetic features, and clinical syndromes. To provide a context for this classification, the large numbers of entities will be grouped into “indolent,” “aggressive,” and “highly aggressive” categories (Table 2).
Table 2 World Health Organization Classification of Lymphoid Neoplasms 2008: selected B- and T-cell neoplasms
| Precursor B- and T-cell neoplasms | ||
| Precursor B-lymphoblastic leukemia/lymphoma Precursor T-lymphoblastic leukemia/lymphoma | ||
| Mature B-cell neoplasms | ||
| Chronic lymphocytic leukemia/small lymphocytic lymphoma | ||
| B-cell prolymphocytic leukemia | ||
| Lymphoplasmacytic lymphoma | ||
| Splenic marginal zone lymphoma | ||
| Hairy cell leukemia | ||
| Splenic B-cell lymphoma, unclassifiable | ||
| Plasma cell neoplasms | ||
| Extranodal marginal zone lymphoma | ||
| Nodal marginal zone lymphoma | ||
| Follicular lymphoma | ||
| Primary cutaneous follicle center lymphoma | ||
| Mantle cell lymphoma | ||
| Diffuse large B-cell lymphoma (DLBCL) | T-cell/histiocyte-rich large B-cell lymphoma Primary DLBCL of the central nervous system Primary cutaneous DLBCL, leg type EBV-positive DLBCL of the elderly | |
| DLBCL associated with chronic inflammation | ||
| Lymphomatoid granulomatosis | ||
| Primary mediastinal large B-cell lymphoma | ||
| Intravascular large B-cell lymphoma | ||
| ALK-positive large B-cell lymphoma | ||
| Plasmablastic lymphoma | ||
| Burkitt’s lymphoma (BL) | ||
| B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL | ||
| B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Hodgkin lymphoma | ||
| Mature T-cell neoplasms | ||
| T-cell prolymphocytic leukemia | ||
| T-cell large granular lymphocytic leukemia | ||
| Adult T-cell leukemia/lymphoma | ||
| Extranodal NK/T-cell lymphoma, nasal type | ||
| Enteropathy-type T-cell lymphoma | ||
| Hepatosplenic T-cell lymphoma | ||
| Subcutaneous panniculitis-like T-cell lymphoma | ||
| Mycosis fungoides | ||
| Sézary syndrome | ||
| Primary cutaneous CD30+ T-cell lymphoproliferative disorders | ||
| Primary cutaneous peripheral T-cell lymphomas, rare subtypes | ||
| Peripheral T-cell lymphoma, not otherwise specified | ||
| Angioimmunoblastic T-cell lymphoma | ||
| Anaplastic large-cell lymphoma, ALK+ | ||
| Anaplastic large-cell lymphoma, ALK- | ||
MALT, mucosal-associated lymphoid tissue; ALK, anaplastic lymphoma kinase; HHV8, human herpes virus-8; NK, natural killer; EBV, Epstein–Barr virus; HIV, human immunodeficiency virus.
Chromosomal translocations and oncogene rearrangements. Given the mechanism of immunoglobulin (Ig) and T-cell receptor (TCR) gene rearrangements in normal lymphoid cells, lymphomas are frequently found to have chromosomal translocations that involve the activation of an oncogene or inactivation of a tumor suppressor gene. The former is more common, whereby a proto-oncogene is brought under the control of a constitutively active promoter, resulting in overexpression of the oncogenic gene and its protein product. Examples include the (8;14)(q24;q32) translocation in BL, involving the MYC proto-oncogene and the IgH gene; the (14;18)(q32;q32) translocation in FL, involving the BCL2 proto-oncogene and the IgH gene; and the (11;14) (q13;q32) translocation in mantle cell lymphoma (MCL), involving the gene encoding cyclin D1 (CCDN1) and the IgH gene. Less commonly, chromosomal translocations produce fusion genes that encode chimeric oncogenic proteins. Examples of this include the (2;5)(p23;q35) translocation involving the ALK (anaplastic lymphoma kinase) and NPM1 genes in anaplastic large-cell lymphoma (ALCL) and the t(11;18)(q21;q21) translocation involving the API2 and MLT genes in MZL lymphoma. These translocations and rearrangements can be detected by polymerase chain reaction (PCR) using probes that span the chromosomal breakpoints, reverse transcriptase polymerase chain reaction (RT-PCR) to detect the RNA product of the fusion gene, or fluorescence in situ hybridization (FISH) using probes to specific chromosomal segments. In cases where the translocation results in expression of a protein or portion of a protein that is never expressed in normal lymphocytes (e.g., ALK kinase), immunohistochemistry can be used to detect the protein.
Indolent lymphomas
The indolent NHLs are generally associated with survival measured in years, even if left untreated, but are typically incurable with conventional treatment. Indolent lymphomas represent 35–40% of the NHLs diagnosed in western countries. The most common subtypes are FL, small lymphocytic lymphoma, and MZL, comprising 22%, 6%, and 5% of all NHLs, respectively. In comparison, lymphoplasmacytic lymphoma, mycosis fungoides/Sézary syndrome, and splenic MZL are rare diseases, comprising 1% or less of all NHLs.
