Neoplasms of the eye
Jasmine H. Francis, MD  Amy C. Schefler, MD, FACS
Amy C. Schefler, MD, FACS  David H. Abramson, MD
David H. Abramson, MD
Overview
Cancers involving the eye are less common compared to lung, prostate and breast cancer. However, they pose a special challenge because they can influence both life and vision. The field of ophthalmic oncology focuses on these two elements: improving efficacy to treat the disease and save life, and limiting the consequence of treatment to maintain vision. The eye itself is composed of a variety of distinct tissues and ophthalmic malignancies can affect a number of these anatomical areas. This makes the topic of neoplasms of the eye heterogeneous in origin, pathogenesis, prognosis, and treatment.
Introduction
This chapter reviews benign and malignant ocular, orbital, and lid tumors in both children and adults. The most common of these are listed in Table 1.
Table 1 Most common ophthalmic neoplasms, benign and malignant
| Benign | Malignant | ||
| Primary | Secondary | ||
| Children | |||
| Ocular | — | Retinoblastoma | Leukemia |
| Orbital | Capillary hemangioma | Rhabdomyosarcoma | Leukemia |
| Adult | |||
| Ocular | Choroid nevus | Uveal melanoma | Metastasis (lung, breast) |
| Orbital | Cavernous hemangioma | Lymphoma | Sinus cancer |
| Lids | Chalazion | Basal cell carcinoma | Lymphoma |
Pediatric ophthalmic oncology: ocular diseases
Benign disease
Benign pediatric ocular lesions are very rare. Choroidal nevi, which are present in more than 10% of the adult population, are rare before puberty and are never seen in the infant. Conjunctival and iris nevi are also extremely rare in prepubertal children. Iris nevi detected in children often represent Lisch nodules, a manifestation of neurofibromatosis type 1.
Benign retinal tumors are also rare. When found they are usually astrocytic hamartomas and are frequently part of the tuberous sclerosis syndrome. When viewed with indirect ophthalmoscopy, astrocytic hamartomas usually have a thin, transparent membrane overlying the retina and typically obscure retinal blood vessels. They may enlarge and calcify with time. They may be confused with myelinated nerve fibers, which are white, follow the distribution of the nerve fiber layer, and obscure retinal vessels.
Hamartomas of the retinal pigment epithelium are rare in children. They are frequently near the optic disc and are pigmented, with distortion of retinal vessels and a slightly opaque appearance. They have no malignant potential.
Primary malignant disease (retinoblastoma)
Introduction to retinoblastoma
The most common primary ocular malignancy of childhood is retinoblastoma.1 Retinoblastoma arises from a cone precursor cell.2 Although retinoblastoma is relatively rare, it has been the subject of great interest because of its well-studied genetic inheritance pattern and molecular biology.
Retinoblastoma has an incidence rate of 1 in 18,000–30,000 live births worldwide. Surveys suggest a relatively constant occurrence in this century. The incidence in the United States is relatively low, at 3.58 cases for each million children under the age of 15 years, and decreases with advancing age. The overall median age at diagnosis in the United States is 18 months, with the median age of diagnosis of bilateral cases occurring at 12 months and of unilateral cases at 24 months. In rare instances, retinoblastoma is detected prenatally via ultrasound or during adulthood.
Survival rates for retinoblastoma patients in the developed world have increased dramatically over the past century. The mortality of retinoblastoma was reported as 83% in 1897 in children who were treated with enucleation, and as 43% in all children in 1916.3 In contrast, cancer registry reports in Europe and the United States have demonstrated 5-year survival rates of 90% and 98%, respectively. The improved survival rate is due to earlier detection of the tumor and improved techniques for local tumor control. In contrast to developed countries, however, developing nations report dramatically low survival rates, as patients in these countries typically present with widespread metastatic disease. Worldwide approximately 50% of patients still die of metastatic retinoblastoma.
There are no differences in incidence by sex, race, or right versus left eye. Some data suggest geographic clustering, but convincing evidence is lacking. Retinoblastoma does appear to occur more commonly in poor patients worldwide.
