INTRODUCTION
SUMMARY
Neutropenia designates a blood absolute neutrophil count that is less than 2 SD below the mean of a normal population. Neutropenia can be inherited or acquired. It usually results from decreased production of neutrophil precursor cells in the marrow. Neutropenia also can result from a shift of neutrophils from the circulating into the marginated cell pools in the circulation. Less commonly, neutropenia results from accelerated destruction of neutrophils or increased egress of neutrophil from the circulation into the tissues. When neutropenia is the sole or dominant abnormality, the condition is called “selective” or isolated” neutropenia, such as severe congenital neutropenia, chronic idiopathic neutropenia, or drug-induced neutropenia. Neutropenia can occur in other inherited or acquired marrow failure syndromes, such as severe aplastic anemia or Fanconi anemia, in which the condition is a bicytopenia or pancytopenia. In some diseases, several cell lineages are mildly affected but the reduction in neutrophil is the most severe, such as Felty syndrome. Neutropenia may be an indicator of an underlying systemic disease, such as early vitamin B12 or transcobalamin deficiency. Neutropenia, particularly severe neutropenia (neutrophil counts <0.5 × 109/L [500/μL]), increases susceptibility to bacterial or fungal infections and impairs the resolution of these infections. Therapy with the hormone primarily responsible for neutrophil production, granulocyte colony-stimulating factor, can increase blood neutrophil counts for most types of neutropenia, although whether its administration makes a clinically useful impact is dependent on the origin, duration and severity of the neutropenia. Clinical guidelines have been published on the rational use of the drug.
Neutrophilia is an increase in the absolute neutrophil count to a concentration greater than 2 SD above the normal population mean value. Neutrophilia contributes to the inflammatory response and to resolution of infections. Inflammatory and infectious diseases are the most frequent causes of neutrophilia. Bacterial infections usually produce neutrophilia, whereas viral infections may not produce neutrophilia or may raise the neutrophil count only slightly. Solid tumors occasionally engender striking neutrophilia. Hereditary neutrophilia can be caused by activating mutations within the CSF3R gene. When the neutrophil count is very high, it may be referred to as a leukemoid reaction. The rare neutrophilic variants of chronic myeloid leukemia and chronic neutrophilic leukemia may result in striking neutrophilia. Demargination of neutrophils or rapid release of neutrophils from a large marrow pool may transiently increase the blood neutrophil count. Sustained increased require increased production of these cells.
Acronyms and Abbreviations:
ANA, antinuclear antibody; BTH, Bruton tyrosine kinase; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; Ig, immunoglobulin; IL, interleukin; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand.
NEUTROPENIA
Neutropenia refers to an absolute blood neutrophil count (total leukocyte count per microliter × percent of neutrophils) that is less than 2 SD below the normal mean of the population. The terms leukopenia, a reduced total white blood cell count, and granulocytopenia, reduced numbers of blood granulocytes (neutrophils, eosinophils, and basophils), sometimes are imprecisely used as synonyms for neutropenia. Agranulocytosis literally means a complete absence of blood granulocytes, but this term often is used to indicate severe neutropenia, that is, counts less than 0.5 × 109/L (0.5 × 103/μL).