FL is the most indolent NHL and morphologically recapitulates normal germinal centers of secondary lymphoid follicles (Figure 1). The WHO classification includes three grades based on the number of large cells per high power field: grade 1 (0–5) (Figure 2), grade 2,6–15 and grade 3 (>15). Grade 3 is subdivided into grade 3A, in which centrocytes predominate, and grade 3B, in which there are sheets of centroblasts. Grade 1 and 2 and many cases of grade 3A FLs are approached similarly. FL grade 3B is an aggressive disease and is grouped with DLBCL.
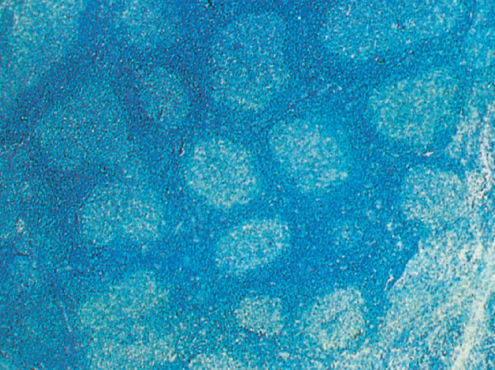
Figure 1 Follicular lymphoma grade I (low power).
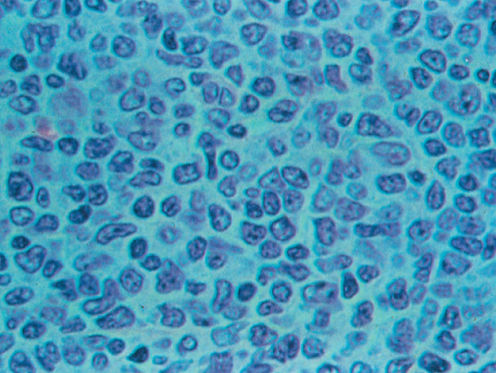
Figure 2 Follicular lymphoma grade I (high power).
FL and normal follicular center B cells express cell surface antigens including monoclonal immunoglobulin and the B-cell antigens CD19, CD20, CD10, and CD79a, but lack CD5. Cytoplasmic bcl-2 protein is overexpressed in essentially all cases of grade 1–2 disease, whereas nuclear bcl-6 is expressed by at least some of the neoplastic cells. The most common cytogenetic abnormality in FL is t(14;18) that leads to overexpression of the antiapoptotic protein bcl-2 in over 85% of cases (see Chapter 8). Recent sequencing studies have found that the most common mutations in FL (90% of tumors) involve MLL2, a gene encoding a histone H3 methylase. Other less common recurrent mutations involve other genes involving epigenetic modifying genes, such as EZH2, CREBBP, and EP300.23, 24
FL accounts for about 22% of NHL.25 Uncommon before the fourth decade, the median age at diagnosis is 60 years. FL is less common in Asians and blacks. Patients usually present with painless peripheral adenopathy, which are often longstanding and may wax and wane. Hilar and mediastinal nodes are often involved, but large mediastinal masses are rare. Patients may present with asymptomatic large abdominal masses. Staging studies usually demonstrates widely disseminated disease with involvement of spleen (40%), liver (50%), and bone marrow (70%). Marrow involvement in FL reveals a unique pattern of paratrabecular infiltration. Few patients present with extranodal extramedullary disease, and only 20% present with B symptoms or lactate dehydrogenase (LDH) elevation. Intestinal-only presentation may occur and has a favorable prognosis.26 CNS involvement is uncommon although peripheral nerve compression and epidural tumor masses causing cord compression may develop.
FL grade 3 was previously called follicular large-cell lymphoma. BCL6 rearrangements are present in a high fraction of grade 3B cases. Most studies have included both FL grades 3A and 3B, which affects the interpretation of the outcomes. Clinically grade 3B more closely approximates DLBCL.22, 27 In contrast many patients with FL grade 3A have a more indolent disease.
The course of FL is quite variable. Some patients can be observed with waxing and waning disease for 5 years or more without the need for therapy.28 Others present with more disseminated disease and rapid growth and require treatment due to organ enlargement, lymphatic obstruction, or organ obstruction.
Histologic transformation to aggressive lymphoma, usually DLBCL, occurs in up to 60%, approximately 2–3% per year, of patients with FL and is characterized by rapid progression of lymphadenopathy, extranodal disease, B symptoms, elevated LDH, and often a poor prognosis.29, 30
Small lymphocytic lymphoma
Small lymphocytic lymphoma and B-cell chronic lymphocytic leukemia are viewed as the same entity by the WHO classification. Although the major population of cells resembles small normal lymphocytes, larger cells resembling those seen in prolymphocytic leukemia are seen in the nodal tissue in areas known as proliferation centers (Figure 3 and Figures 2, 3 of Chapter 116). The small lymphocytic lymphomas are phenotypically identical to B-cell chronic lymphocytic leukemias but have fewer than 5000 circulating malignant B cells. They express HLA-DR, B-cell antigens CD19, CD20, CD23, weak surface immunoglobulin, and CD5. Cytogenetic abnormalities include trisomy 12 present in about 40% of cases, 13q in 45–55%, 11q abnormalities in 17–20%, and 17p abnormalities in 7–10% of cases. Cases with 13q deletions have the most favorable prognosis, whereas those with 11q or 17p abnormalities have an unfavorable prognosis.31
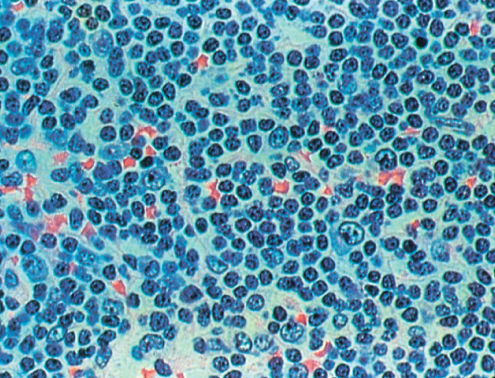
Figure 3 Small lymphocytic lymphoma.
