Advances in tumor genetics and drug development over the last decade have led to the generation of a wealth of anti-cancer targeted therapies. These drugs aim at targeting a particular vulnerability in the tumor generated in most cases as a result of dependence on an oncogene and/or loss of a tumor suppressor. Several recent examples indicate that these drugs are mainly, if not exclusively, active against tumors of a particular genotype that can be identified by a diagnostic test, usually detecting a somatic alteration in tumor DNA. However, for the majority of targeted therapies in development, there are still no clinical tools to determine which patients are most likely to benefit or, alternatively, be resistant de novo to these novel agents or drug combinations. In this chapter, we will review some of the newer (molecule) targeted therapies, the molecular targets and/or pathways they engage, the rationale for their use in breast cancer, pharmacodynamics and predictive biomarkers indicative of drug target modulation and clinical response, respectively, and the latest completed and planned clinical trials with them.
PHOSPHATIDYLINOSITOL-3 KINASE (PI3K)/AKT INHIBITORS
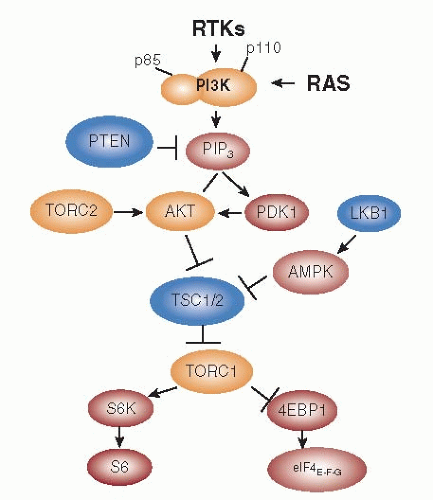
Multiple PI3K families exist in higher eukaryotes. To date only class I
A PI3K has been implicated in cancer. Class I
A PI3Ks are heterodimers consisting of a p85 regulatory subunit and a p110 catalytic subunit. Growth factor receptor tyrosine kinases (RTKs) such as EGFR, HER2, insulin receptor, IGF-IR, MET, etc. also signal via class I
A PI3K (
1). These transmembrane receptors do not bind PI3K directly but activate this enzyme by phosphorylating adaptor proteins such as GAB1/2, IRS-1/2, and HER3 (ErbB3), which via YXXM motifs bind the amino terminal domain of p85. This binding relieves the inhibition of p110 by p85 and recruits the p85-p110 heterodimer to its substrate, the lipid phosphatidylinositol-4,5-bisphosphate (PIP2), at the plasma membrane. PI3K (p110) then phosphorylates PIP2 to produce the second messenger phosphatidylinositol-4,5-trisphosphate (PIP3). A Ras-binding domain (RBD) in p110α also mediates activation by Ras. PTEN (phosphatase and tensin homologue) dephosphorylates PIP3, thus, negatively regulating the product of PI3K (
2). Several pleckstrin homology (PH) domain-containing proteins, including Akt, SGK, and PDK1, bind to PIP3 at the plasma membrane. The phosphorylation of Akt at Thr
308 by PDK1 and at Ser
473 by a complex involving mTOR/Rictor (TORC2) results in full activation of this enzyme. Akt phosphorylates a host of cellular proteins, including GSK3α, GSK3β, FoxO transcription factors, MDM2, BAD, and p27
KIP1 to facilitate survival and cell cycle entry (
3). In addition, Akt phosphorylates and inactivates Tuberin, a GTPase-activating protein (GAP) for the Ras homologue Rheb. Inactivation of Tuberin allows GTP bound-Rheb to accumulate and activate the mTOR/Raptor (TORC1) complex, which ultimately regulates protein synthesis and cell growth and inhibits autophagy (
4) (
Fig. 75-1).