The concentration of neutrophils in blood is influenced by age, activity, and genetic and environmental factors (Chap. 2). For children from 1 month to 10 years old, neutropenia is defined as a blood neutrophil count less than 1.5 × 109/L. For individuals older than age 10 years, neutropenia is defined as a count less than approximately 1.8 × 109/L (see Chap. 7 regarding levels in newborns). Healthy older persons have the same blood neutrophil counts as younger individuals (Chap. 9). Some racial and ethnic groups, such as Africans, African Americans, and Yemenite Jews, have lower mean neutrophil counts than persons of Asian or European ancestry (see Chap. 2, Table 2–2). The mean differences in neutrophils are modest and have no recognized health consequences.1,2
Severe neutropenia is a predisposing factor for infections. The organisms normally are found on the skin, in the nasopharynx, and as part of the intestinal flora. The risk of infections is inversely related to the severity of the neutropenia (Chap. 24). Individuals with neutrophil counts of 1.0 to 1.8 × 109/L are at little risk of infection. In general, neutrophil counts between 0.5 and 1.0 × 109/L are associated with only slight risk of infection unless other contributing factors are present. Individuals with neutrophil counts less than 0.5 × 109/L are at substantially greater risk, but the frequency of infections varies considerably, depending on the cause and duration of neutropenia. Severe acute neutropenia (i.e., developing over a few hours or days) usually is associated with greater risk of infection than severe chronic neutropenia (usually present for months or years). Neutropenia resulting from disorders of production that affect early hematopoietic precursor cells (e.g., aplastic anemia, severe congenital neutropenia) leads to greater susceptibility to infections than do conditions with adequate neutrophil precursors in the marrow and neutropenia attributed to accelerated turnover in the blood (e.g., rheumatoid arthritis, Felty syndrome, autoimmune neutropenia). For patients made severely neutropenic by cancer chemotherapy, the risk is greater when the neutrophils are decreasing than with similar counts when neutrophils are increasing. Neutropenia accompanied by monocytopenia, lymphocytopenia, or hypogammaglobulinemia is more serious than isolated neutropenia. Other factors, such as the integrity of the skin and mucous membranes, the vascular supply to tissues, and the nutritional status of the patient, also influence the risk of infections.
Neutropenia occurs because of (1) hypoplastic neutropoiesis, (2) ineffective neutropoiesis (resulting from exaggerated apoptosis of late precursors), (3) accelerated removal or utilization of circulating neutrophils, (4) shifts of cells from the circulating to the marginal blood pools, or (5) a combination of these mechanisms (Fig. 65–1). Some production disorders are caused by intrinsic abnormalities of hematopoietic progenitor cells (Chap. 83). Other disorders in cell production are caused by extrinsic factors, including changes in the marrow environment, such as tumor infiltration, fibrosis, or irradiation (Chap. 45). Myelotoxic chemotherapeutic drugs commonly cause neutropenia because of the high proliferative activity of neutrophil precursors in the marrow and short half-life (4 to 8 hours) of neutrophils in the blood. Production of neutrophils is defined as ineffective when, under a steady state of hematopoiesis, a relative abundance of early neutrophil precursors, a paucity of late-maturing cells, and neutropenia occur. This condition has often been referred to as “maturation arrest,” but it is almost always explained by either the apoptotic loss of late precursors in the marrow as an intrinsic defect in cell maturation or rapid release of segmented neutrophils because of exaggerated tissue demands.
Figure 65–1.
Mechanisms of neutropenia are shown schematically. The size of each pool is represented by the size of the cross-hatched areas. The rate of flow of cells through each compartment is represented by the size of the arrows. CGP, circulating granulocyte (neutrophil) pool; MGP, marginated granulocyte (neutrophil) pool; Mi, mitotic; MSP, maturation (marrow storage) pool.