Recent studies suggest that 30–50% of these diseases have nonmutated immunoglobulin variable region genes and correspond to naïve B cells. These cases often express CD38 and the tyrosine kinase ZAP-70 and have a worse prognosis.32 The remaining 50–70% of cases have mutated immunoglobulin variable region genes, are derived from germinal center or postgerminal center B cells,33 and have a favorable prognosis.34 Deep sequencing studies of chronic lymphocytic leukemia have revealed a number of recurrent mutations, including NOTCH1, MYD88, and SF3B1 genes.35
Small lymphocytic lymphomas makes up about 6% of all NHLs.25 The clinical presentation is similar to FL. Unlike B-cell chronic lymphocytic leukemia, the peripheral blood may be normal or reveal only a mild lymphocytosis although the bone marrow is positive in 70–90% of cases. A serum paraprotein is found in about 20% of cases and hypogammaglobulinemia is present in about 40%. Small lymphocytic lymphomas and B-cell chronic lymphocytic leukemia can convert to DLBCL or less commonly HL (Richter syndrome, Figure 4).36
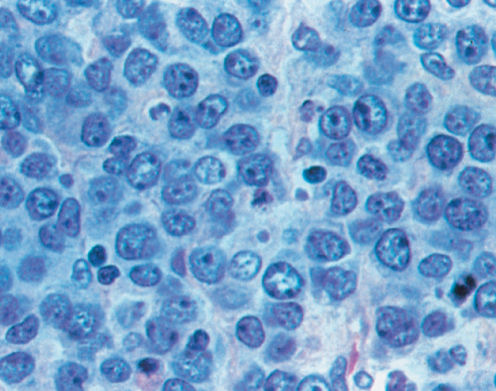
Figure 4 Diffuse large B-cell lymphoma.
Lymphoplasmacytic lymphoma
Lymphoplasmacytic lymphoma is an indolent lymphoma composed of diffuse proliferation of small lymphocytes with evidence of maturation to plasma cells.37 Evidence of immunoglobulin is seen in these cells by special stains or inclusions. These tumors express B-cell antigens, CD19 and CD20, and surface immunoglobulin M isotype, and in general do not express CD5, CD10, or CD23. Deletions of 6q21 have been identified in 40–60% of patients with lymphoplasmacytic lymphoma and the syndrome associated with this is known as Waldenström’s macroglobulinemia. Activating mutations in MYD88, a protein involved in signaling pathways downstream of the Ig receptor, are present in close to 100% of cases.38
Lymphoplasmacytic lymphoma represents about 1% of all NHLs. Clinically, this disease is similar to small lymphocytic lymphomas. The median age is early 60s, and virtually all patients have stage IV disease by virtue of bone marrow involvement, and lymph nodes and spleen are commonly involved. B symptoms and elevated serum LDH are rare. A serum M component is common. As with B-CLL, the paraprotein may have autoantibody or cryoglobulin activity. However, most cases with mixed cryoglobulinemia have been shown to be related to concurrent hepatitis C virus (HCV) infection39 and may respond to therapy directed at hepatitis C. In the WHO clinical study, 5-year overall survival (OS) (58%) and failure-free survival (25%) were identical to that of small lymphocytic lymphomas.
Marginal zone lymphomas
MZLs are a group of distinct entities including nodal MZL; extranodal MZL also known as the lymphomas of mucosal-associated lymphoid tissues (MALTs); and the splenic MZL.40, 41 In the nodal MZL, the tumor cells cytologically resemble “normal” monocytoid B cells and often involve lymph node sinuses. Phenotypically, tumor cells express surface immunoglobulin M and B-cell antigens (CD19, CD20). Similar to other indolent lymphomas, MZL can transform into a higher grade lymphoma. The nodal MZLs constitute 1% of all NHLs. Over 70% of patients present with stage III/IV disease and the majority are asymptomatic. Bone marrow involvement is less common than in most indolent lymphomas. The 5-year survival for patients with nodal MZL is 55–79%.
