Fig. 1.1
Model depicting the role of ARC KNDy neurons in the generation of pulsatile GnRH release. KNDy neurons in the ARC nucleus form an interconnected population with their axons and/or dendrites. In the presence of low levels of Dyn, individual KNDy neurons (insert, 1–3) begin constitutively increasing NKB secretion leading to a synchronous excitation and Kiss release which stimulates GnRH secretion (graphs in insert). Moments later, Dyn levels begin to increase and apply a break to NKB, Kiss and GnRH secretion. Increased local concentration of Dyn will inhibit KNDy neurons, resulting in an interpulse interval, until a lower threshold is reached and the cycle can reset
As shown in Fig. 1.2, the release of GnRH is influenced by multiple neurotransmitters including glutamate, gamma-aminobutyric acid (GABA), neuropeptide Y, opiates, dopamine, norepinephrine, cAMP and nitric oxide [33]. The presence of receptors on GnRH neurons for most of these substances implies that they influence GnRH neurons directly. NMDA receptors that mediate glutamate activation of GnRH may involve the nitric oxide signaling pathway. Neurotransmitters with receptors that are not expressed on GnRH neurons may regulate GnRH via synaptic connections between GnRH neurons and other interneurons. Regulation of GnRH secretion may also occur directly on neuronal axon terminals that abut on capillaries in the median eminence.

Fig. 1.2
Diagram showing activation of GnRH neurons by neurotransmitters and the relation to the anterior pituitary
A second form of GnRH, GnRH-2 [34, 35] that was initially identified in non-vertebrates, is also found in the primate brain [36]. GnRH-2 appears to activate a unique GnRH-II receptor [37], but this receptor does not appear to be expressed in humans [38]. Thus, the significance of GnRH-2 in humans is not yet known, and there is little evidence for a role in reproduction.
There has been resurgence in research on a gonadotropin-inhibiting hormone (GnIH) which appears to have a functional role in regulating GnRH neurons and gonadotropin secretion in seasonal breeders and lower mammals (for review, see [39]). However, the role of the primate GnIH ortholog, RFRP-3, has not been elucidated. In a recent study in rhesus macaque, an antagonist to RFRP-3 was demonstrated to prevent the decline in circulating testosterone levels that accompanies acute fasting [40]. This result suggests the possibility that a GnIH hormone exists in primates and may have a role in modulating reproductive function in response to varying metabolic states. Further research is necessary to validate this hypothesis.
Gonadotrophs and GnRH Receptors
Gonadotrophs account for 6–10% of the cells of the normal anterior pituitary [41]. Gonadotrophs may be small and round, or larger and ovoid, and are difficult to identify by morphological criteria. Instead, immunostaining using specific antibodies for LH-β and FSH-β proteins are used to identify gonadotrophs. In primates [42], as in rodents [41], the great majority of gonadotrophs are bihormonal, i.e., they express both LH-β and FSH-β subunit genes. A small fraction of cells appear to express LH or FSH selectively, and some gonadotrophs also produce growth hormone. The proportions of each of the hormone-producing gonadotrophs are developmentally regulated [43]; however, the biological significance of these observations remains unclear.
GnRH activates gonadotrophs through both short-term and long-term mechanisms that are illustrated in Fig. 1.3. Upon reaching the pituitary, GnRH binds to and activates a cell-surface G-protein coupled receptor (GnRH-R) [44]. This receptor is a structurally unique member of the seven-transmembrane G-protein-linked receptor family that lacks the long C-terminal intracellular tail that is typical of most G-protein-coupled receptors. This tail is important in the rapid desensitization of other G-protein-coupled receptors, whereas down-regulation of the GnRH receptor by GnRH is a relatively delayed event that occurs over hours rather than minutes. Binding of GnRH to its receptor facilitates binding of a G-protein to the receptor’s third intracellular loop. The bound G-protein exchanges GDP for GTP and dissociates into its constituent α and βγ subunits. The α-subunits are unique to each G-protein, whereas the β- and γ-subunits of the different G-proteins are similar. The dissociated G-protein α-subunit activates downstream signaling pathways [45]. Gαq/11, the major G-protein that associates with the GnRH-R, activates membrane-associated phospholipase C to hydrolyze membrane phosphoinositides and increase intracellular inositol phosphates (Ips) including inositol triphosphate (I1,4,5)P3. IP3 rapidly mobilizes calcium from intracellular stores, and L-type voltage-gated calcium channels open following which extracellular calcium enters the cell [45]. The rise in intracellular free calcium is primarily responsible for the immediate release of LH and FSH [46]. GnRH receptors also interact with other G-proteins including Gα i and Gα s . The GnRH receptor can couple to Gα s in rat pituitary cells [47] and subsequently activate cAMP production and the PKA signaling pathway [48]. Interaction with Gα i occurs primarily in non-pituitary tissues.