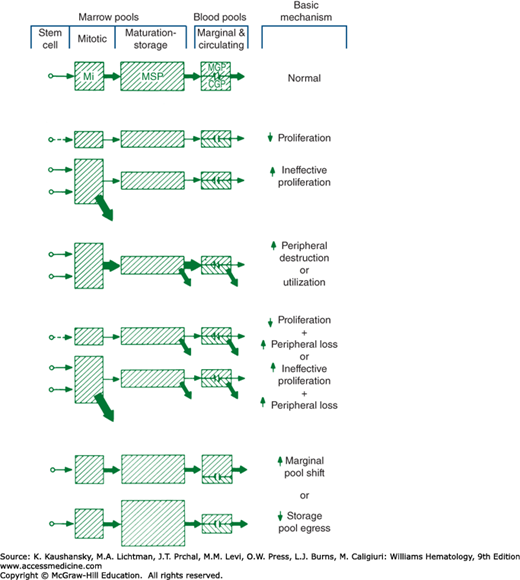
Accelerated neutrophil utilization occurs with autoimmune neutropenia and acute bacterial infections. When rapid neutrophil utilization and impaired production occur, acute severe neutropenia often develops. The condition is illustrated by the abrupt and sustained fall in neutrophils when an alcoholic patient develops pneumococcal pneumonia. Alcohol suppresses the marrow, and the infection consumes the available neutrophil supply. After myelotoxic cancer chemotherapy, the abrupt fall in blood neutrophils at the onset of infections reflects a similar mechanism: high demand and limited supply. With idiosyncratic drug-induced neutropenia, the counts may fall abruptly because both blood and marrow cells are simultaneously damaged. Acute neutropenia that develops because of a shift of blood neutrophils from the circulating to the marginal blood pools, that is, increased margination (e.g., after injection of endotoxin, with exposure of blood to dialysis membranes, or after intravenous granulocyte colony-stimulating factor [G-CSF] or granulocyte-macrophage colony-stimulating factor [GM-CSF]) usually is a transient event. The marginated cells reenter the circulating pool, and the blood supply of neutrophils is rapidly restored from the large reserves of marrow neutrophils entering the blood.
Our understanding of the mechanisms of neutropenia at the cellular and molecular levels is increasing rapidly because of advances in molecular genetics and cell biology. For many inherited forms of neutropenia, the genetic mutations causing these diseases are now known, and the mutant protein products have been identified. Some mutations and acquired defects shorten the survival of the precursor cells, that is, they accelerate apoptosis. This form of cell loss now is thought to be the mechanism for “maturation arrest” in several diseases. Examples of increased apoptosis causing neutropenia include vitamin B12 or transcobalamin deficiency,3 clonal cytopenias (myelodysplasia),4 myelokathexis,5 congenital and cyclic neutropenia,6,7 and the Shwachman-Diamond syndrome.8 Neutrophils also can be depleted from the blood and the marrow as a result of extrinsic factors such as antineutrophil antibodies and toxic cytokines generated by other cells.9,10 Some disorders that cause neutropenia also perturb neutrophil function, such as glycogen storage disease type 1b,11 Chédiak-Higashi syndrome,12 and HIV infection.13 Susceptibility to infection in these conditions relates to the combination of defects.
Causes of neutropenia are classified physiologically as disorders of production, distribution, or turnover. Not every condition fits neatly into this scheme, but it provides a framework for understanding these diverse disorders.
Cytotoxic drugs given for cancer chemotherapy and as immunosuppressive agents regularly cause neutropenia by decreasing cell production (Chap. 22). These drugs now are probably the most frequent cause of neutropenia in the United States. Neutropenia as a result of impaired production is a common feature of several diseases affecting hematopoietic stem cells, such as acute leukemia (Chaps. 88 and 91), the myelodysplastic syndromes (Chap. 87), and aplastic anemia (Chap. 35). The selective causes of impaired production, progressing from disorders of early precursors to disorders presumed to involve defective maturation (ineffective production), are described briefly as follows.
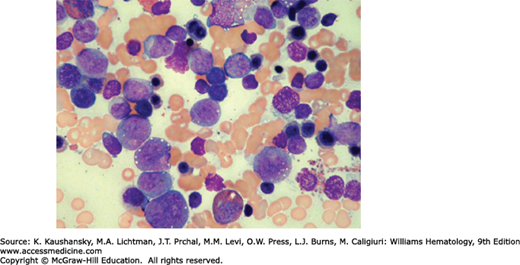
In 1956, Kostmann described congenital neutropenia (agranulocytosis) as an autosomal recessive disease occurring in an extended family in northern Sweden.14 Phenotypically similar sporadic cases and families with autosomal dominant congenital neutropenia have been reported.15,16 In severe congenital neutropenia, symptoms and signs of otitis, gingivitis, pneumonia, enteritis, peritonitis, and bacteremia usually begin in the first months of life. At diagnosis, the neutrophil count usually is less than 0.2 × 109/L.17 Monocytosis, mild anemia, thrombocytosis, and splenomegaly frequently are present. Characteristically, the marrow shows early neutrophil precursors (myeloblasts, promyelocytes) but few or no myelocytes or mature neutrophils (Fig. 65–2). Marrow eosinophilia is common. In vitro marrow culture studies show poor growth in response to various growth factors and with reduced numbers of marrow neutrophil and monocyte progenitor cell colonies.18 Usually blood lymphocyte numbers are normal, immunoglobulin levels are normal or increased, and lymphocyte functions are intact.