The extranodal MZL tumor cells resemble monocytoid B cells, express CD19, CD20, and surface immunoglobulin M, and are thought to arise from memory B cells. Lymphoepithelial lesions may be seen associated with centrocytes. The disease does not form follicles; rather the malignant cells surround reactive follicles. When extranodal MZL spreads to lymph nodes, the neoplastic cells involve the marginal zones. The most common cytogenetic abnormality is trisomy 3, occurring in up to 60% of cases (particularly the gastric extranodal MZL), and t(11;18), occurring in 25–40% of cases.42
Extranodal MZLs constitute about 5% of all NHLs and almost 50% of all gastric lymphomas. B symptoms are uncommon and most patients present with stage I or II disease. There is no age predilection. The gastrointestinal tract (most commonly stomach), lung, dura, lacrimal and salivary glands, skin, thyroid, and breast may be involved. MZL is associated with autoimmune diseases and infections with H. pylori, B. burgdorferi, C. psittaci, C. jejuni, and HCV.43–46 Fewer than 25% of cases have lymph node or bone marrow involvement. Patients can present with peptic ulcer disease, abdominal pain, and sicca syndrome, or a mass at the site of involvement. These lymphomas can disseminate to other MALT sites or bone marrow in about 30% of cases, typically later in the course of the disease. This is more commonly seen in nongastric MZLs.47 Complete remission rates are high, and OS up to 80% at 10 or more years.48 Like all indolent NHLs, these can transform to DLBCL.
Splenic MZL constitutes <2% of all NHLs, with a median age of 65, and is uncommon before age 50.25 Histologically, there is expansion of marginal zones in the spleen. Bone marrow and peripheral blood involvement (referred to as splenic lymphoma with villous or nonvillous lymphocytes) can also be present. In splenic MZL, trisomy 3 is present in 39% of cases. The survival of patients is in excess of 70% at 10 years. Sequencing studies demonstrate recurrent somatic mutations in genes involved in the NOTCH, NF-κB, and B-cell receptor pathways, as well as mutations in TP53.49
Aggressive lymphomas
Mantle cell lymphoma
MCL is generally an aggressive disease.50, 51 The neoplastic cells are counterparts of naive “mantle zone” B cells and are medium sized with irregular nuclei. The disease may have either diffuse architecture or a vaguely nodular appearance. Some cases of MCL have a predominance of “blastoid” cells with a high mitotic rate. The cells express B-cell antigens, surface immunoglobulin M with or without immunoglobulin D, CD5, and CD43, but lack CD10 and CD23, respectively, and overexpression of cyclin D1. Approximately 70% of MCLs have t(11;14)(q13;q32) that creates rearrangements of the bcl-1 (cyclin D1) gene. Eight percent of MCL cases are cyclin D1-negative and overexpress cyclin D2 and 4 or cyclin D3, without chromosomal rearrangements52 and are clinically similar to cyclin D1-positive cases.53 Deep sequencing54 has identified NOTCH1 mutations in a minority of cases, which may be associated with poor prognosis. SOX11 overexpression is also associated with a worse prognosis.52, 55
MCL constitutes about 7% of all NHLs. About 75% of patients are males, with median age of 63. Approximately 70% of patients have stage IV disease, and B symptoms are observed in approximately one-third of patients. Typical sites of involvement are lymph nodes, spleen, liver, Waldeyer’s ring, and bone marrow. Peripheral blood involvement is present in 25–50% of patients at presentation. MCL can involve any region of the gastrointestinal tract, occasionally presenting as multiple intestinal polyposis. The median survival of patients with MCL is 3–6 years and patients with the blastic variant at diagnosis have a median survival of 18 months. Blastic transformation occurs in 35% of patients, with a risk of 42% at 4 years; the median survival is 3.8 months.56 Mutations and deletions of p53 are also associated with a worse prognosis.57
Diffuse large B-cell lymphoma
DLBCL consists of a diffuse proliferation of large cells with a high mitotic rate. The cells have a moderate amount of cytoplasm with either cleaved or noncleaved nuclei often with multiple nucleoli, although there can be great variability in the morphology (Figure 4). DLBCL represents many distinct disease entities.22 (Table 2) Gene expression profiling has been applied to DLBCL.58–61 These studies subdivided DLBCL into distinct genetic entities. DLBCLs correspond to germinal center B cells or activated B cells. The tumor cells generally express B-cell antigens (CD19 and CD20), monoclonal surface immunoglobulin M, and occasionally other heavy chain isotypes. CD5-positive cases are uncommon and may have a worse prognosis.62 CD10 and bcl-6 support a germinal center origin, whereas expression of MUM1 supports a nongerminal center origin. Approximately 70% express bcl-6 protein, consistent with a germinal center origin.63
Several chromosomal abnormalities have been observed in DLBCL. Bcl-6 is associated with chromosomal rearrangements involving 3q27.64 Rearrangements of the gene occurs in 20–40% of diffuse aggressive lymphomas. t(14;18) has been observed in approximately 30% of patients with DLBCL. Some of these cases may represent histologic transformations of prior FL. By gene expression profile (GEP), the germinal center B-cell (GCB) type is often associated with the t(14;18) and amplifications of the REL oncogene on chromosome 2. In contrast, the activated B-cell (ABC) type is associated with loss of 6q21, trisomy 3, gains of 3q and 18q21–22, and mutations of EZH2.65, 66 ABC cases also have high-level activation of NF-κB.67 MYC is rearranged in 10% of DLBCLs, with the partner gene being one of the Ig genes in 60% of cases and alternative genes in 40% of cases. Approximately 20% of MYC-rearranged cases have concurrent BCL2 or BCL6 rearrangements, a combination referred to as “double-hit lymphoma.”68 Amplification and/or overexpression of MYC independent of rearrangements or amplification has also been described and is also associated with a poor prognosis.69, 70
DLBCL constitutes 31% of all NHLs and is the most common histologic subtype. Patients who are generally middle-aged or older (median age 64 years) present with either nodal enlargement or extranodal disease. DLBCL presents in a localized (stage I or IE) manner approximately 20% of the time and 30–40% of patients will have I or II disease. Stage IV disease is seen in approximately 40% of patients. B symptoms occur in 30% of patients, and unlike most NHLs, LDH is elevated in over half the patients. During the course of the disease, the liver, the kidneys, the lung, the bone, and the peripheral nerves may be involved. Bone marrow involvement is initially found in 10–20% of patients. Extranodal diseases, specifically testicular, bone marrow, paranasal sinus, multiple extranodal sites, and elevated LDH, are other risks for CNS dissemination.71 Rare cases of DLBCL present with a disseminated intravascular proliferation of large lymphoid cells, involving small blood vessels, without an obvious tumor mass72–74 most commonly involving the CNS, kidneys, lungs, and skin.