Fig. 1.3
Signaling mechanisms initiated by activation of the GnRH receptor stimulate gonadotropin synthesis and secretion
The long-term stimulatory effects of GnRH are to increase transcription of the genes for the gonadotropin subunits and the GnRH receptor, among other genes. These effects occur primarily through liberation of membrane diacylglycerol (DAG) that in turn activates protein kinase C. The subsequent phosphorylation of subfamilies of mitogen-activated protein kinases (MAP kinases), including members of the ERK and JNK families, initiates nuclear translocation of proteins that bind directly to the 5′ regulatory regions of gonadotropin subunit and the GnRH-receptor genes or serve as cofactors for promoter activation [49]. Increased intracellular calcium also causes increased activation of calmodulin-dependent protein kinases which contributes to the transcriptional effects of GnRH [50].
GnRH receptors are up-regulated by pulsatile GnRH [51]. Accordingly, when pulsatile GnRH secretion increases, as in castration or primary testicular failure, GnRH receptors increase. Gonadotrophs become more responsive to GnRH, and the LH response to GnRH stimulation is amplified [52]. With continuous GnRH treatment on the other hand, GnRH receptors decline, followed by suppression of LH-β and FSH-β mRNAs. This “homologous desensitization” of the GnRH-R is regulated by several serine-threonine protein kinases including protein kinase A (PKA) and protein kinase C (PKC), as well as by G-protein-coupled receptor kinases (GRKs) which combine to cause a reduction in the efficiency with which Ins(1,4,5)P3 mobilizes Ca2+ from intracellular storage [53]. GnRH receptors also decline with GnRH deficiency.
GnRH receptors undergo a low level of agonist-independent constitutive internalization which is not influenced by binding to GnRH agonists [54]. Accordingly, a wide variety of postreceptor modifications have been proposed for the observed receptor desensitization that disconnects GnRH binding from LH secretion after prolonged exposure [55]. The primate GnRH-R is different than other mammalian GnRH-R due to the presence of a primate-specific Lys191 [56], the lack of a second glycosylation site near the NH2 terminus [57] as well as the absence of any COOH tail [58]. These differences lead to a comparatively significant reduction in the percentage of membrane-bound receptors and suggest that differential expression of intracellular chaperones or membrane scaffolding proteins may play an important role in GnRH signaling and desensitization. Evidence for this is the recent discovery that the proto-oncogene SET, also called I2PP2A (inhibitor 2 of protein phosphatase 2A), interacts with the first and third intracellular loops of the GnRH receptor and leads to inhibition of calcium signaling while increasing the cAMP signaling pathway [59].
The Gonadotropic Hormones
LH and FSH are members of the glycoprotein family of hormones that also includes thyrotroph-stimulating hormone (TSH) and chorionic gonadotropin (hCG). These heterodimers are composed of a common α-subunit and unique β-subunits. The subunits have N-linked oligosaccharide chains that are associated with asparagine residues. Each subunit is encoded by a unique gene that is found on a separate chromosome: The human α-subunit gene is on 6p21.1–23, LH-β is on 19q13.3, and FSH-β is on 11p13 (see Chap. 6).
The production of LH and FSH is directly influenced by the level of the gonadotropin subunit mRNAs. This relationship seems especially strong for FSH-β mRNA and FSH secretion. Each of the gonadotropin subunit mRNAs is increased by GnRH, and stimulation of the β-subunit genes by GnRH is primarily transcriptional. Complexes of transcription factors including SF-1, EGR-1, and SP1 are activators of the LHβ-subunit gene [60], whereas the AP1 proteins fos/jun are important for up-regulation of FSH-β transcription by GnRH [61]. GnRH stimulation must be pulsatile for LH-β and FSH-β mRNA levels to be increased. Transcriptional regulatory proteins that control α-subunit expression include cAMP response element binding protein (CREB), MAP kinase/ERK1 and GATA-binding proteins [62]. α-Subunit gene expression is increased robustly both by pulsatile and by continuous GnRH, and GnRH not only stimulates α-subunit transcription but also prolongs the half-life of the α-subunit mRNA [63]. Thus, the requirements for α-subunit mRNA up-regulation by GnRH seem less stringent than are those for the β-subunit genes. These factors partly explain why α-subunit is synthesized in excess of β-subunits and why β-subunits are rate limiting for gonadotropin synthesis.
