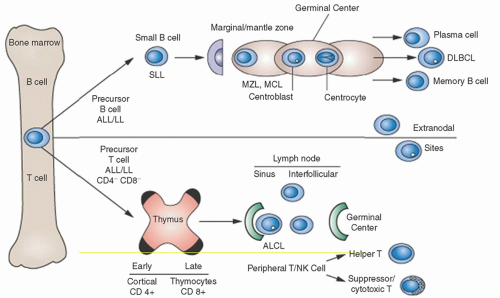
 FIGURE 88.1. Cellular origins of non-Hodgkin lymphoma by B and T cell differentiation pathways. B and T cells originate in the marrow, where they are antigen independent. The follicular center is a normal site of antigen-dependent B cells which go through different stages: (a) proliferation of centroblasts (large noncleaved cells), (b) selection of centrocytes (small cleaved cells) or cell death via apoptosis, (c) differentiation into postgerminal center memory B lymphocytes or plasma cells, often in association with increased antigen affinity and an immunoglobulin isotope switch. The T cells depend upon the thymus for early differentiation before becoming peripheral T cells. The most immature T cell precursors are negative for both CD4 and CD8. Thymocytes can become committed to either the gamma-delta or alpha-beta lineage. As thymocytes within the alpha-beta lineage mature they gain CD4 and then CD8 to become double positive thymocytes. Normal thymocytes downregulate the expression of CD4 or CD8 to become mature peripheral T cells, which are subdivided into helper (CD4+) and suppressor/cytotoxic (CD8+) subsets. Corresponding lymphomas are based on stage of cellular differentiation and site(s) of nodal and/or extranodal origin. BCL, precursor B cell lymphoblastic lymphoma; SLL, small lymphocytic lymphoma; MZL, marginal zone lymphoma (MALT types, postgerminal center); MCL, mantle cell lymphoma (pre-germinal center); FL, follicular lymphoma; DLBCL, diffuse large B cell lymphoma; TCL, precursor T cell lymphoblastic lymphoma; ALCL, anaplastic large cell lymphoma (sinus involvement). Peripheral T/NK lymphomas tend to involve the interfollicular zone of lymph nodes or specific extranodal sites. |
TABLE 88.1 EPIDEMIOLOGIC FACTORS THAT ARE ASSOCIATED WITH AN INCREASED RISK OF NON-HODGKIN LYMPHOMA | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
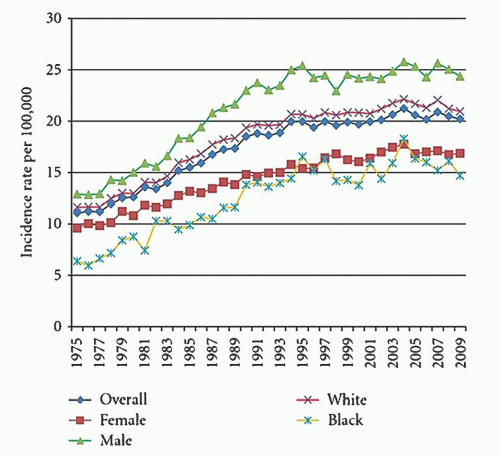 FIGURE 88.2. Temporal trends in the age-adjusted incidence rates of NHL in the United States (SEER data, 1979 to 2009). |
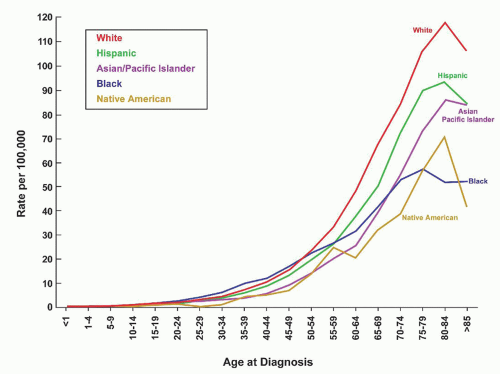 FIGURE 88.3. Age-specific incidence rates of NHL according to race (SEER data, 1979 to 2009). |
and geography.46 There are at least two mechanisms in which infections contribute to lymphomagenesis. The best described is direct lymphocyte transformation by microbial agents. Lymphotropic viruses include Epstein-Barr virus (EBV), human T-leukemia virus-1 (HTLV-1), and human herpes virus 8 (HHV8). An alternative mechanism is an agent, such as Helicobacter pylori (H. pylori), infecting host tissues and eliciting a lymphoproliferation response that remains dependent upon the presence of the agent, or antigen. The former does not usually respond to antimicrobial therapy, while the latter does. There are an expanding number of infections associated with specific types of NHL (Table 88.3).