Within the DLBCL group is a distinct clinical entity known as primary mediastinal large B-cell lymphoma (PMLBCL) (7% of all cases of DLBCL).75 Histologically, the cellular infiltrate is heterogeneous, and sclerosis is frequently present. The immunophenotype includes B-cell antigens (CD19, CD20), but they are often negative for surface and cytoplasmic immunoglobulin. GEP suggests that this is a distinct entity from germinal center or ABC types of DLBCL. Recent studies suggest that the GEP pattern of PMLBCL closely resembles the Reed–Sternberg cells of HL.76, 77 Copy number gains in the region on chromosome 9p containing the genes for JAK2 and programmed cell death ligand 1 and 2 (PDL1 and PDL2) and ligands for the programmed cell death receptor-1 (PD-1), which has a role in suppressing T-cell function, are common.65 PMLBCL resembles classical HD by GEP.76 PMLBCL has a female predominance, with a median age of 40. Over 70% of these patients present with stage I/II bulky disease involving the mediastinum. Pleural and pericardial effusions are seen in about one-third of patients. Superior vena cava syndrome is common. Similar to DLBCL, an elevated LDH is present in the majority, whereas bone marrow involvement is infrequent. The prognosis of patients with PMLBCL is similar to patients with DLBCL.
Peripheral T-cell lymphomas
Peripheral T-cell lymphomas (PTCLs) include a large number of entities that constitute 15% of all NHLs in adults.78 Among these, in decreasing frequency, are PTCL, not otherwise unspecified (NOS); ALCL; angioimmunoblastic T-cell lymphoma (AITL), extranodal NK/T-cell lymphoma, nasal type; and much rarer entities, panniculitis-like T-cell lymphoma, enteropathy-type T-cell lymphoma, and hepatosplenic γ/δ T-cell lymphoma.
PTCLs can be nodal- or extranodal-based diseases. The diffuse cellular infiltrates range from a mixture of small and large cells; infiltrates of pleomorphic cells, often with a background of epithelioid histiocytes, plasma cells, eosinophils, and Reed–Sternberg-like cells; or predominantly large cells. In contrast to B-cell lymphomas, the pattern of expression of T-cell surface antigens is highly variable. The majority express CD2, CD3, and CD4, with a subset of expressing CD8.79 In most cases, one or more “mature” T-cell antigens, such as CD5 or CD7, are lost. Many cases of PTCL express EBV, especially the extranodal NK/T-cell lymphomas, nasal type.80 EBV positivity is associated with a poor prognosis.
Abnormal metaphases are seen in 90% of T-cell lymphomas. The most commonly seen translocations in PTCLs are t(7;14), t(11;14), inv(14), and t(14;14). AITL is associated with trisomy 3 and/or 5.81 These translocations involve genes for the TCR at 14q11, 7q34–35, and 7p15. Young patients with ALCL have t(2;5) and less commonly t(1;2).82 ALCL in adults generally lack t(2;5). Hepatosplenic γ/δ T-cell lymphomas are associated with isochromosome 7q and trisomy 8.83
Patients with PTCL have a similar median age as patients with DLBCL. In contrast, 80% of patients with PTCL have stage III/IV disease and more frequently have B symptoms, hepatosplenomegaly, and extranodal disease, such as the skin. PTCL generally has a worse prognosis than DLBCL.84, 85 A number of uncommon subtypes of PTCL have unique histologic features. AITL, in addition to a pleomorphic heterogeneous cellular infiltrate, displays increased amounts of high endothelial venules, giving a hypervascular appearance. In this subtype, Ig heavy chains may be rearranged in 10% cases, and EBV genomes are detected in most cases and may be in either T or B cells.86 AITL typically affects older adults who present with the acute onset of generalized lymphadenopathy, hepatosplenomegaly, skin rash, and B symptoms.86 Immunologic abnormalities are common and include plasmacytosis, polyclonal hypergammaglobulinemia, and a positive Coombs test. The median survival is 30 months. Infection is the most common cause of death, followed by the T-cell lymphoma or development of EBV-positive DLBCL.