The PI3K/AKT pathway is the most frequently mutated pathway in breast cancer, with mutation and/or amplification of the genes encoding the PI3K catalytic subunits p110α (
PIK3CA) and p110β (
PIK3CB), the PI3K regulatory subunit p85α (
PIK3R1), receptor tyrosine kinases (RTKs) such as HER2 (
ERBB2) and FGFR1, the PI3K activator K-Ras, the PI3K effectors AKT1, AKT2, and PDK1, and loss of the lipid phosphatases PTEN and INPP4B (
5). The three genes
PIK3CA, PIK3CB, and
PIK3CD encode the homologous p110α, p110β, and p110δ isozymes, respectively. Expression of p110δ is largely restricted to immune and hematopoietic cells, whereas p110α and p110β are ubiquitously expressed. The p110α isozyme is essential for signaling and growth of
tumors driven by
PIK3CA mutations, RTKs, and/or mutant Ras, whereas p110β lies downstream of GPCRs and has been shown to mediate tumorigenesis in PTEN-deficient cells.
PIK3CA mutations are the most common genetic alterations of this pathway, where ≥80% occur within the helical (E542K and E545K) and kinase (H1047R) domains of p110α. Such mutations confer increased catalytic activity through different mechanisms, but both induce characteristics of cellular transformation including growth factor- and anchorageindependent growth, and resistance to anoikis.
Drug Resistance and Rationale for Combinations
Activation of the PI3K/AKT pathway has been shown to confer resistance to antiestrogen in various experimental models, including PTEN- and INPP4B-deficient cells and in cells overexpressing HER2, IGF-IR, and mutant AKT1 (
6). Tumor cells with acquired endocrine resistance have been shown to upregulate IGF-IR, InsR, HER2, and EGFR levels as well as PI3K/AKT/mTOR activity (
7). Inhibitors of the PI3K pathway trump this adaptation of ER+ cells to estrogen +deprivation. A recent study showed that in ER+ breast cancer cells treated with the PI3K/TOR inhibitor BEZ235 or with p110 siRNAs, exogenous estradiol prevents BEZ235- and siRNAinduced apoptosis (
8). Because most breast cancers that adapt to antiestrogen therapy retain ER, these data imply that unopposed estrogen will protect ER+ tumors from the action of PI3K inhibitors used as single agents.
Clinical evidence suggests that activation of PI3K as a result of overexpression of HER2 or FGFR1 or loss of INPP4B also confer resistance to antiestrogens. Of interest, the activating mutations in PIK3CA correlate with good patient prognosis in some retrospective series. However, PI3K has been shown to interact with ER directly and indirectly, resulting in ER phosphorylation and an increase in estrogen- and tamoxifen-induced as well as ligand-independent ER transcription. In turn, estrogen can rapidly activate PI3K via Ins/IGF-IR, EGFR, and Src. Further supporting cross-talk between these two pathways, Guo et al. reported that a constitutively active mutant of AKT reduces ERα expression, whereas AKT inhibition results in an increase in ERα. Upon inhibition of PI3K/AKT, the transcription factor FoxO3a translocates to the nucleus where it transactivates ERα mRNA; expression of dominant negative FoxO3a reduces ERα levels in MCF-7 cells (
9,
10). This co-regulation supports the need of combined inhibition of both pathways in ER+ breast cancers with aberrant activation of PI3K/AKT. Indeed, preclinical evidence with ER+/PI3K mutant breast cancer cells suggests that simultaneous inhibition of PI3K and ER is synergistic (
8,
11), providing a rationale for combinations of antiestrogens with PI3K pathway inhibitors. In breast cancers with
HER2 gene amplification, HER2/HER3 dimers potently activate PI3K which, in turn, is critical for tumor cell progression and survival. Indeed, sustained inhibition of PI3K is required for the antitumor action of the HER2 inhibitors trastuzumab and lapatinib. A significant fraction of HER2+ tumors also harbor somatic alterations in the PI3K pathway that further dysregulate PI3K/AKT signaling and, as a result, confer resistance to HER2 inhibitors (
12). In a large-scale siRNA genetic screen, Berns et al. identified PTEN as the only gene whose knockdown conferred trastuzumab resistance (
13). Further, induced overexpression “hot spot”
PIK3CA mutants similarly conferred trastuzumab resistance. Patients with “hot spot”
PIK3CA mutations and undetectable or low PTEN measured by IHC exhibited a poorer outcome after treatment with chemotherapy and trastuzumab compared to patients without those alterations. An earlier study had also shown that loss of PTEN correlates with a lower response to trastuzumab. More recently, Esteva et al. found that PI3K pathway activation, defined as PTEN loss and/or
PIK3CA mutations, was associated with a poor response to trastuzumab as well as a poorer overall survival (
14). In addition, human breast cancer cell lines containing endogenous mutations in
PIK3CA are intrinsically resistant to trastuzumab. Human breast cancer cell lines in which
PIK3CA mutations are ectopically expressed exhibit an attenuated response to lapatinib. On the other hand, loss of PTEN has not been consistently associated with resistance to lapatinib.