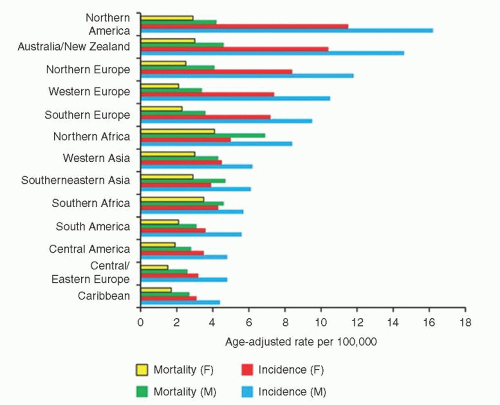 FIGURE 88.4. Age-adjusted incidence and mortality rates of NHL in selected world regions. From International Agency for Research on Cancer GLOBOCAN 2008 data, from Bassig BA, Lan Q, Rothman N, et al. Current understanding of life-style and environmental factors and risk of non-Hodgkin lymphoma: an epidemiological update. J Cancer Epidemiol 2012: 978930, with permission. |
TABLE 88.2 CLINICOPATHOLOGIC DIFFERENCES BETWEEN CHILDHOOD AND ADULT NON-HODGKIN LYMPHOMAS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 88.3 INFECTIONS AND ASSOCIATIONS WITH LYMPHOMA | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||
Yoshida et al. in Hinuma’s lab in Japan independently discovered an unique retrovirus, HTLV-1, as the etiologic agent of ATL.57, 58 HTLV-1 was shown to be endemic to certain geographic areas: southwestern Japan, in which 6% to 20% of the population is seropositive for HTLV-1; the Caribbean Islands, New Guinea, parts of Central Africa and South America, parts of West Africa, the Middle East and Melanesia (Fig. 88.6).59, 60
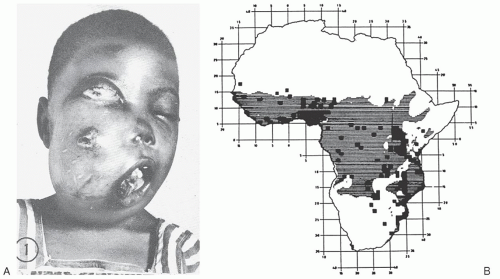 FIGURE 88.5. A: Burkitt lymphoma involving mandible, maxilla, and orbit. From O’Conor GT. Significant aspects of childhood lymphomas in Africa. Cancer Res 1963;23:1514-1527. B: Lymphoma belt of Africa: Burkitt lymphoma occurred only in areas below 3,000 feet (above sea level), with mean temperature above 15.6°C, and with annual rainfall over 50 cm. Shaded area, where Burkitt lymphoma would be expected to occur; black squares, sites of cases identified by Burkitt. From Haddow AJ. An improved map for study of Burkitt lymphoma syndrome in Africa. E Afr Med J 1963;40:429-432. |
patient with an unusual T cell variant of hairy cell leukemia.68 It has subsequently been isolated only rarely in lymphoid neoplasia and in HTLV-I negative tropical spastic paraparesis, but it is prevalent in intravenous drug abusers.69 Two other related viruses, HTLV-3 and HTLV-4, were reported in 2005 in Central Africa but have not been linked to human disease.70 Salahuddin et al. identified HHV-6 in B lymphocytes from 6 patients with various lymphoproliferative disorders;71 it was identified subsequently as the etiologic agent for exanthem subitum (roseola infantum) and as a cause for pneumonia in immunocompromised hosts.72 A role for HTLV-2 or HHV6 in lymphoproliferation has not been detected. KSHV, also referred to as HHV-8, has been identified in AIDS-related body-cavity based B cell lymphomas and multicentric Castleman disease and has been associated with EBV genome in the absence of MYC rearrangement.73, 74 Theoretically, KSHV acts synergistically with EBV to transform B cells and causes a unique clinical presentation.