Molecular biology of retinoblastoma
The traditional view of retinoblastoma genetics, widely held until recently, was that the disease occurs in two forms, germinal and nongerminal. Both forms occur as a result of loss or mutation of both alleles of the retinoblastoma gene (rb1). In nongerminal cases, both rb1 alleles are inactivated somatically in a single developing retinal progenitor cell, whereas in germinal cases, the first mutation occurs in the germline and only the second mutation is somatic. Nongerminal retinoblastoma is always unilateral and unifocal, although the tumor may break apart resulting in hundreds of tiny intraocular seeds. Recent evidence has indicated that nearly all retinoblastoma patients probably demonstrate a degree of mosaicism for the rb1 mutation. Furthermore, evidence suggests that genomic instability, microsatellite instability, defects of the DNA mismatch repair system, and alterations in DNA methylation and acetylation/deacetylation may also be necessary for the malignant transformation of retinoblastoma after the loss of pRB.4
The proposed retinoblastoma gene was localized to chromosome 13ql4 through deletion studies and linkage analysis. The protein is a regulator at the cell cycle checkpoint between G1 and entry into the S-phase (Figure 1). Loss of normal rb1 function, as in the case of the tumors, allows for uncontrolled entry into the S-phase and more rapid cell division. Recently, it was discovered that Rb1 wild-type tumors may occur in the context of MYCN oncogene amplification.5
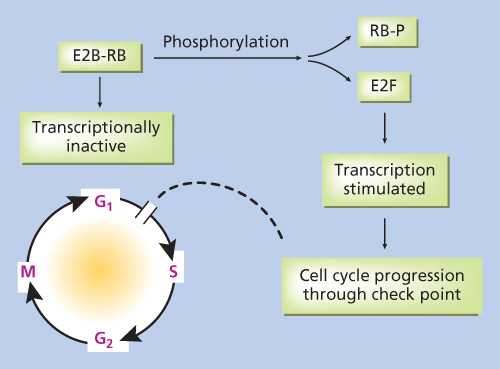
Figure 1 Artistic rendition of molecular mechanism of retinoblastoma gene action.
Rb1 was the first tumor suppressor gene to be identified. The tumor-suppressive function of the rb1 gene was confirmed in studies that demonstrated the loss of both alleles of the gene in tumor tissue specimens. Later studies showed that a germinal rb1 mutation was present in virtually all retinoblastoma kindreds and that inheritance of the mutant rb1 allele predicted disease.6 The tumor-suppressive function of the gene has furthermore been demonstrated in transfection studies showing that the introduction of wild-type expression in pRB-defective cell lines partially reverses the malignant phenotype.
Genetic testing
