INTRODUCTION
Neuroendocrine tumors (NETs) originate from enterochromaffin cells distributed throughout the body. This chapter focuses on low- to intermediate-grade gastroenteropancreatic NETs (GEP-NETs), although the term neuroendocrine tumor also denotes diseases such as small cell carcinoma, medullary thyroid carcinoma, neuroblastoma, and Merkel cell tumor. Pancreatic NETs (PNETs), previously known as islet cell carcinomas, arise from pancreatic ductal progenitors. Extrapancreatic low- to intermediate-grade NETs are generally called carcinoids and most often originate along the aerodigestive tract. These tumors share the capacity for hormone production and usually have an indolent clinical course. Presenting symptoms are caused by secreted hormones, local tumor growth, and/or metastasis. Surgical resection is the curative approach for localized disease. In unresectable or metastatic disease, long-acting somatostatin analogues improve quality of life and progression-free survival. In PNETs, recent randomized studies support the use of targeted therapies such as everolimus and sunitinib, with older prospective and retrospective data supporting the use of alkylating chemotherapy such as streptozocin and temozolomide. This chapter presents a comprehensive overview of the diagnosis and management of pancreatic and extrapancreatic NETs.
EPIDEMIOLOGY
The overall incidence of NETs in the United States is rising and presently estimated at 5.25 cases per 100,000 (1). Most NETs progress slowly and may remain undiagnosed for years or even a person’s entire natural life. Small bowel NETs are found in 0.65% to 1.2% of patients during unselected necropsy (2,3). These tumors are usually diagnosed in the sixth and seventh decades of life (1,4). The gastrointestinal tract is the most common primary site of NETs, accounting for 58% of NETs (1). The distribution of NETs is illustrated in Table 26-1.
PROGNOSIS
The prognosis for patients with NETs varies by histologic grade, stage, and primary site. High-grade NETs demonstrate similar biology and prognosis to small cell lung carcinoma. Low- to intermediate-grade NETs have a more favorable prognosis. The median overall survival of patients with localized low- to intermediate-grade NET is 223 months, according to a recent analysis of the Surveillance, Epidemiology, and End Results (SEER) database of patients registered from 1973 to 2004. For patients with regional disease, defined as involvement of regional lymph nodes, extension to adjacent tissue, or both, the median overall survival is 111 months. For metastatic disease, the median overall survival is 33 months (1). The prognoses of NETs by anatomic site are discussed subsequently.
PATHOGENESIS AND MOLECULAR BIOLOGY
Sporadic and hereditary tumors have yielded insights into NET biology. The MEN1 gene, mutated in multiple endocrine neoplasia, type 1, was mutated in 44% of patients with resected sporadic PNETs in a recent exome sequencing study, with 43% having mutations in the DAXX/ATRX complex and 14% harboring mutations in the mammalian target of rapamycin (mTOR) pathway (5). Menin, the protein product of the MEN1 gene, suppresses tumorigenesis by multiple mechanisms, including transcription regulation via interaction with histone methyltransferases, direct cell cycle regulation via interaction with the genetic loci of cyclin dependent kinase inhibitors, and facilitation of apoptosis by increased caspase 8 production (6). Genomic analyses of small intestinal NETs revealed rare mutation events, with alterations in CDKN2B observed in nearly 10% of cases (7). Multiple older techniques, such as comparative genomic hybridization (8) and single-nucleotide polymorphism–based array technology (9), have confirmed chromosome 18 loss in up to 43% of midgut NETs. Therefore, the somatic genomic alterations in pancreatic and extrapancreatic NETs are qualitatively and quantitatively different, aligning their genetic and clinical heterogeneity. By immunohistochemistry, vascular endothelial growth factor (VEGF) has been associated with poorer clinical outcomes in GEP-NETs (10), generating significant interest in antiangiogenic therapies, as discussed later.
A significant minority of NETs (5%-10%) occur in the context of multiple endocrine neoplasia, type 1 (MEN1), an autosomal dominant disorder characterized by pituitary tumors, hyperparathyroidism, and PNETs. Neuroendocrine tumors related to MEN1 demonstrate a unique pattern of symptomatic and asymptomatic lesions. Duodenal gastrinomas, causing the Zollinger-Ellison syndrome of peptic ulcers, gastroesophageal reflux, and diarrhea, afflict nearly 60% of MEN1 patients. Characteristic asymptomatic lesions include small duodenal foci of somatostatin expression and pancreatic adenomas secreting glucagon or pancreatic polypeptide. Roughly 20% of MEN1-associated pancreatic macroadenomas are “insulinomas,” causing hyperinsulinemic hypoglycemic syndrome. Notably, approximately 10% of PNETs are associated with MEN1 syndrome (11). Neurofibromatosis type 1, von Hippel-Lindau syndrome, and tuberous sclerosis complex 2 also predispose to NETs, but with very low penetrance, and these diseases account for far fewer NETs than MEN1 (11).