ALCL is a T-cell NHL that can present as primary systemic ALCL, ALK-positive; primary systemic ALCL, ALK-negative; and primary cutaneous ALCL. When involving nodes, ALCL characteristically fills the sinusoids of lymph nodes with bizarre large cells. Neoplastic cells derived from patients with ALCL also generally express the phenotype of mature activated T cells (HLA-DR, CD30, CD25). The ALK protein is detected in 40–60% of cases using the ALK1 monoclonal antibody, showing both nuclear and cytoplasmic stainings in cases with the t(2;5).87 The resulting fusion gene encodes a chimeric NPM-ALK fusion protein with constitutive tyrosine kinase activity. ALK-positive cases are more common in children and younger adults and have a better prognosis than ALK-negative cases.88
ALCL constitutes 2% of all NHLs in adults, but is the second most common T-cell lymphoma. The median age of patients with ALCL is 34 with a male predominance. There is a bimodal distribution of this disease, with peaks in childhood, young adults, and late adulthood. In adults, B symptoms, peripheral adenopathy, and retroperitoneal adenopathy are common. Skin is a frequent site of extranodal disease (about 25% of patients), whereas bone marrow involvement is uncommon. An unusual form of ALCL arises within the breast associated with breast implants.89 The prognosis of adult patients with systemic ALK-positive ALCL is similar to patients with DLBCL.90 When ALCL, PTCL NOS, and AITL are compared, ALCL has the highest OS, whereas AITL had the lowest.91
Gray zone lymphoma
The gray zone lymphomas include B-cell lymphoma unclassifiable with features intermediate between BL and DLBCL (B-UNC/BL/DLBCL) and between DLBCL and cHL (B-UNC/cHL/DLBCL).22
In B-cell lymphoma unclassifiable with features intermediate between BL and DLBCL (B-UNC/BL/DLBCL), the neoplastic cells are intermediate to large size, may have a very high Ki67 index, and are CD10+. Cells are more variable in size than BL cells, often BCL2 positive, may be BCL6 negative, and may have a Ki67 index lower than 100%. GEP has shown these to be heterogeneous.92 Between 30% and 45% cases have chromosomal translocations involving c-myc and bcl-2, known as “double-hit” lymphomas.68 B-UNC/BL/DLBCL often presents with extranodal disease with high IPI and generally has a poor prognosis. Double-hit lymphomas, with this histology, have a very poor prognosis, with a median OS of 4 months.93
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and classical HL (B-UNC/cHL/DLBCL) has features intermediate between PMBCL and classical HL, or B-UNC/cHL/DLBCL. This rare disease presents commonly in men with a mediastinal mass. Histologically, cells resemble both the Reed–Sternberg cell of classical HL and the large cells of DLBCL or PMLBCL. Fibrous stroma and an inflammatory infiltrate are seen. Malignant cells express CD45, CD20, CD79a, and CD30, but often are CD15(−); other B-cell markers such as PAX5, OCT-2, and BOB1 are often positive. The prognosis of these patients is inferior to both cHL and PMLBCL; the optimal therapy has not been demonstrated.94
Highly aggressive lymphomas
Precursor T- or B-lymphoblastic leukemia/lymphoma
LBL and acute lymphoblastic leukemia ALL (see Chapter 116) represent two presentations of the same disease with LBL defined as having <25% bone marrow involvement. The neoplastic cells have a high nuclear to cytoplasmic ratio, scant cytoplasm, and nuclei with fine chromatin with multiple small nucleoli and have a high mitotic rate. The nuclei can have folds or convolutions. Typically, nodes involved with LBL are effaced by malignant cells (Figure 5).
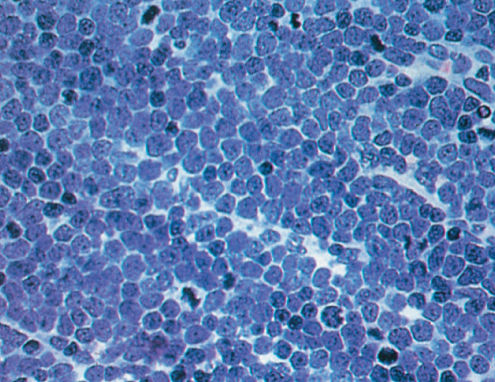
Figure 5 T-lymphoblastic lymphoma.
The vast majority of LBLs are of T-cell lineage. Several investigators have noted that most T-cell LBLs correspond to stages of thymocyte differentiation (see Chapter 116).
Although LBLs represent a major subgroup of childhood NHLs, they are unusual in adults (2% of adult NHLs). Patients are usually males in their 20s or 30s, who present with lymphadenopathy in cervical, supraclavicular, and axillary regions (50%) or with a mediastinal mass (50–75%). This entity may be associated with superior vena cava syndrome, tracheal obstruction, and pericardial effusions. Less commonly, patients present with extranodal disease (e.g., skin, testicular, or bony involvement). More than 80% of patients present with advanced stage disease, almost 50% have B symptoms, and the majority have elevated LDH. Although the bone marrow is frequently normal at presentation, virtually all patients develop bone marrow infiltration and a subsequent leukemic phase. Patients with bone marrow involvement have a very high incidence of CNS infiltration. B-cell LBL is a very rare variant, affecting patients with a median age of 39.95 B-cell LBL presents without a mediastinal mass but instead involves lymph nodes and extranodal sites.