The majority of patients with sporadic or autosomal dominant severe congenital neutropenia have heterozygous mutations of the gene for neutrophil elastase (also called ELANE). Its product is a protease found normally in the neutrophil’s primary granules and lead to the induction of the unfolded protein response in the endoplasmatic reticulum.15,19,20 A variety of mutations in exons 2 through 5 as well as in introns III and IV are the cause of this disease.20,21,22,23 In the original Kostmann family, and some other families with autosomal recessive disease, neutropenia is caused by mutations in the HAX-1 gene.24 HAX-1 is a mitochondrial protein, and the mutations lead to accelerated apoptosis of myeloid cells, as well as neurologic abnormalities. In addition, mutations in HAX-1 lead to defective G-CSF receptor signaling via HCLS1 and LEF-1.25 Mutations in the gene for glucose-6-phosphatase catalytic subunit 3 (G6PC3) also cause severe neutropenia as a result of apoptosis of neutrophil precursors, as well as congenital cardiac and urogenital abnormalities.26 Additional autosomal-dominant, autosomal-recessive, X-linked, and sporadic forms have been described, with mutations in other genes, including GFI1,27 WAS,28 p14,29 TAZ,30 JAGN1,31 TCIRG1,32 and many others, although the genetic causes in many patients with severe congenital neutropenia remain unidentified.
Mutations in the gene for the receptor for G-CSF also occur in patients with severe congenital neutropenia33,34; however, most of these receptor mutations have caused truncations of the distal portion of the cytoplasmic domain of the receptor, an abnormality associated with altered sensitivity to G-CSF. G-CSF receptor mutations are part of the evolution to myelodysplasia or acute myelogenous leukemia and are not the primary cause of this neutropenia. An exception may be a patient identified with a mutation in the external domain of the G-CSF receptor who responded to treatment with G-CSF and glucocorticoids and has not developed leukemia over several years of observation.35 There are rare cases of biallelic mutations within the extracellular domain of the G-CSF receptor which lead to nonresponse to treatment with G-CSF.36
G-CSF is a very effective therapy for all of the recognized subtypes of severe congenital neutropenia, increasing the neutrophil counts and reducing recurrent fevers and infections.37 G-CSF acts to increase the neutrophil counts by enhancing expression of a critical transcription factor for granulopoiesis, C/EBPβ (CCAAT/enhancer binding protein β), and the “emergency” pathway of myelopoiesis (as during steady state C/EBPα is not functional).38 Approximately 5 percent of patients do not respond to G-CSF. Hematopoietic transplantation is the only other therapy known to improve the clinical course for these patients.39,40 Untreated patients and patients treated with G-CSF are at risk for developing acute myelogenous leukemia. The risk increases with time on treatment with G-CSF, particularly in poorly responsive patients.41 A novel molecular pathway of leukemogenesis was recently identified: mutations in the hematopoietic cytokine receptor (G-CSFR) in combination with the second mutations in the downstream hematopoietic transcription factor (RUNX1), which could be used as a marker for identifying severe congenital neutropenia patients with a high risk of progressing to leukemia or myelodysplastic syndrome.42
Neutropenia is a feature of the congenital immunodeficiency diseases and a contributing factor to their susceptibility to infections (Chap. 80). In most of these conditions, neutropenia is attributed to a production disorder based largely on histologic examination of the marrow. In X-linked agammaglobulinemia, which is attributed to defective B-cell development and a mutation in a cytoplasmic (Bruton) tyrosine kinase (BTK), severe neutropenia is present in approximately 25 percent of patients.43 Children with common variable immunodeficiency often have neutropenia associated with thrombocytopenia and hemolytic anemia.43 Neutropenia occurs in almost half of patients with the X-linked hyperimmunoglobulin-M syndrome, a disorder caused by a mutation in the gene encoding the CD40 ligand.44 In severe combined immunodeficiency, neutropenia is not always present. The neutropenia varies over time in individual patients. Neutropenia is particularly prominent in the rare immunodeficiency state, reticular dysgenesis.43 Neutropenia is a less-common feature of adenosine deaminase deficiency, the T−B+, T−B−, Wiskott-Aldrich, and Omenn syndromes.43,45,46 Neutropenia also occurs on an autoimmune basis in some cases of the Wiskott-Aldrich syndrome.34,46 Mutations in the genes for growth factor independent protein-1 (GFI 1) can also cause neutropenia.47
G-CSF therapy is effective in most patients with neutropenia associated with these immunodeficiency syndromes.