PATHOLOGIC CLASSIFICATION
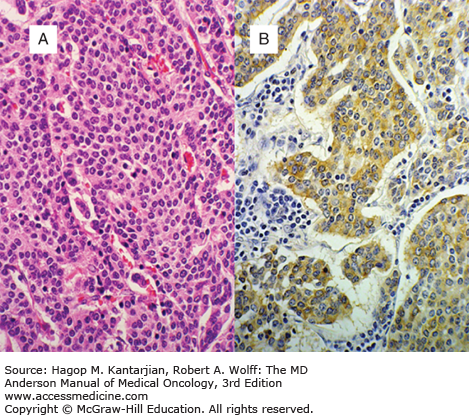
NETs are characterized by monotonous sheets of small round cells with uniform nuclei and cytoplasm (Fig. 26-1). Neuroendocrine cells store secreted substances in membrane-bound vesicles. Malignant neuroendocrine cells have minimal mitotic activity, cytologic atypia, or nuclear polymorphism. Immunohistochemical markers used to confirm a NET diagnosis include neuron-specific enolase, CD56, chromogranin A (CgA), and synaptophysin (Table 26-2).
| Marker | Significance |
|---|---|
| Neuron-specific enolase | Cytoplasmic glycolytic enzyme, a less specific neuroendocrine marker |
| Synaptophysin | Presynaptic vesicle membrane glycoprotein, present on normal and neoplastic neuroendocrine cells |
| Chromogranin A | Acidic protein, universal marker for neuroendocrine tissue |
| CD56 | Neural adhesion molecule |
| Cytokeratin(s) | Lack of cytokeratin expression suggests the tumor is either an anaplastic neoplasm or may not be a carcinoma |
Assessing tumor grade is critical in all NETs. Modern grading schemes distinguish principally between high-grade (grade 3) tumors, with mitotic count >20/10 high-powered fields or Ki-67 proliferation index >20%, and low- or intermediate-grade (grade 1-2) tumors. Clinical behavior and therapy are largely determined by this distinction (12). However, a large retrospective analysis revealed a Ki-67 index of 55% or greater to better predict responsiveness to the platinum-based chemotherapy recommended for high-grade NETs (13). Therefore, it is likely that grading systems will evolve with our biological understanding and therapeutic options.
CLINICAL PRESENTATION, DIAGNOSTIC WORKUP, AND CLINICAL STAGING
Although NETs are well known for their hormonal syndromes, many malignant PNETs are nonfunctional, and carcinoid syndrome is typically present only in the setting of metastases. Symptoms can be insidious and present for years before diagnosis. Symptoms of local and regional extrapancreatic and nonfunctional pancreatic NETs are often vague, relating to obstruction, mesenteric fibrosis, or vascular compromise.
Although general serum and urine biomarkers are useful for tumor monitoring, they are insufficient for NET diagnosis, which requires pathologic evaluation. Frequently measured tumor markers in carcinoid disease include serum CgA and 5-hydroxyindoleacetic acid (5-HIAA) levels in a 24-hour urine sample. False-positive results occur with consumption of tryptophan- or serotonin-rich foods (bananas, butternuts, kiwis, mockernuts, pecans, pineapples, plantains, plums, shagbark, sweet pignuts, tomatoes, and walnuts). Common medications affecting urinary 5-HIAA levels include guaifenesin, acetaminophen, and salicylates. Serum CgA level is a sensitive, but nonspecific, marker for NETs, and is elevated among patients receiving proton pump inhibitors or with impaired renal, hepatic, or cardiac function.
In addition to CgA and 5-HIAA, NETs can synthesize other bioactive amines and peptides including 5-hydroxytryptamine (5-HTP), 5-hydroxytryptophan (5-HT), serotonin, insulin, gastrin, glucagon, somatostatin, vasoactive intestinal polypeptide (VIP), adrenocorticotropic hormone, melanocyte-stimulating hormone, pancreatic polypeptide, and pancreastatin (14).