A causal role of aberrant activation of the PI3K pathway with
de novo and acquired drug resistance is further supported by the effects of inhibitors of PI3K/AKT. For example, combinations of trastuzumab with the PI3K inhibitor XL147, or trastuzumab or lapatinib with the dual PI3K-mTOR inhibitor BEZ235, inhibit growth of
PIK3CA mutant xenografts resistant to anti-HER2 therapies (
15). In a model of trastuzumab resistance caused by PTEN loss, targeting mTOR or AKT was able to at least partially overcome resistance. Evidence is also emerging from the clinic that targeting the PI3K axis in addition to HER2 may be a strategy to overcome resistance. For example, in a phase II study, the combination of the TORC1 inhibitor everolimus with trastuzumab and chemotherapy showed a partial response rate of 19% and a clinical benefit rate of 81% in patients with HER2+ metastatic breast cancer that had previously shown progression on trastuzumab plus a taxane (
16).
Therapeutic Inhibitors
Several drugs targeting multiple levels of the PI3K network (i.e., PI3K, AKT, mTOR) have been developed (
Tables 75-1 and
75-2). A number of ATP-mimetics that bind competitively and reversibly to the ATP-binding pocket of p110 are in early clinical development. These include the pan-PI3K inhibitors BKM120, XL-147, PX-866, PKI-587, and GDC-0941, the p110α-specific inhibitors BYL719, GDC-0032, and INK-1117, the p110δ-specific inhibitor CAL-101, and the dual PI3K/mTOR inhibitors BEZ235, BGT226, PF-4691502, GDC-0980, and XL-765. The pan-PI3K and p110α-specific inhibitors are equally potent against oncogenic p110α mutants. The rationale for the development of isozyme-specific antagonists is to allow higher doses of anti-p110α and anti-p110β drugs to be delivered without incurring side effects caused by pan-PI3K inhibitors. Interim results from a phase I trial with the p110δ-specific inhibitor CAL-101 in patients with hematologic malignancies showed that treatment reduced P-AKT levels >90% in peripheral blood lymphocytes and induced objective clinical responses. Recently completed phase I trials with BKM120, BEZ235, and XL-147 showed that treatment partially inhibited PI3K as measured by levels of P-S6 and P-AKT in patients’ skin or tumors, and 2-deoxy-2-[
18F]fluoro-D-glucose (FDG) uptake measured by PET. Main toxicities were rash, hyperglycemia, diarrhea, fatigue, and mood alterations (
17). Few clinical responses were observed in patients with and without detectable PI3K pathway mutations, although screening for genetic lesions in this pathway was not comprehensive.
Both allosteric and ATP-competitive pan-inhibitors of the three isoforms of AKT are also being developed. AZD5363, GDC-0068, GSK2141795, and GSK690693 are ATPcompetitive compounds that have shown antitumor activity in preclinical models and recently entered phase I trials. Allosteric inhibitors such as MK-2206 bind to the AKT PH domain and/or hinge region to promote an inactive conformation of the AKT protein that is unable to bind to the plasma membrane. MK-2206 inhibits AKT signaling
in vivo, and suppresses growth of breast cancer xenografts harboring
PIK3CA mutations or
ERBB2 amplification (
18). Phase I data showed that treatment with MK-2206 decreases levels of P-AKT, P-PRAS40, and P-GSK3β in tumor cells, peripheral blood mononuclear cells, and hair follicles.
Another approach to block this pathway has been the development of ATP-competitive inhibitors of the mTOR kinase, which block both mTORC1 and mTORC2. Several dual TORC1/2 inhibitors have been identified, including INK128 (Intellikine),CC223 (Celgene), OSI-027 (OSI Pharmaceuticals), AZD8055 (AstraZeneca), AZD2014 (AstraZeneca), and Palomid 529 (Paloma Pharmaceuticals).