Proteins that are destined for secretion are synthesized on ribosomes bound to the endoplasmic reticulum. During the translational process, preformed oligosaccharide chains are linked to the side chain amino group of asparagines on the gonadotropin subunits. As translation continues, sugar moieties are trimmed, and the subunits change configuration allowing for their combination. A region of the β-subunit, termed the “seatbelt,” wraps around the α-subunit loop 2 [64]. Dimeric LH and FSH are segregated in the ER and are transferred to the Golgi where they are concentrated in secretory granules. These protein-rich vesicles subsequently fuse with the plasma membrane following stimulation by GnRH. This process is termed “exocytosis.” There is evidence that granules containing FSH are also exported directly to the plasma membrane independent of GnRH pulsatile stimulation. This mode of secretion allows for FSH secretion between pulses. Gonadotrophs also secrete uncombined α-subunit both in pulsatile fashion and continuously.
Pulsatile Gonadotropin Secretion
Experiments in ovariectomized rhesus monkeys rendered gonadotropin-deficient with hypothalamic lesions lead to the proposal that an intermittent pattern of GnRH secretion was necessary for normal LH secretion [65]. In those animals, GnRH administered in pulses stimulated LH secretion, but GnRH administered continuously was much less effective. The pulsatile nature of LH secretion was subsequently established in all species, including man [66]. Accordingly, LH secretion is stimulated when GnRH is administered in pulses to gonadotropin-deficient patients but not when administered continuously [67, 68]. An understanding of these physiological principles leads to the use of pulsatile GnRH as a treatment to stimulate fertility and to the development of long-acting GnRH analogs that initially stimulate LH/FSH secretion but then deplete membrane GnRH-R and inhibit gonadotropin production to produce a biochemical gonadectomy that is used as a treatment for patients with prostate cancer and other androgen-dependent disorders [69].
With current assays, GnRH is undetectable in the peripheral circulation. Therefore, GnRH secretion cannot be studied directly in humans. Instead, changes in circulating LH levels are used as a surrogate marker for the activity of the GnRH pulse generator. LH secretion is determined by the frequency, amplitude, and duration of its secretory pulses. Presumably because of its longer circulating half-life, FSH pulses are less clearly defined in peripheral blood than are LH pulses because FSH pulses are clearly evident in jugular blood (in ewes) where clearance effects are minimized [70].
Cultured pituitary cells that are perifused with pulses of GnRH are a powerful model yielding important information on the actions of GnRH and other factors that regulate gonadotropin secretion under controlled conditions. Using this experimental approach, episodes of LH as well as FSH secretion are distinct and short-lived with a rapid upstroke and abrupt termination. LH pulse amplitude is directly proportional to the dose of GnRH administered, and the median duration of an LH pulse about 25 min.
In contrast to the regularity of LH pulses produced by a constant dose of GnRH in vitro, Fig. 1.4 illustrates that LH pulses in the peripheral circulation in man vary in amplitude and that interpulse intervals are inconstant. LH pulses in vivo are also characterized by a less rapid upstroke and a slower decline from the peak than are pulses in vitro. This presumably reflects dilution of secreted hormone by plasma in the general circulation and the influence of clearance of LH by the liver and kidney. Whether LH is released in the basal interval between GnRH-initiated secretory episodes has been debated, but this mode of secretion is small and is probably not biologically important.

Fig. 1.4
Circulating LH and alpha subunit levels in a normal adult man. Blood samples were drawn every 10 min for 12 h beginning at 0800 h and measured for LH using Nichols Allegro LH 2-site assay, and a specific assay double antibody immunoassay for α-subunit
Hormone pulse detection has been standardized by the development of computer algorithms [71]. Using this approach, objective assessment of the frequency and amplitude characteristics of hormone pulses has been possible. Pulses of LH secretion occur throughout the day and night in normal adult men. However, estimates of the frequency of LH (GnRH) pulses in men have varied based on the intensity and duration of the blood sampling protocol, the assay used to measure LH and the algorithm used to identify pulses. Most investigators have proposed an average frequency of 1 LH pulse every 1–2 h for normal men, but interestingly there is a large between-individual variation [72]. Because of variability in pulse amplitude and frequency, the distinction between true and artifactual pulses can be difficult. One approach is to co-analyze LH and uncombined α-subunit pulses [31] because α-subunit is released into the circulation by GnRH together with LH and FSH (Fig. 1.4). According to this logic, concordant LH and α-subunit fluctuations presumably reflect true GnRH pulsatile signals. There is generally a positive relationship between LH pulse amplitude and the preceding interpulse interval in part because a longer interval allows for the circulating level to decline to a lower baseline value.