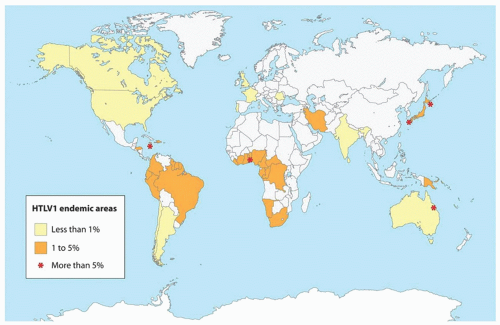 FIGURE 88.6. Geographic distribution of HTLV-1 in countries where the disease is endemic. The stars emphasize high-prevalence areas. The country boundaries shown in the map are not coincidental with the areas of endemicity, reflecting the cluster nature of HTLV infection. From Goncalves DU, Proietti FA, Ribas JG, et al. Epidemiology, treatment, and prevention of human T cell leukemia virus type 1-associated diseases. Clin Microbiol Rev 2010;23:577-589, with permission. |
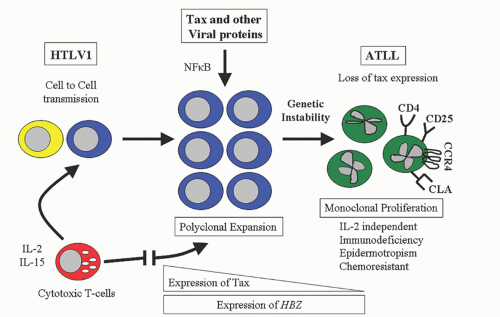 FIGURE 88.7. Course of HTLV-1 infection. After infection of a mature T helper cell, there is a long latency period (decades) which can be controlled by cytotoxic T cells and an autocrine IL-2 loop. Clonal proliferation is promoted by pleiotropic actions of Tax and other viral proteins that inhibit apoptosis and induce IκBα, which activates the NFκB pathway. HBZ promotes T cell proliferation and the HBZ protein suppresses TAX-mediated viral transcription. As TAX expression is lost, there is the emergence of a monoclonal T cell population that is independent of IL-2.CC chemokine receptor 4 (CCR4) and cutaneous lymphocyte antigen (CLA) on ATL cells interact with endothelial cells in the skin and contribute to epidermotropism. ATL follows a multistep process of worsening genetic instability and is subdivided into clinical syndromes characterized by immunodeficiency and chemoresistance. |
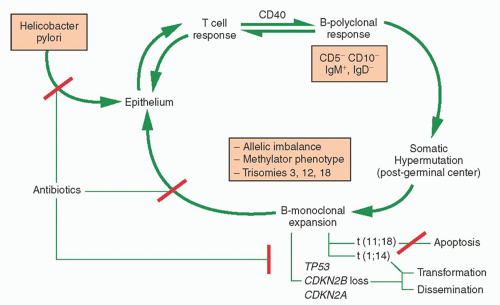 FIGURE 88.8. MALToma of the stomach: model for lymphomagenesis. Helicobacter pylori infects the epithelial cells and there is recruitment of both T and B cells. Contact-dependent T cell help is mediated by CD40 and CD40 ligand interaction. B cells undergo a T cell-dependent polyclonal response which can develop into a postgerminal center monoclonal B cell lymphoma. Three-fourths of gastric MALTomas have allelic imbalance, methylator phenotype, and/or trisomies and respond to antibiotics that eradicate Helicobacter pylori. Alternatively, there can be clonal evolution that involves a specific translocation (i.e., t[11;18] or t[11;14]) and/or a loss of tumor suppressor genes (i.e., TP53, CDKN2B/p15, CDKN2A/p16), which are associated with dissemination of disease. The t(11;18) rarely is associated with transformation while the t(11;14) may progress to a diffuse large B cell lymphoma. |
of BCL2, blocks apoptosis of B cells, and leads to antibiotic resistance, while translocation-negative cases involve inflammatory and immune responses, which maintain B and T cell interaction and respond to H. pylori eradication.105 The oncoprotein of the CAGA gene has been shown to be directly delivered into B cells by H. pylori, and it likely contributes to lymphomagenesis.106
with an increased risk of NHL.155, 156, 157, 158 The epidemiology of NHL continues under investigation and requires carefully designed studies with large cohorts and prolonged follow-up to determine the validity of an association between a factor and NHL. InterLymph, an international consortium of NHL studies, is providing large databases to assess the impact of environmental risk factors.159