Dual PI3K/mTOR inhibitors have also been developed in the hope of overcoming the loss of feedback inhibition or PI3K activation observed with rapalogs. The mTORC1 pathway is one of the prominent negative feedback regulators of the PI3K pathway; inhibition of mTORC1 can release this feedback inhibition and activate the PI3K pathway (
19). BEZ235, a dual PI3K/mTOR inhibitor, showed higher antiproliferative activity than rapamycin in a preclinical study trastuzumab-resistant and -sensitive human breast cancer cell lines (
15). In breast cancer, dual PI3K/mTOR inhibitors are being combined with everolimus, endocrine therapies (exemestane and letrozole), chemotherapy (paclitaxel), and anti-HER2 therapy (trastuzumab).
Overall, the profile of PI3K inhibitors regarding adverse events has been acceptable, with no unexpected toxic effects. Toxic effects have been primarily mild to moderate and manageable with supportive medication. Dose-limiting toxic effects reported with multiple agents include hyperglycemia, maculopapular rash, gastrointestinal intolerance (anorexia, nausea, vomiting, dyspepsia, diarrhea), and stomatitis. Although some of these toxic effects are “off-target” effects, others may be related to target engagement and directly related to mechanisms of action.
Clinical Trials
Several early-stage clinical trials as both single agents and in combination have already been reported or are underway with AKT inhibitors (allosteric such as MK-2206, and catalytic inhibitors such as GDC-0068 and GSK690693) and all three categories of PI3K inhibitors: (i) pan-PI3K inhibitors that target all p110 isoforms (GDC-0941, XL147, BKM120; the irreversible pan-PI3K inhibitor PX-866) (ii) isoform-specific PI3K inhibitors that target a specific p110 isoform (such as the p110δ-selective inhibitor CAL-101, and the p110α-selective INK1117 and BYL719); and (iii) PI3K/mTOR dual inhibitors that inhibit all p110 isoforms as well as mTOR (SF1126, BEZ235, XL765, and GSK1059615).
PI3K Inhibitors and Endocrine Therapy
A phase Ib trial of letrozole and BKM120 (pan-PI3K inhibitors that target all p110 isoforms) for postmenopausal patients with ER+/HER2- metastatic breast cancer (MBC) evaluated safety and preliminary anti-tumor activity of two different schedules of BKM120 administration in combination with letrozole: continuously or intermittently (5 on/2 off days) (
20). Fifty-one patients were accrued in total; 40/51 (78%) had previously progressed on an aromatase inhibitor in the metastatic setting. The dose-limiting toxicity (DLT) in the continuous BKM120 schedule was transaminitis, and in the intermittent BKM120 schedule, mood disorders. Of note, the overall rate of grades 1-3 toxicities was higher in the continuous schedule arm. Six of 20 (30%) patients in the continuous BKM120 schedule arm and 10/31 (32%) of patients in the intermittent BKM120 schedule arm received therapy for >6 months. Interestingly, 45% of evaluable patients with a PI3K mutant cancer maintained stable disease for ≥6 months but duration of therapy. However, this clinical benefit has also been observed in a similar number of patients with PIK3CA wild-type tumors. A large phase II-III neoadjuvant trial with letrozole with or without BKM120 or BYL719 in patients with ER+ operable breast cancer is under way.
PI3K Inhibitors and HER2-targeted Therapy
Early clinical studies have also combined dual PI3K/mTOR inhibitors with trastuzumab. A phase I/Ib dose-escalation study of BEZ235, a dual PI3K/mTOR inhibitor, with trastuzumab aimed to enrich for patients with PI3K pathway alterations by limiting the study to patients with mutations in PIK3CA or PTEN or loss of PTEN by IHC in tumor samples. The maximum tolerated dose (MTD) for BEZ235 is estimated to be 600 mg per day in combination with trastuzumab, and this dose is being carried forward to the dose-expansion cohort. The combination also appears to be clinically active, with a clinical benefit rate (CBR) of 27%. In this study, there does not appear to be an association between PI3K pathway alteration and response (
21). Given the short half-life of BEZ235, a twice-daily schedule was investigated in a phase Ib/II study in combination with trastuzumab, but found to be too toxic.