In addition to moment-to-moment pulsatile pattern of LH secretion, there is a diurnal rhythm in circulating LH as well as testosterone levels in pubertal boys with increased LH pulsatile amplitude during sleep and increased testosterone levels in the early morning hours [73]. The diurnal variation in plasma testosterone in adults is coincident with diurnal rhythms for LH [74, 75]. The diurnal testosterone variation in men is disrupted by fragmented sleep [76], while LH rhythms are not affected [75] and therefore the mechanism for this alteration has not been established. The diurnal variation in testosterone is blunted in older men [77] and in young men with testicular failure [78].
LH Control of Testosterone Synthesis
Testosterone, a C19 3-keto, 17β-hydroxy Δ4 steroid, is synthesized from cholesterol through a series of cytochrome P450- and dehydrogenase-dependent enzymatic reactions [79]. The conversion of cholesterol to pregnenolone occurs within mitochondria and is catalyzed by P450scc, the cytochrome P450 side chain cleavage enzyme. Pregnenolone exits the mitochondria and can be converted to testosterone by two alternative routes that are referred to as the Δ4-pathway or the Δ5-pathway, based on whether the steroid intermediates are 3-keto, Δ4 steroids (Δ4) or 3-hydroxy, Δ5 steroids (Δ5). Classical experiments using human testicular microsomes incubated with radiolabeled steroids revealed that the Δ5-pathway predominates in the human testis. In that pathway, C17 hydroxylation of pregnenolone to form 17α-hydroxypregnenolone is followed by cleavage of the C17–C20 bond of 17α-hydroxypregnenolone to produce dehydroepiandrosterone (DHEA). Oxidation of the 3β-hydroxy group and isomerization of the C5–C6 double bond of DHEA by 3β-hydroxysteroid dehydrogenase/Δ5–Δ4 isomerase (3βHSD) forms androstenedione. The C17 keto group of androstenedione is oxidized to a hydroxyl group by 17β-hydroxysteroid dehydrogenase type III (17β HSD)-producing testosterone (see Chap. 3).
LH stimulates testosterone biosynthesis in Leydig cells through a G-protein-associated seven-transmembrane receptor [80]. LH binding to the receptor initiates a signaling cascade by activating Gs that stimulates adenylate cyclase activity and increases intracellular cAMP levels to activate cAMP-dependent protein kinase A (PKA). cAMP-dependent PKA stimulates testosterone synthesis in at least two ways. An acute response, within minutes of hormonal stimulation, is characterized by an increase in cholesterol transport into the mitochondria and is mediated by the steroidogenic acute regulatory (StAR) protein [81]. StAR acts on the outer mitochondrial membrane, where it undergoes conformational changes through interactions with protonated phospholipids causing opening and closing of a sterol-binding pocket, permitting uptake and discharge of cholesterol [82]. The chronic response to LH, which requires several hours to days, involves transcriptional activation of the genes encoding the steroidogenic enzymes of the testosterone biosynthetic pathway, P450scc, P450c17, 3βHSD and 17βHSD.
Other factors that stimulate testosterone synthesis directly include PRL, GH, T3, PACAP, VIP and inhibin, whereas glucocorticoids, estradiol, activin, AVP, CRF and IL-1 have been reported to reduce testosterone production by Leydig cells. Although controversial for many years, experiments with recombinant FSH suggest that FSH is not an important regulator of Leydig cell function [83, 84]. However, it can indirectly stimulate Leydig cells through a Sertoli cell-mediated paracrine loop. In an elegant experiment, however, selective genetic ablation of Sertoli cells in neonatal or adult mice resulted in a significant decrease in Leydig cell number with normal mean testosterone levels but fewer high level pulses of circulating testosterone and higher levels of LH [85], suggesting that a Sertoli cell factor may regulate pulsatile testosterone biosynthesis.
The blood production rate of testosterone in normal adult men has been estimated to range from 5000 to 7500 μg/24 h [86], and levels of total testosterone among normal men range from 250 to 1000 ng/dl (10–40 nmol/L) in most assays. The level of testosterone in adult men declines by more than 95% if the testes are removed. The remainder of the circulating testosterone is derived from the production of androstenedione and dehydroepiandrosterone (DHEA) by the adrenal cortex. Intratesticular testosterone concentrations in normal men are 100-fold higher than are serum levels [87]. The higher concentration is due to local production as well as sequestration by the Sertoli cell-derived androgen binding protein [88]. High intratesticular concentrations of testosterone are believed to be important for spermatogenesis; however, concentrations tenfold above serum levels are sufficient for normal spermatogenesis [89, 90].