This rare autosomal recessive disorder is characterized by short-limbed dwarfism, hyperextensible digits, very fine hair, neutropenia, lymphopenia, and recurrent infections.43 The genetic locus is at 9p13 and affects a gene coding for an endoribonuclease. The degree of neutropenia is variable, with blood counts ranging from 0.1 to 2.0 × 109/L. An accompanying defect in T-cell proliferation results from an abnormality in the transition from the G0 to the G1 phase of the mitotic cycle. Patients have frequent bacterial and viral respiratory infections. Hematopoietic stem cell transplantation can correct the neutropenia and immune deficiency.48,49
This autosomal recessive disorder combines short stature, pancreatic exocrine deficiency, and marrow failure with neutropenia beginning early in the neonatal period.50,51,52,53 Thrombocytopenia and anemia may be severe (Chap. 35). The chromosomal locus of the mutation is at 7qll, and the mutation affects the SBDS gene.50 The mutation causes a proliferative defect and increased apoptosis of early myeloid progenitor cells.51 A chemotactic defect also occurs in mature neutrophils.52 The patients are malnourished, but the neutropenia is not corrected by improving the patients’ nutritional status. Treatment with G-CSF raises blood neutrophil levels, and hematopoietic stem cell transplantation corrects the hematologic abnormalities.53 Without transplantation, the risk of evolution to myelodysplastic syndrome and acute myelogenous leukemia is 20 percent or greater.53
Neutropenia is a rare complication of hereditary hypoplastic anemia.54 Other features include congenital anomalies of the head and upper limbs. Two genetic loci have been identified: 19q13.2 and 8p23.55,56 The varying severity of neutropenia may reflect genetic heterogeneity among patients with this diagnosis (Chap. 36).
This rare autosomal recessive disorder is characterized by pigmentary dilution and variable degrees of cellular immunodeficiency. The syndrome consists of three types. Neutropenia is a feature of type 2 but not types 1 or 3. In type 2, the neutropenia is relatively mild and associated with pancytopenia. These hematologic abnormalities are attributable to a mutation located at 15q21 affecting the RAB27a gene.57 The gene product is a guanosine triphosphatase (GTPase). The mutation also causes abnormal release of granule proteins and hematophagocytosis.58 As in the Chédiak-Higashi syndrome (Chap. 66), type 2 patients may develop an acute phase of uncontrolled lymphocyte and macrophage activation leading rapidly to death.59 Hematopoietic stem cell transplantation can correct the hematologic features. Evolution to myelodysplasia has been reported.60
This rare autosomal recessive disorder is characterized by partial oculocutaneous albinism, giant granules in many cells (including granulocytes, monocytes, and lymphocytes), neutropenia, and recurrent infections (Chap. 66). This syndrome now is attributable to a chromosomal mutation at 1q43 affecting the LYST gene.61 The product of this gene regulates lysosomal trafficking. In Chédiak-Higashi syndrome, the neutropenia usually is mild, and susceptibility to infection is attributed to neutropenia and defective microbicidal activity of the phagocytes.62