The safety and efficacy of a novel PI3K inhibitor (XL147) administered in combination with trastuzumab or with paclitaxel and trastuzumab are being evaluated in patients with HER2-positive metastatic breast cancer that progressed during previous trastuzumab treatment (NCT01042925).
Combinations of MEK and PI3K pathways inhibitors are being explored in attempts to block escape pathways, which may become prominent when the PI3K pathway is inhibited. Several new generation PI3Kα-selective inhibitors are currently being evaluated in phase I clinical trials, including BYL719 (NCT01219699), INK-1114 (NCT01449370), and GDC-0032 (NCT01296555), as a single agent or in combination with endocrine therapies (letrozole and fulvestrant) or HER2-targeted agents (trastuzumab).
INHIBITORS OF MTOR
mTOR kinase is a serine-threonine kinase and member of the PI3K-AKT pathway that integrates signals from growth factors and nutrients to regulate key metabolic and macromolecular processes, such as mRNA translation, ribosome biogenesis, protein synthesis and cell motility. mTOR nucleates two protein complexes, TORC1 and TORC2, that mediate different functions in part through mRNA translational control (
4). TORC1 also contains the regulatory protein Raptor, which recruits substrates to TORC1 for phosphorylation, including the eIF4E binding/inhibiting protein (4E-BP, inhibited by phosphorylation) and ribosomal protein S6 kinases 1,2 (S6K1,2). S6K1,2 phosphorylate S6 and the initiation factor eIF4B, stimulating its RNA binding activity and cap-dependent translation. The second complex, TORC2 contains the rapamycin-insensitive component of mTOR (Rictor). It regulates cytoskeletal organization in response to growth factors and lead to phosphorylation of AKT (in Ser473) and its activation. AKT activates TORC1/Raptor via inactivation of TSC1 (
22).
Support for the combination of mTOR inhibitors with either tamoxifen or an aromatase inhibitor (AI) has been demonstrated in preclinical models of antiestrogen-sensitive and -resistant ER+ breast cancer (
23). MCF-7 cells expressing constitutively active Akt were able to proliferate under reduced estrogen conditions and were resistant to the growth inhibitory effects of tamoxifen, both
in vitro and
in vivo (
23). However, co-treatment with temsirolimus inhibited mTOR and restored sensitivity to tamoxifen, primarily through induction of apoptosis, thus suggesting that Aktinduced tamoxifen resistance may in part be mediated by signaling through the mTOR pathway. Further, everolimus has also been shown in
in vitro (
24) and
in vivo (
25) models to restore sensitivity of cancer cells to letrozole.
Rapamycin and its analogs form complexes with FK506-binding protein (FKBP12). This complex then binds to mTOR and inhibits the kinase activity of TORC1 but not TORC2. Formulation problems of rapamycin prompted the development of analogs such as CCI-779 (temsirolimus), RAD001 (everolimus), AP-23573 (deferolimus), and MK-8669 (ridaferolimus). These rapalogs have shown cytostatic activity in preclinical models and clinical trials, particularly in patients with renal cell cancer and in patients with mutations in the TSC complex (upstream of TORC1) who harbor renal angiolipomas. Compounds that target the ATP-binding cleft of mTOR (i.e., OSI-027, AZD8055, INK-128), and are thus active against both TORC1 and TORC2, are also in phase I trials. Of note, treatment with rapalogs results in compensatory stimulation of AKT-TORC1 through activation of TORC2 (
19). This has stimulated the interest on development of small molecule catalytic inhibitor of TORC1/2.
 Breast Cancer Screening
Breast Cancer Screening
 Ductal Carcinoma In Situ and Other Intraductal Lesions: Pathology, Immunohistochemistry, and Molecular Alterations
Ductal Carcinoma In Situ and Other Intraductal Lesions: Pathology, Immunohistochemistry, and Molecular Alterations
 Postmastectomy Radiation Therapy
Postmastectomy Radiation Therapy
 Preoperative Chemotherapy for Operable Breast Cancer
Preoperative Chemotherapy for Operable Breast Cancer
 Palliative Care in Breast Cancer
Palliative Care in Breast Cancer
 Nursing Care in Patient Management and Quality of Life
Nursing Care in Patient Management and Quality of Life