Estrogens in Males
Normal men produce about 40 μg of estradiol and 60 μg of estrone per day. Estradiol is produced from testosterone, and estrone from androstenedione, by aromatase P450, the product of the CYP19 gene [91]. This microsomal enzyme oxidizes the C19 angular methyl group to produce a phenolic A ring. Aromatase mRNA is expressed in adult Leydig cells where it is activated by LH/hCG [92]. However, most of the estrogen in men is derived from aromatase in adipose and skin stromal cells, aortic smooth muscle cells, kidney, skeletal cells and the brain. The promoter sequences of the extra-gonadal and testicular P450 aromatase genes are distinct and tissue-specific due to differential splicing, but the translated protein appears to be the same in all tissues. There is evidence in vitro that cytokines such as IL-6 stimulate aromatase in stromal cells, but the factors that regulate extra-testicular aromatase are not well understood.
Estrogens are known to exert important physiological effects on reproduction as well as other organ systems in men. The two forms of the estrogen receptor, ER-α and ER-β, are each known to play a role in reproduction [93]. ER-α is the dominant form in the pituitary and hypothalamus, whereas both ER-α and ER-β are found in the testis, prostate and epididymis [94]. Studies in one man with an inactivating ER-α mutation and four men deficient in aromatase (Table 1.1) reveal effects of estrogens on bone metabolism, insulin sensitivity, hepatic lipid metabolism and reproduction. Studies in normal men using pharmacologic inhibitors and steroid add-back protocols support these findings [95]. Dilatation and atrophy of the seminiferous tubules in ER-α-deficient mice [96] imply an effect of estrogen to regulate aquaporin expression in the efferent ductules of the testis. Aromatase-deficient mice, although fertile in early life, develop infertility. Thus, estradiol is essential for male reproduction.
Table 1.1
Hormone levels in a man with a mutation of the ERα (1), and four men deficient in aromatase (2–5)
Age (yrs) | Testosterone | Estradiol (pg/ml) | LH (mIU/ml) | FSH (mIU/ml) | Testis size | Sperm count | Reference | |
|---|---|---|---|---|---|---|---|---|
1 | 28 | 44 (ng/dL) | 119 | 37 (2.0–20) | 33 (2.0–15) | 20–25 mL | 25 mil/ml | [176] |
2 | 24 | 2015 (ng/dL) | <7 | 26.1 (2.0–9.9) | 28.3 (5.0–9.9) | R 6 × 3.5 L 5.5. × 3.2 cm | ND | [177] |
3 | 31 | 523 (ng/dL) | <10 | 5.6 (1.4–8.9) | 17.1 (1.7–6.9) | 8 mL | 1 mil/ml | [178] |
4 | 27 | 829 (ng/dL) | <20 | 8.3 (1.2–8.6) | 18.8 (1.3–19.3) | Ambiguous genitalia 20 mL | 56 mil/ml | [110] |
5 | 24 | 20.l5 (nmol/L) | UD | 4.8 (<11.1) | 14.4 (<20.3) | 15 mL | 129 mil/ml | [109] |
Testicular Control of Gonadotropin Secretion
Gonadotropin secretion, while up-regulated by GnRH, is maintained at physiological levels through testicular negative feedback mechanisms summarized in Fig. 1.5. Accordingly, plasma LH and FSH levels decrease following the administration of testosterone or estradiol and rise when negative feedback is disrupted by castration.

Fig. 1.5
Diagram of the negative feedback control of gonadotropin secretion by testicular hormones
The process of negative feedback control of LH and FSH by gonadal steroids in males is partly species-specific. There is considerable evidence in primates that androgens suppress LH synthesis and secretion primarily through an action on the GnRH pulse generator. For example, LH pulse frequency and amplitude are elevated in castrates and are suppressed by testosterone replacement [97]. Furthermore, when male rhesus monkeys were rendered gonadotropin-deficient with radio-frequency lesions, and stimulated with pulses of GnRH, LH secretion increased little after bilateral orchidectomy until GnRH pulse frequency was increased [98]. In addition, expression of the mRNAs for GnRH [3], as well as pituitary GnRH receptors, and the gonadotropin subunit genes are increased in orchidectomized monkeys [99]. Moreover, when pituitary cells from adult male monkeys were stimulated with pulses of GnRH, no inhibition of GnRH-induced LH secretion by testosterone or DHT was found. In pituitary cells from rats, on the other hand, the gonadotroph is a direct site of testosterone negative feedback control since GnRH-stimulated LH pulses were suppressed in amplitude by testosterone, and α-subunit gene expression was reduced (Fig. 1.6 [100]).