Endoscopic techniques localize tumors and facilitate biopsy. Esophagogastroduodenoscopy (EGD) can often locate gastric and duodenal NETs. Colonoscopy can identify colorectal NETs. Double-balloon enteroscopy and capsule endoscopy may assist in localizing small bowel NETs. Disadvantages of endoscopy include the requirement for patient sedation and the difficulty in visualizing small submucosal lesions. Endoscopic ultrasound is useful in the assessment, visualization, and biopsy of pancreatic and some small duodenal NETs.
Computed tomography (CT) and magnetic resonance imaging (MRI) are the preferred initial imaging studies for localizing NETs. However, the utility of cross-sectional imaging for diagnosing typical small bowel NETs is limited; usually their presence must be inferred from luminal narrowing, adenopathy, and mesenteric fibrosis. Computed tomography and MRI technologies are far more useful in detection of hepatic metastases, which frequently present convenient sites for diagnostic biopsy. Computed tomography and MRI are helpful in the detection of PNETs, for which the sensitivities of CT and MRI are up to 82% and 100%, respectively (15).
Somatostatin receptor scintigraphy (SRS) has improved the visualization of NETs. Somatostatin receptor scintigraphy uses a somatostatin analogue, 111In-labeled diethylenetriamine penta-acetic acid octreotide (DTPA-D-Phe1-octreotide), to visualize tumors expressing somatostatin receptors 2 and 5. Compared with CT or MRI, SRS detects additional metastases in about one-third of patients. Moreover, SRS may help to identify small tumors when conventional scans are unrevealing. The overall sensitivity of SRS is 80% to 90% (16).
Because 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET) identifies tumors with significant proliferative activity, it is unreliable in NETs. Gallium-68 (68Ga)-DOTA-NOC PET, which uses labeled octreotide to detect somatostatin-avid tumors, had a sensitivity of 78.3% and a specificity of 92.5% in an initial study (17). Ongoing studies are evaluating alternative 68Ga-labelled somatostatin analogues, and this technology may emerge as more convenient than SRS, with potentially improved test characteristics.
Metaiodobenzylguanidine (MIBG) is absorbed by carcinoid tumor cells. Iodine-131-labeled MIBG (131I-MIBG) has an overall sensitivity of 55% to 70% in detecting NETs (18). Although 131I-MIBG is less sensitive than SRS, it may be used in patients receiving long-acting octreotide, which competitively inhibits uptake of radiolabeled somatostatin analogues.
The American Joint Committee on Cancer (AJCC) has proposed site-specific staging systems for grade 1 to 2 NETs using the TNM system (19). The details vary, particularly with respect to the T stage, which can depend on depth of invasion or involvement of critical structures. This staging is prognostic, but not predictive of the benefit with any therapy. The relevant distinction is therefore between resectable and unresectable/metastatic disease. High-grade NETs are described with a TNM stage according to guidelines for carcinomas of the primary site but are summarized as limited stage or extensive stage disease, defined by the ability of the cancer to be treated in a single radiation port, based on the staging of the biologically analogous small cell lung cancer (20).
CARCINOID CLINICAL BEHAVIOR BY SITE
Gastric carcinoid tumors are divided into three types. Type 1 (75%) is associated with chronic atrophic gastritis, type 2 (5%-10%) is associated with with Zollinger-Ellison syndrome, and type 3 (15%-25%) is sporadic (21). Conceptually, type 1 and 2 gastric carcinoids arise as a physiologic response to excessive gastrin secretion, with the gastrin being physiologic in type 1 gastric carcinoids and pathologic in type 2 gastric carcinoids. Because they are independent of known stimuli, type 3 gastric carcinoids have the worst prognosis, frequently presenting with metastatic disease. A SEER analysis revealed a median overall survival of 13 months for patients with metastatic gastric carcinoid (1). Clinical features of the three groups of gastric carcinoid are summarized in Table 26-3.
Small intestine NETs, most frequently associated with carcinoid syndrome, are often found in the distal ileum within 60 cm of the ileocecal valve. At diagnosis, multiple putative “primary” lesions may be present. Analysis of SEER data from 1973 to 2004 demonstrates that jejunum and ileum NETs (30%) were far more likely than duodenal (9%), rectal (5%), or appendiceal (9%) lesions to be metastatic at diagnosis. The median overall survival times of patients with duodenal and jejunum/ileum carcinoids were 107 and 111 months, respectively, for localized disease and 57 and 56 months, respectively, for metastatic disease (1).
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