Burkitt’s lymphoma
BL cells resemble the small noncleaved cells within normal germinal centers of secondary lymphoid follicles. Because of the high mitotic rate, frequent mitotic figures are seen and, analogous to normal germinal centers, tingible body macrophages are seen, giving the classical “starry sky” appearance. It is generally agreed that the fraction of Ki-67 (proliferating cells) in BL should be 99% or greater.22
BL is a tumor of B-lineage derivation identified by the expression of a variety of B-cell-restricted antigens including CD19, CD20, surface immunoglobulin M, CD10, and nuclear bcl-6 protein.96 The endemic BL is EBV positive, whereas the vast majority of nonendemic BL is EBV negative. BL cells lack bcl-2 protein.
BL involves a translocation of chromosome 8q24 in over 95% of the cases with chromosome 14, 2, or 22.22 Pathologically identified Burkitt or histologically atypical Burkitt has a GEP that is associated with overexpression of myc target genes, differential expression of normal germinal center genes, and decreased expression of MHC class I and NF-κB target genes. These studies will help refine the histologic diagnosis of difficult to classify cases.92, 97, 98
BL, in general, is a pediatric tumor that has three major clinical presentations. The endemic (African) form presents as a jaw or facial bone tumor that spreads to extranodal sites including ovary, testis, kidney, breast, and especially to the bone marrow and meninges. The nonendemic form has an abdominal presentation with massive disease, ascites, renal, testis, and ovarian involvement and, like the endemic form, also spreads to the bone marrow and CNS. Immunodeficiency-related cases more often involve lymph nodes and peripheral blood. BL has a male predominance and is typically seen in patients <35 years of age. These tumors have a high propensity to invade the bone marrow and CNS.
Adult T-cell leukemia/lymphoma
ATLL is a rare disease associated with infection by the HTLV-1 in 100% of cases.99–101 ATLL is endemic in southern Japan, the Caribbean basin, Africa, and the southeastern United States. The normal counterparts of the neoplastic cells are activated CD4+ T cells expressing CD2, CD3, CD5, and CD25. The median age of patients is 60.102 The disease can present as four variants: acute (most common and highly aggressive), lymphomatous, chronic, and smoldering. The median survival of these variants is 6, 10, 24 months, and “not reached,” respectively.103 Patients present with BM and peripheral blood involvement, high white blood cell count, hypercalcemia (owing to PTH-related protein, TGF-β, and RANK ligand), lytic bone lesions, lymphadenopathy, hepatosplenomegaly, skin lesions, and interstitial pulmonary infiltrates.
Differential diagnosis and sites of disease at presentation
More than two-thirds of patients with NHL present with persistent painless peripheral lymphadenopathy. At the time of presentation, differential diagnosis of generalized lymphadenopathy includes infectious etiologies. A firm lymph node larger than 1.5 × 1.5 cm not associated with a documented infection, persisting longer than 4–6 weeks, and progressing should be considered for biopsy. However, lymph nodes in indolent NHLs frequently wax and wane. In teenagers and young adults, infectious mononucleosis and HL should be placed high in the differential diagnosis. Involvement of Waldeyer’s ring, epitrochlear, and mesenteric nodes are more frequently observed in patients with NHL than HL. 40% of all patients with NHL present with systemic complaints. B symptoms are more common in patients with aggressive subtypes approaching 50%. Less frequent presenting symptoms, occurring in <20% of patients, include fatigue and malaise.
NHLs also present with thoracic, abdominal, and/or extranodal symptoms. Approximately 20% of patients with NHL present with mediastinal adenopathy. These patients most frequently complain of persistent cough and chest discomfort and rarely display superior vena cava syndrome. Differential diagnosis of mediastinal presentation includes infections, sarcoidosis, HL, and other neoplasms. Involvement of retroperitoneal, mesenteric, and pelvic nodes is common in most subtypes of NHL, and the majority of patients are asymptomatic. Aggressive NHLs can present with primary cutaneous lesions, testicular masses, acute spinal cord compression, solitary bone lesions, and rarely lymphomatous meningitis. Symptoms of primary NHL of the CNS include headache, lethargy, focal neurologic symptoms, seizures, and paralysis.
When NHL presents in an extranodal site, the differential diagnosis is more difficult. NHL uncommonly presents in the lung.104 Between 25% and 50% of patients with NHLs present with hepatic infiltration, although relatively few have large hepatic masses; these are almost always associated with aggressive lymphoma. Of the advanced stage indolent lymphomas, nearly 75% of patients have microscopic hepatic infiltration at the time of diagnosis. Primary lymphoma of bone occurs in <5% of patients, presenting as a painful bony site, most commonly involving the femur, pelvis, and vertebrae. Approximately 5% of NHLs present as primary gastrointestinal lymphoma. These patients may have hemorrhage, pain, or obstruction as the stomach is most frequently infiltrated followed by the small intestine and the colon, respectively. Most gastrointestinal lymphomas are of the aggressive subtypes, specifically DLBCL, MCL, and the intestinal T-cell lymphoma and may present rarely with bleeding, obstruction, or perforation. The most common site for extranodal MZL is the stomach. A subset of MCLs presents as multiple intestinal polyposis involving any sites in the gastrointestinal tract. Two to fourteen percent of NHL present with renal infiltration, and even less common has localized presentation in the prostate, the testis, or the ovary. The typical histologic subtypes of these sites are DLBCL and BLs. Rare sites of primary lymphoma include the orbit, the heart, the breast, the salivary glands, the thyroid, and the adrenal gland.