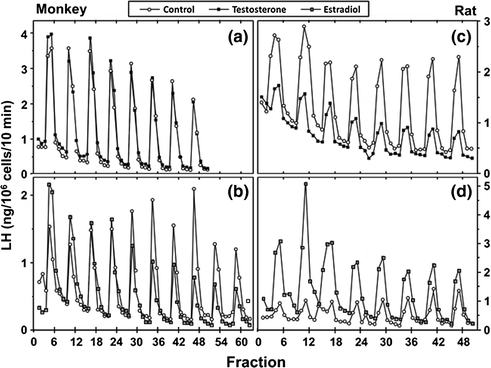
Fig. 1.6
Effects of testosterone or estradiol on GnRH-stimulated LH secretion from pituitary cell cultures from adult male monkeys and rats. From Kawakami and Winters [100]. Reprinted with permission from Endocrine Society
GnRH-deficient men represent a human model to examine the effects of testicular steroids on the hypothalamus and pituitary. When such patients were treated with fixed doses of GnRH [101], testosterone suppressed LH secretion less than in normal men implying that testosterone controls GnRH secretion. Moreover, suppression by testosterone was blocked by the aromatase inhibitor testolactone [101], and in separate experiments, dihydrotestosterone had no effect [102]. Together these data imply that the pituitary effect of testosterone to inhibit LH was through bioconversion to estradiol.
Whether it is testosterone, itself, and/or the estradiol derived from testosterone by aromatase in the central nervous system, pituitary system, or peripheral tissues that controls LH secretion, has been a subject of considerable interest. The finding that dihydrotestosterone (DHT), a non-aromatizable androgen, decreases LH pulse frequency strongly supports a role for androgens in the regulation of the GnRH pulse generator. Furthermore, LH pulse frequency is increased in patients with non-functional androgen receptors (AR) in the complete androgen insensitivity syndrome, indicating that AR signaling regulates GnRH pulse frequency [103].
Estradiol also plays an important physiological role in the negative feedback control of gonadotropin secretion in men. This control mechanism was suggested by pharmacological studies using the estrogen antagonist clomiphene [104], or the aromatase inhibitor, testolactone [105]. When those drugs were administered to normal men, circulating LH and FSH levels rose together with plasma testosterone concentrations. This finding was confirmed and extended in studies using the aromatase inhibitor, anastrazole [106, 107]. Variable LH and FSH levels in an adult man with an inactivating mutation of the ER-α and four men with mutations of the CYP19 aromatase gene (reviewed [108–110]) are shown in Table 1.1. Estradiol treatment also decreases the LH response to GnRH stimulation in men [111] and in primate pituitary cells perifused with pulses of GnRH (Fig. 1.6, [100]) indicating an additional direct negative feedback effect of estradiol on the pituitary.
GnRH neurons appear by autoradiography and immunocytochemistry to lack both androgen and estrogen receptors and numerous animal models have shown that kisspeptin neurons mediate the negative feedback effects of gonadal steroid hormones (for review, see [112]). Both estrogen and androgen receptors are localized to kisspeptin neurons in the hypothalamus of male mice [113]. Furthermore, testosterone administration to castrated male monkeys decreased kisspeptin mRNA in the medial basal hypothalamus coincident with suppression of circulating levels of LH [114]. A comparison of kisspeptin neurons in the hypothalamus of young and elderly men dying a sudden death revealed an age-related increase in the number of kisspeptin neurons and fibers and an enhancement of afferent contacts they established onto GnRH neurons [115]. These studies support the concept that kisspeptin neurons mediate the negative feedback effects of testosterone in primates including man and are consistent with studies showing that androgen negative feedback responsiveness increases with aging [116]. However, many other hypothalamic neuromodulators are responsive to castration and testosterone replacement, and therefore, the role of kisspeptin neurons may be partial with regard to steroid hormone negative feedback.
Inhibin, Activin and FSH
Although LH and FSH are both produced within the same cell type, the synthesis and secretion of the gonadotropins are, in some circumstances, differentially regulated. Both LH and FSH are stimulated by GnRH and are suppressed by gonadal steroid hormones. However, FSH levels may rise selectively in men with seminiferous tubular failure, and FSH synthesis, and secretion, can be selectively regulated through the actions of two paracrine factors, pituitary activin and follistatin, and by testicular inhibin-B. Activin selectively up-regulates FSH secretion by stimulating FSH-β gene expression [117] with negligible effects on LH synthesis. Inhibin and follistatin prevent activin from binding to its receptor through separate mechanisms, effectively antagonizing the stimulatory effects of activin on FSH synthesis.