TABLE 88.4 PRELYMPHOMATOUS CONDITIONS | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||
after solid organ transplantation and in patients with Crohn disease, the majority of whom were on anti-TNF therapy.119, 233 PTCL comprise only 3% of all AIDS lymphomas.234 Patients with gluten-sensitive enteropathy or celiac disease have an increased incidence of intestinal T cell lymphoma.235 Although Isaacson et al. initially reported that these lymphomas were variants of malignant histiocytosis, subsequent studies with gene rearrangement techniques indicated a T cell origin and they are referred to as enteropathy-associated T cell lymphoma (EATL).236 Primary anaplastic large cell lymphoma has been reported after breast implant surgery.237
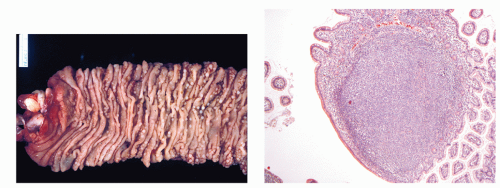 FIGURE 88.9. Mantle cell lymphoma. A: Intestine involved by multiple lymphomatous polyposis. Multiple small nodules extensively involve the bowel mucosa. Image provided by Dr. Lawrence Weiss, City of Hope National Medical Center. B: Aggregates of small B cells from multiple nodules just beneath the mucosa express CD5 and cyclin D1. Image provided by Dr. Dan Arber, Stanford University, Palo Alto, CA. |
as nodules involving head, neck, or trunk.256, 257 The major types of primary cutaneous B cell lymphomas and their distinguishing immunotypes include marginal zone (MALT-type) (CD20+, CD5–, CD10–), primary cutaneous follicular lymphoma (PCFL) (CD20+, CD10+), and primary cutaneous large B cell lymphoma (PCBCL) (CD20+; usually CD10–). Other T cell lymphomas with unique skin involvement include anaplastic large cell lymphoma (ALCL), which can be a primary cutaneous type or a systemic disease;258 subcutaneous panniculitis-like PTCL, αβ subtype; and 3 provisional entities, cutaneous γδ T cell lymphoma (TCL), aggressive epidermotropic CD8+ cytotoxic TCL, and primary cutaneous CD4+ small/medium-sized pleomorphic TCL.
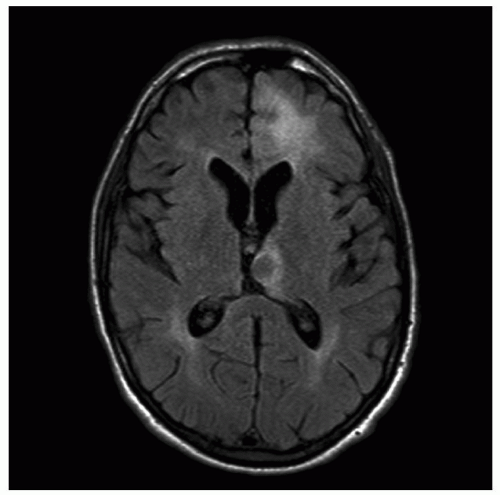 FIGURE 88.10. Primary CNS lymphoma. This FLAIR sequence MRI demonstrates a small periventricular mass lesion in the left thalamus. The lesion displays a signal intensity that is isointense compared with normal gray matter structures. This feature is consistent with a highly cellular lesion, a finding that suggests lymphoma but is not specific for that diagnosis. In addition, there are multiple areas of high signal intensity with ill-defined borders, most pronounced in the left frontal region. This appearance is also nonspecific, reflecting increased water content or decreased myelin in those regions, as may occur with any cause of inflammation. It is consistent with a multifocal, widely infiltrative process, and supportive of the diagnosis of lymphoma. Description provided by Dr. Paul Moots, Vanderbilt University, Nashville, TN. |
Modification in clinical prognostic indices have been made for different types of NHL and biologic parameters can further subdivide groups (see section on “Prognostic Factors”).