Myelokathexis is a rare autosomal dominant or sporadically occurring disorder in which patients have severe neutropenia and lymphocytopenia, with total white cell counts often less than 1.0 × 109/L.63 WHIM syndrome, characterized by warts, hypogammaglobulinemia, infections, and myelokathexis, now is attributable to a mutation in the gene encoding the receptor for the CXC chemokine CXCL12 (previously termed stromal cell-derived factor-1), termed CXCR-4.64,65 The ligand–receptor pair CXCL-12/CXCR-4 is important for regulating the trafficking of all type of blood and marrow cells, including hematopoietic stem cells, from the marrow to the blood and tissues. In these syndromes, the marrow usually shows abundant precursors and developing neutrophils. Neutrophils in the marrow and the blood show hypersegmentation with pyknotic nuclei and cytoplasmic vacuoles. These morphologic changes and some molecular studies suggest cell loss in the marrow and blood caused by accelerated apoptosis. Favorable responses to G-CSF and GM-CSF occur, as does evolution to the myelodysplastic syndrome. A myelokathexis-like variant of myelodysplastic syndrome has been reported.66
Cohen syndrome is another rare cause of neutropenia. Mental retardation, postnatal microcephaly, facial dysmorphism, pigmentary retinopathy, myopia, and intermittent neutropenia are characteristic features. Patients with Cohen syndrome of diverse origins have mutations in the COH1 gene.67 Current studies suggest that COH1 plays a role in vesicle-mediated sorting and transport of proteins within many types of cells.
These autosomal recessive disorders are characterized by hypoglycemia, hepatosplenomegaly, seizures, and failure to thrive in infants. Only type 1b is associated with neutropenia.68 The genetic defect in type 1b maps to chromosome 11q23 and is attributed to a defect in an intracellular transport protein for glucose.69 The marrow appears normal despite severely reduced blood neutrophils. The neutrophils have a reduced oxidative burst when stimulated and defective chemotaxis.70,71 Treatment with G-CSF is effective for correcting the neutropenia and improving the associated inflammatory bowel disease, but has been associated with evolution to acute myelogenous leukemia.72
Cyclic neutropenia is an autosomal dominant or sporadically occurring disease characterized by regularly recurring episodes of severe neutropenia, usually every 21 days.73 Regular oscillations of other white cells, reticulocytes, and platelets are sometimes observed. Cyclic neutropenia now is attributable to mutations in the gene for neutrophil elastase (ELANE) at locus 19q3. Most mutations in the ELANE gene are in the regions of exons 4 and 5, but there are also mutations in exons 2 and 3, as well as in the introns II and IV.74,23 The diagnosis usually is made in the first year of life, especially in the presence of a family history of the condition.75 The neutropenic periods last for 3 to 6 days and often are accompanied by fever, malaise, anorexia, mouth ulcers, and cervical lymphadenopathy. A few cases of acquired cyclic neutropenia in adults, some of whom have an associated clonal proliferation of large granular lymphocytes (Chap. 94), have been reported.76
The diagnosis of cyclic neutropenia can be made only by serial differential white cell counts, at least two or three times per week for a minimum of 6 weeks. Sequencing of the gene may be helpful in confirming the diagnosis.77 Most affected children survive to adulthood, with symptoms often milder after puberty. Fatal clostridial bacteremia has been reported in several cases, and careful observation is warranted with each neutropenic period in untreated patients. Treatment with G-CSF is very effective.78 G-CSF does not abolish cycling, but it shortens the neutropenic periods sufficiently to prevent symptoms and infections. In contrast to severe congenital neutropenias, cyclic neutropenia patients have no risk to develop leukemias.