Activin, a member of the TGF-β family of growth factors, is a dimeric peptide consisting of two similar subunits that were designated as β-subunits because the identification of activin followed the cloning of inhibin, an α–β heterodimer. There are at least four forms of the β subunit: βA, βB, βC and βD. Activin-B (βBβB) is the predominant form in the pituitary, whereas most other tissues produce activin-A. Activin functions in neural and mesodermal morphogenesis, wound healing, vascular remodeling and inflammation, as well as in reproduction [118–121]. Activins (and follistatin) primarily function as local paracrine or autocrine factors with no observable changes in the general circulation except in pregnancy and advanced stages in cancer patients [122, 123]. Therefore, activin-stimulated FSH synthesis and secretion is tightly and selectively regulated within the pituitary milieu.
Like other members of the TGFβ superfamily, activins signal through a serine-threonine receptor hetero-oligomeric complex that activates intracellular SMAD proteins (Fig. 1.7, [124]). Initially, the activin ligand binds to a specific type II receptor subunit, either ActRII or IIB [125–127]. Subsequent to ligand binding, the type II subunit pairs with a type I receptor subunit (either ActRI or IB) and forms a heteromeric complex at the cell surface [128–130]. The serine/threonine kinase of the type II subunit is responsible for phosphorylation of the type I subunit, thereby initiating postreceptor signaling/phosphorylation of SMAD2 and SMAD3 proteins which then dissociate from the receptor complex, bind SMAD-4 and translocate from cytoplasm to nucleus. Activated SMAD complexes interact with cofactor proteins and bind to SMAD binding elements on gene promoters, including FSH-β, driving immediate gene activation.

Fig. 1.7
A diagram for activin signaling through Smad proteins
The actions of activin are antagonized by follistatin that binds to and neutralizes the bioactivity of activin, as well as by inhibin that competes with activin for binding to the activin RII receptor [131]. Inhibin is an antagonist of activin because it fails to initiate intracellular SMAD signaling. Inhibin has a lower affinity for Act RII than activin, and betaglycan, a membrane proteoglycan, appears to function as an accessory receptor binding protein to potentiate inhibin binding, as well as for TGF-β and bone morphogenic protein [132]. Follistatin is structurally unrelated to activin and inhibin, but like activin, is found in all tissues examined. In the rat pituitary, follistatin is up-regulated by activin, GnRH and PACAP, and is suppressed by testosterone and by follistatin itself, no doubt through binding to activin. In primate pituitary cultures, on the other hand, GnRH is ineffective, and testosterone, as well as activin, increases follistatin expression [133]. Pituitary follistatin influences the FSH and LH response to castration. Follistatin expression increases following orchidectomy in adult male rats [134], and there is a reciprocal relationship over time between follistatin and FSH-β gene expression implying that follistatin attenuates the FSH castration response in that species. In male primates including man, by contrast, the level of follistatin mRNA is relatively unaffected by bilateral orchidectomy, and FSH-β mRNA increases about 50-fold (Fig. 1.8, [99]). Thus, follistatin appears to function as a brake on FSH production in male rodents but not in primates.
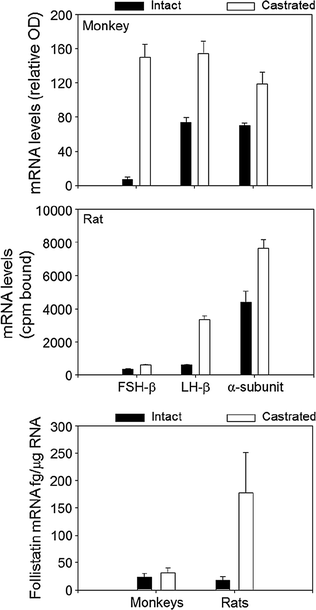
Fig. 1.8
A comparison of the effects of orchidectomy on gonadotropin subunit and follistatin mRNA levels in adult male rats and monkeys. From Winters et al. [99]. Reprinted with permission from Endocrine Society
Inhibin is produced by Sertoli cells and by fetal Leydig cells and plays a fundamental role in the selective regulation of FSH [135, 136]. Inhibin also regulates ovarian steroidogenesis and follicular development while its paracrine function in testis is not well understood. The term inhibin was first applied in 1932 [137] to the aqueous extract of bull testis that prevented the development of castration cells within the anterior pituitary, and was distinguished from androitin, that was present in the ether-extract and stimulated prostate growth, and is now known as testosterone.
Inhibin is a heterodimer of an α-subunit (20 kDa) and one of two β-subunits, βA and βB (13 kDa). Of these, only the βB subunit is expressed by the testis in most species, and therefore testicular inhibin is inhibin-B. The inhibin α-subunit is expressed in Sertoli cells and somewhat in Leydig cells and is up-regulated by FSH. The βB subunit is expressed in Sertoli cells and germ cells, but the factors regulating the βB subunit gene are not well understood. The level of inhibin/activin βB mRNA in the rat testis is unaffected by hypophysectomy or by FSH treatment [138]. Transcription factors of the GATA-binding protein family regulate both the inhibin β- and α-subunit promoters [139]. Exogenous testosterone treatment results in suppression of FSH and a parallel decrease in circulating inhibin-B [140].