Both population data gathered from families and our current understanding of the molecular mechanisms of inheritance of the disease have enabled us to predict the likelihood that new offspring in families affected by retinoblastoma will develop the disease (Table 2). Karyotypic studies, which analyze the morphology of entire chromosomes, are generally not useful for the clinical diagnosis of retinoblastoma because they can only identify deletions spanning 2–5 million base pairs and only 3–5% of retinoblastoma patients carry deletions this large.7 Instead, more sophisticated indirect and direct techniques that detect smaller mutations are used. At the present time, a single genetic test is unlikely to detect all germline RB gene mutations in patients with retinoblastoma because of the variety of types and locations of mutations that occur. However, adaptation of a routine clinical protocol including a series of complementary tests based on the observation that most mutations alter the protein size and disrupt the large pocket domain may be able to rapidly detect the majority of mutations.8
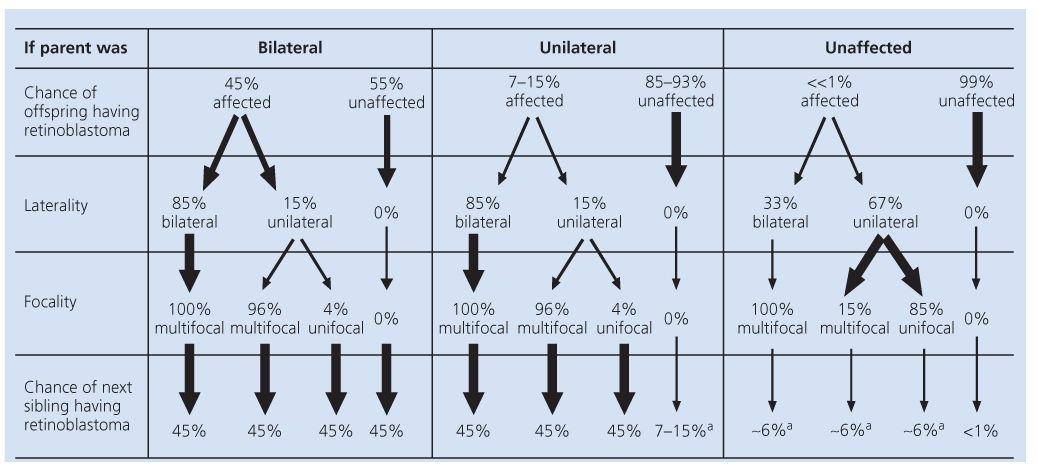
a If parent is a carrier, then the chance of next sibling having retinoblastoma is 45%.
Presenting signs and symptoms of retinoblastoma
The most common presenting signs and symptoms of retinoblastoma vary depending on the socioeconomic conditions in which the patient presents. In developing countries, children have often developed extraocular disease before they are diagnosed and frequently present with proptosis and an orbital mass (Figure 2). These children are older at diagnosis (age 4–6 years) compared patients in the United States and few survive. Many large retrospective studies over the past quarter-century have examined the most common presentations of retinoblastoma in large developed nations.8 In the United States, the most common presenting sign (60% of cases) is leukocoria, a white pupillary reflex (Figure 3). The reflex is caused either by the reflection of incoming light off the tumor or by the retinal detachment caused by the underlying tumor.
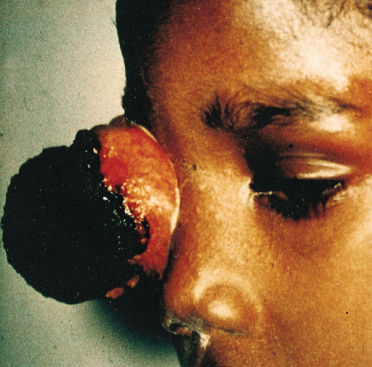
Figure 2 Advanced orbital presentation of retinoblastoma.
Source: Courtesy of A. Wachtel, M.D., Lima, Peru.
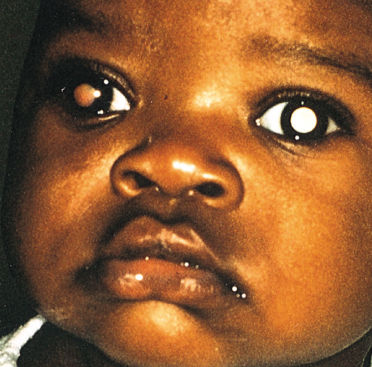
Figure 3 Leukocoria (white pupillary reflex) caused by retinoblastoma. The tumor can be seen in the vitreous. There are seeds in the anterior chamber, anterior to the iris.