Staging and disease detection
The Lugano classification105 is the currently used staging system. It is derived from the Ann Arbor staging system, which was originally developed for HL in 1971. Table 3 summarizes the essential features of the Ann Arbor system. Because NHLs most frequently disseminate hematogenously, this staging system has proven to be much less useful than for HL. The Lugano modification requires documentation of size of bulk disease.
Table 3 Revised staging system for primary nodal lymphoma
| Stage | Involvement | Extranodal (E) status |
| Limited | ||
| I | One node or a group of adjacent nodes | Single extranodal lesions without nodal involvement |
| II | Two or more nodal groups on the same side of the diaphragm | Stage I or II by nodal extent with limited contiguous extranodal involvement |
| II bulky* | II as above with “bulky” disease | Not applicable |
| Advanced | ||
| III | Nodes on both sides of the diaphragm; nodes above the diaphragm with spleen involvement | Not applicable |
| IV | Additional noncontiguous extralymphatic involvement | Not applicable |
*Whether stage II bulky disease is treated as limited or advanced disease may be determined by histology and a number of prognostic factors
Systemic “B” symptoms (fever, sweats, and weight loss) are no longer included into the staging system for NHL, because these symptoms are not independent prognostic factors for these patients.
The concept of staging has less impact in NHL than in HL. Multiple studies demonstrate that prognosis is more dependent on lymphoma subtype and clinical parameters than stage at presentation. Staging is undertaken in NHLs to identify the small number of patients who can be treated with local therapy or combined modality treatment and to stratify within subtypes to determine prognosis and assess the impact of treatment.
Diagnosis and initial evaluation
Staging must be undertaken in the context of the histology. After the initial biopsy, blood tests should be obtained, including complete blood count, routine chemistries, renal function, liver function tests, and serum protein electrophoresis to document the presence of circulating monoclonal paraproteins. HIV, hepatitis B (which may reactivate with lymphoma therapy), and hepatitis C serologies should be performed. Serum beta-2 microglobulin can be useful as a surrogate marker of disease burden in indolent NHLs. Serum concentrations of LDH are an important independent predictor of survival. Isolated Waldeyer’s ring involvement is associated with intestinal involvement in 20% of cases and endoscopy should be considered. Chest, abdominal, and pelvic computed tomography scan is essential for accurate staging. Unilateral bone marrow biopsies should be performed in most subtypes of lymphoma, as the likelihood of lymphomatous involvement of the marrow is relatively high, especially in most indolent lymphomas. Recently, the role of BM biopsy in DLBCL is being questioned. In a recent analysis of patients with DLBCL undergoing initial staging with PET-CT scanning, the sensitivity and specificity of PET scanning for marrow involvement was 89% and over 99%, respectively. BM biopsy should still be performed in patients with a negative PET scan, but of limited utility in patients with a positive scan showing BM involvement.106 In patients with aggressive lymphomas with marrow involvement, paranasal sinus involvement, paraspinal masses, testicular, or if clinically indicated, examination of the cerebral spinal fluid (CSF) by lumbar puncture should be performed. Positron emission tomography, PET, using 18F-fluorodeoxyglucose is a highly sensitive and specific scanning modality for detecting NHL in both nodal and extranodal sites. PET scanning is very useful for DLBCL, MCL, and FL and is now recommended in the initial staging of these entities. PET scanning is not as sensitive in SLL and MZL.107
The International Harmonization Project has provided consensus recommendations for PET scanning. Among the recommendations are the following: PET should be used for DLBCL and HL; scanning during therapy should still be only part of clinical trials; and the scan after all therapy is completed can be done at least 3 but preferably 6–8 weeks after chemotherapy and 8–12 weeks after radiation or chemoradiotherapy. There is no evidence that long-term follow-up should include PET scanning.108 Magnetic resonance imaging is most valuable for evaluation of the brain and spinal cord.
Immunologic and molecular studies
Biologic studies including cell surface markers, cytogenetics, and molecular techniques are used in diagnosis, staging, and minimal disease detection. Monoclonal antibodies directed against cell surface antigens expressed on lymphoid cells and molecular techniques to define immunoglobulin and TCR gene rearrangements are sensitive tools with which to assess tumor cell infiltration. Immunophenotypic and cytogenetic studies can help to determine histologic subtypes of lymphomas. For those NHLs with known chromosomal translocations, it is possible to identify unique chromosomal breakpoints that can be studied with FISH, cytogenetics, and PCR. Studies of minimal disease may provide important prognostic information.
Disease parameters that influence prognosis and assessment of disease response
Prognostic factors in NHL
Aggressive NHLs
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