Conversely, inhibin-B levels increase in men treated with FSH for infertility [140, 141]. Control of inhibin βB by a germ cell factor is suggested by the decline in plasma inhibin-B levels, but not inhibin-α subunit levels, that follows destruction of germ cells by cancer chemotherapy [142]. Inhibin-B is a marker of Sertoli cell function and is positively correlated with sperm count and testicular volume [143].
Inhibin-B is a selective negative regulator of FSH. While initially shown in vitro using pituitary cells from rodents [144], subsequent experiments in adult male monkeys revealed suppression of circulating FSH with no effect on the levels of LH or testosterone [145]. Inhibin can also augment the suppressive effect of testosterone on FSH synthesis, because inhibin suppresses FSH secretion [146]. On the other hand, very large doses of FSH are required to increase circulating inhibin-B levels [147]. Consequently, plasma inhibin-B and FSH concentrations are inversely related among normal men and are more strongly inversely correlated when values from men with varying degrees of primary testicular failure are included in the analysis [147, 148]. The relationship between inhibin-B and FSH differs from that between LH and testosterone which is bidirectional, i.e., LH stimulates testosterone, and testosterone suppresses LH, such that there is no comparable strong correlation between circulating LH and testosterone levels among normal men. The different relationships between plasma LH with testosterone compared to FSH with inhibin-B levels were clearly demonstrated in experiments conducted by Ramaswamy et al. [149]. These investigators removed one testis from adult male rhesus monkeys following which the plasma levels of both testosterone and inhibin-B decreased. The decline in testosterone was brief and was restored to normal by a rise in LH, whereas inhibin-B levels remained at approximately 50% of baseline values for up to 6 weeks even though FSH levels rose. Thus, LH and testosterone form a classical feed-forward/feedback loop whereas inhibin-B controls FSH but is less dependent on FSH stimulation.
Inhibin-B levels increase during the neonatal phase of development, sometimes called mini-puberty, and again at puberty. Although plasma LH and FSH levels rise at these developmental stages, the number of Sertoli cells also increases. Furthermore, studies in adult monkeys showed a strong positive correlation between circulating inhibin-B levels and Sertoli cell number [150]. Thus, circulating inhibin-B appears to reflect the number and function of Sertoli cells and is less dependent on FSH stimulation.
Activin, inhibin and follistatin may also act locally in the testes to regulate reproductive function. Activin receptor gene expression has been detected in Sertoli cells, primary spermatocytes and round spermatids, and radiolabeled activin-A binding has been demonstrated in the latter cell types [138, 151, 152]. Both proposed inhibin receptors, betaglycan and InhBP/p120 are present in rat Leydig cells [132, 153], and betaglycan is expressed in germ cells [154]. In general, activin treatment reduces hCG-driven Leydig cell production of androgens [155] while inhibin action can prevent activin effects [155], and has been shown to stimulate testosterone release in some [156] but not all [157] studies. In terms of actions on gamete production, activin and inhibin tend to have opposing actions with activin stimulating [158] and inhibin reducing [159, 160] spermatogenesis. Inhibin-α-deficient mice have large testes and develop gonadal stromal tumors that appear to occur due to activin excess [161] and over-expression of the α-subunit in transgenic mice results in hypospermatogenesis [162]. Thus, the balance of activin and inhibin (and potentially follistatin), directly in the testes, may impact both endocrine signals via altered testosterone production as well as gamete maturation.
Related posts:
 Male Hypogonadism Due to Disorders of the Pituitary and Suprasellar Region
Androgen Therapy for Hypogonadism in Men with Chronic Illnesses
Sex Hormone-Binding Globulin and the Metabolic Syndrome
Stimulation of Spermatogenesis in Hypogonadotropic Men
Congenital Hypogonadotropic Hypogonadism in Males: Clinical Features and Pathophysiology
The Undescended Testis
Male Hypogonadism Due to Disorders of the Pituitary and Suprasellar Region
Androgen Therapy for Hypogonadism in Men with Chronic Illnesses
Sex Hormone-Binding Globulin and the Metabolic Syndrome
Stimulation of Spermatogenesis in Hypogonadotropic Men
Congenital Hypogonadotropic Hypogonadism in Males: Clinical Features and Pathophysiology
The Undescended Testis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