TABLE 88.5 STAGING OF NON-HODGKIN LYMPHOMA | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 88.6 STAGING STUDIES IN NON-HODGKIN LYMPHOMA | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 88.7 CLINICAL AND PATHOLOGIC FEATURES THAT AFFECT TREATMENT OUTCOME IN NON-HODGKIN LYMPHOMA | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
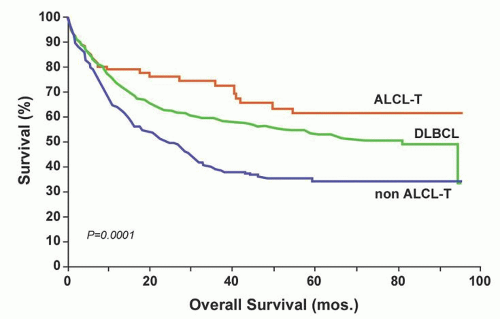 FIGURE 88.11. Overall survival of 228 PTCL (non-ALCL) and 60 T-ALCL compared with 1,595 DLBCL patients. From Gisselbrecht C, Gaulard P, LePage E, et al. Prognostic significance of T cell phenotype in aggressive non-Hodgkin’s lymphoma. Blood 1998;92:76-82, with permission. |
is more relevant to current patients, however, as it is based on initial therapy with rituximab-containing regimens (Fig. 88.13). The acronym “BABA6” can be used for the 5 FLIPI2 markers: Beta-2 microglobulin > normal, Anemia with hemoglobin <120 g/L, Bone marrow positive for lymphoma, Age > 60 years, and one or more nodal masses ≥6 cm in diameter. Five- and ten-year overall survival rates had a strong inverse correlation with low-, intermediate-, and high-risk FLIPI2 scores.305
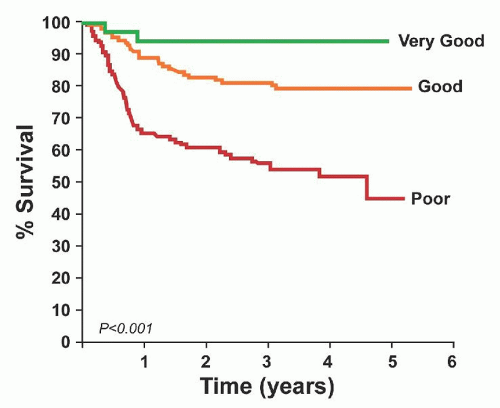 FIGURE 88.12. Progression-free survival according to the revised International Prognostic Index. From Sehn LH, Berry B, Chhanabhai M, et al. The revised International Prognostic Index (R-IPI) is a better predictor of outcome than the standard IPI for patients with diffuse large B cell lymphoma treated with R-CHOP. Blood 2007;109:1857-1861, with permission. |
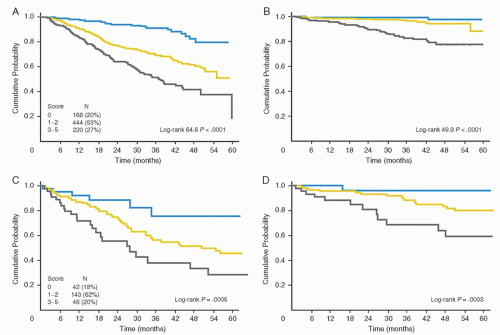 FIGURE 88.13. Progression-free survival are overall survival according to the Follicular Lymphoma International Index 2. A and B are the PFS and OS for the training sample (832 patients) and C and D are for the validation sample (231 patients). FLIPI2: low risk (blue line), score 0; intermediate risk (yellow line), score 1-2; high risk (grey line), score 3-5. From Federico M, Bellei M, Marcheselli L, et al. Follicular lymphoma international prognostic index 2: A new prognostic index for follicular lymphoma developed by the International Follicular Lymphoma Prognostic Factor Project. J Clin Oncol 2009;27:4555-4562, with permission. |
the t(2;5) and its variants in ALCL.310 BCL-6 expression has been associated with an improved outcome in DLBCL, but its absence may be less deleterious in the rituximab era.311
TABLE 88.8 PROGNOSTIC BIOMARKERS BY IMMUNOHISTOCHEMISTRY IN AGGRESSIVE LYMPHOMA | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
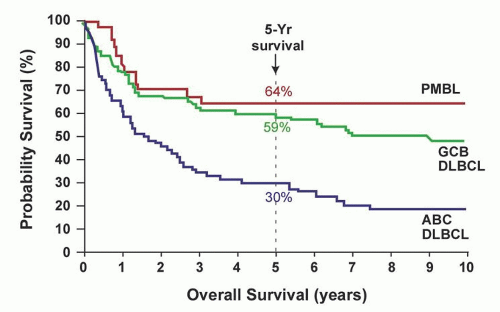 FIGURE 88.14. Gene expression arrays can stratify diffuse large B cell lymphoma (DLBCL) into at least three subtypes, primary mediastinal B cell lymphoma (PMBL), germinal center B cell (GCB), and activated B cell-like (ABC). This Kaplan-Meier curve indicates differing survival among the groups. From Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 2003;198:851-862, with permission. |
TABLE 88.9 2007 RESPONSE CRITERIA FOR NON-HODGKIN LYMPHOMA | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
adversely affects outcome in therapy is controversial, but comorbid illnesses and biologic differences of lymphomas can contribute to higher mortality in the elderly. Treatment-related toxicities are greater in elderly patients, but deaths from unrelated causes are also increased.342, 343 Biologic differences of NHL between young and old patients are implicated by a greater lymphomarelated mortality in some series of elderly patients compared with younger cohorts344 (see section on “Therapy in the Elderly”).