Neutropenia caused by genetic defects of folate, cobalamin, and transcobalamin IIA varieties of congenital disorders lead to disturbed function of methylmalonyl coenzyme A mutase and methionine synthetase, the two cobalamin-requiring enzymes. Each of these disorders causes neutropenia, anemia, and thrombocytopenia as a result of ineffective hematopoiesis (Chap. 41).79,80,81
Several disorders, currently with only descriptive names, may be genetically determined forms of neutropenia. These cases often are called familial (benign) neutropenia and probably are autosomal dominant disorders.82,83,84 Some cases of chronic benign neutropenia of childhood (usually a negative family history) may represent new mutations, and patients with chronic idiopathic neutropenia of adulthood may be childhood cases escaping early detection. Until better information is available, these conditions probably are best referred to as “idiopathic neutropenias.”
Hypertensive women often have low-birth-weight infants with low neutrophil counts, attributed to decreased production.84 The neutropenia often is severe with a high risk of infection, particularly during the first few weeks of life. The neutropenia usually resolves within a few weeks. G-CSF elevates the neutrophil count in this form of neonatal neutropenia, but the clinical benefit of treatment remains to be determined.85
Neutropenia is an early and consistent feature of megaloblastic anemias resulting from vitamin B12 or folate deficiency. When present it usually is accompanied by macrocytic anemia and mild thrombocytopenia (Chap. 41). Copper deficiency can cause neutropenia in patients on total parenteral nutrition, with a history of gastrectomy, and in malnourished children86,87,88 and the bicytopenia or tricytopenias with a marrow showing dysplastic precursors can masquerade as myelodysplastic syndrome.
Pure white cell aplasia is a rare acquired disorder causing severe selective neutropenia. The marrow is devoid or nearly devoid of neutrophils and their precursors.89 Ibuprofen, chlorpropamide, excessive zinc, and various infectious and inflammatory diseases are considered possible causes of this syndrome. Differential diagnosis includes aplastic anemia, myelodysplasia, hairy cell leukemia, and neutropenia associated with the large granular lymphocyte syndrome. Immunosuppressive therapy with antithymocyte globulin, glucocorticoids, and cyclosporine has been used in individual cases.
This is a distinct syndrome predominantly affecting young adult women ages 18 to 35 years; the female-to-male ratio is approximately 8:1.90 The medical history (lack of episodes of fever, gingivitis, mouth sores, or other infections) and previous blood counts suggest the condition is acquired in most cases. Erythrocyte, reticulocyte, and platelet counts usually are normal. Mild leukopenia and lymphocytopenia may be present, and the spleen is normal or only minimally enlarged. The patients have no chromosomal abnormalities or other evidence of myelodysplasia.91,92 Marrow examinations show a spectrum of abnormalities, ranging from normal cellularity to selective hypoplasia of the neutrophilic series. In most cases, quantitative marrow studies show the ratio of immature to mature cells is increased, suggesting loss of cells during the maturation process, that is, ineffective granulocytopoiesis.93 Antineutrophil antibodies, autoantibodies, including antinuclear or antimitochondrial antibodies, are absent.94 Chronic idiopathic neutropenia in adults is the result of accelerated apoptosis of neutrophils and their precursors mediated via the Fas ligand or interferon-γ.95 The disease mechanism, that is, activation of the extracellular apoptotic pathway, is similar to the mechanism described for patients with systemic lupus erythematosus.96
For most patients, the clinical course can be predicted from the level of blood neutrophils, marrow examination, and prior history of fevers and infections. In general, patients with the lowest levels of blood and marrow neutrophils have the most frequent problems. Long-term observations show, however, that some patients have very low blood neutrophil levels for long periods with few or no infections. Evolution to acute leukemia or aplastic anemia generally does not occur. G-CSF increases neutrophils in most patients and is a useful therapy for patients with recurrent fever and infections.37,97
Immune disorders primarily alter the distribution of neutrophils in the blood and accelerate neutrophil turnover. Antineutrophil antibodies cause transfusion reactions, alloimmune neonatal neutropenia, and autoimmune neutropenia. Antigen–antibody complexes, autoantibodies, and cytokine-mediated cellular injury are possible contributors to neutropenia of systemic lupus erythematosus and Felty syndrome. The association of neutropenia with increased numbers of circulating large granular lymphocytes demonstrates that cellular and humoral immune mechanisms can cause neutropenia (Chap. 94).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