TABLE 88.10 CLINICAL SCHEMA FOR LYMPHOID NEOPLASMS | ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||
T/NK lymphomas/leukemias which can have an indolent phase include large granular T/NK leukemia, smoldering and chronic HTLV-1+ ATL, T-prolymphocytic leukemia, and, more controversially, some types of PTCL. FLs formerly included follicular small cleaved (FSCL), mixed (FML), and large cell (FLCL), and roughly correlated with cytologic grades 1, 2, and 3, respectively. Cytologic grading of FL is under scrutiny due to poor reproducibility among pathologists.
TABLE 88.11 THERAPEUTIC OPTIONS FOR INDOLENT LYMPHOMA | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
A paraprotein may be present and is usually IgM. Despite disseminated disease, splenectomy can alleviate symptoms and improve cytopenias.389, 390 Median survival has been recorded to be 10.5 years, but is shorter in the presence of a paraprotein, elevated β-2m, or lymphocytosis (>9 × 109/L).391, 392 A simple prognostic scoring system is based on 3 factors: hemoglobin <120 g/L, LDH level greater than normal, and albumin <35 g/L. The 5-year cause-specific survival was subdivided into 3 groups: 88% for no adverse factor, 73% (one factor), and 50% (two or more factors).388 Complete or partial trisomy 3 is the most frequent (˜85%) cytogenetic abnormality.393 The abnormality characteristic of SMZL is a deletion or translocation of chromosome 7q32, present in 40% of patients.384 Adverse cytogenetics include del(17p), del(8p), and del(7q) with unmutated genes.394, 395
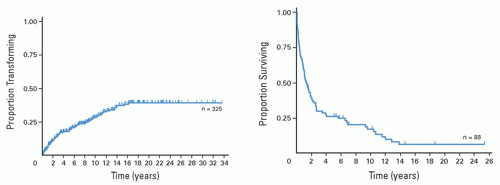 FIGURE 88.15. Histologic transformation of follicular lymphoma. A: Cumulative incidence of transformation. B: Survival (median = 1.2 years) in patients after transformation. From Montoto S, Davies AJ, Matthews J, et al. Risk and clinical implications of transformation of follicular lymphoma to diffuse large B cell lymphoma. J Clin Oncol 2007;25:2426-2433, with permission. |
in the rituximab era.416, 417 In a study that evaluated the role of reduced field of radiation (involved regional field or involved nodal radiation therapy), the 10-year PFS and OS were 49% and 66%, respectively. By reducing the field of radiation, there was no difference in the recurrence rate.418
Related posts:
 Clinical Flow Cytometry
Lymphocytes and Lymphatic Organs
Endothelium: Angiogenesis and the Regulation of Hemostasis
Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane
Thalassemias and Related Disorders: Quantitative Disorders of Hemoglobin Synthesis
Anemias Unique to the Fetus and Neonate
Clinical Flow Cytometry
Lymphocytes and Lymphatic Organs
Endothelium: Angiogenesis and the Regulation of Hemostasis
Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane
Thalassemias and Related Disorders: Quantitative Disorders of Hemoglobin Synthesis
Anemias Unique to the Fetus and Neonate
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