Presenting signs in the United States that occur less commonly include strabismus (misalignment of the eyes), inflammatory signs (mimicking orbital cellulitis), anisocoria (different sized pupils), heterochromia (different colored irides), hyphema (blood in the anterior chamber), tumor hypopyon (tumor in the anterior chamber), and nystagmus. Two large retrospective studies on large populations of retinoblastoma patients have recently examined the patterns of detection of retinoblastoma in the United States. The vast majority of patients’ disease was discovered by a family member (80%) rather than by a pediatrician (8%) or an ophthalmologist (10%).8
Diagnostic testing
The differential diagnosis of retinoblastoma includes lesions that can simulate a solitary ocular tumor such as astrocytic hamartomas and toxocara canis and lesions that can cause a total retinal detachment such as Coats’ disease, retinopathy of prematurity, and persistent hyperplastic primary vitreous [persistent fetal vasculature (PFV) syndrome] (Table 3). Patients suspected of having retinoblastoma should undergo indirect ophthalmoscopy and fundus photography as well as ophthalmic ultrasonography. Ultrasonography is useful as it demonstrates masses with high reflectivity that block sound, causing characteristic shadowing behind the tumor. Needle biopsies are rarely performed for suspected retinoblastoma, as puncturing the eye and aspiration can lead to tumor seeding with orbital invasion and even death from metastatic disease.
Table 3 Lesions simulating retinoblastoma
| Solitary ocular tumor |
| Astrocytic hamartoma |
| Toxocara canis |
| Total retinal detachment |
| Coats’ disease |
| Retinopathy of prematurity |
| Persistent fetal vasculature (PFV) |
Staging of intraocular retinoblastoma
The Reese–Ellsworth classification scheme was the worldwide gold standard for describing intraocular tumors (Table 4a).9 It is not a true staging scheme (for untreated patients do not progress from group I to higher groups), but it has served as an excellent ocular reference for comparison of different series and treatment schemes. A higher numeric classification signified that a tumor was more anterior and that there was a decreased success rate in treating the lesion with lateral port external-beam radiation. Its usefulness has been questioned by many ophthalmic oncologists, who feel that it is no longer appropriate given the current trend away from the use of external-beam radiation. An alternate classification system was developed and has been used clinically by several centers in a collaborative study (Table 4b).10 It has been shown to correlate with the response to treatment of intraocular disease with systemic chemotherapy combined with focal therapy. As with the Reese–Ellsworth classification, this system may also become antiquated as treatment trends inevitably change in the future.
Table 4 (a) Reese–Ellsworth scheme for intraocular retinoblastoma. (b) New international classification for retinoblastoma
| (a) | |
| Group I |
|
| Group II |
|
| Group III |
|
| Group IV |
|
| Group V |
|
| (b) | |
| Group A | Rb ≤ 3 mm in basal dimension or thickness |
| Group B | Rb > 3 mm or with one or more of the following:
|
| Group C | Retinoblastoma tumor with one of the following:
|
| Group D | Retinoblastoma tumor with one of the following:
|
| Group E | Extensive retinoblastoma occupying >50% of globe or any of the following:
|
Treatment of intraocular retinoblastoma
Survival rates in excess of 95% are currently achieved for most patients treated at major centers in developed countries with a multimodal treatment approach that may include enucleation, external-beam radiation, brachytherapy, transpupillary thermotherapy (TTT), cryotherapy, and chemotherapy (systemic, intra-arterial, periocular, and intravitreal). The treatment strategy for each patient depends on several factors: involvement of one or both eyes, heredity of the disease, age of the patient, tumor volume and localization, stage of disease (including Reese–Ellsworth classification), and presence of extraocular disease.1
Enucleation
Enucleation is surgical removal of the eye without resecting the lids or extraocular muscles.1 Patients considered for enucleation include those with advanced retinoblastoma in one or both eyes, active tumor in a blind eye, and painful glaucoma from tumor invasion.
External-beam radiation
External-beam radiation, once employed in many patients with intraocular retinoblastoma, has fallen out of favor due to its contribution to the development of second nonocular cancers in germinal retinoblastoma patients.1 When radiation is given, it is often used as a last resort salvage therapy after other therapies have failed.
Transpupillary thermotherapy (TTT)
TTT is a treatment modality for retinoblastoma that is delivered using modifications to the hardware and software of the infrared diode laser.1 TTT is used to treat selected small retinoblastomas, typically those located in the posterior aspect of the globe. A study examining the use of TTT as single modality therapy for small tumors found that 92% of tumors 1.5 disc diameters or less in base diameter were successfully cured with TTT alone.
Cryotherapy
Cryotherapy is used as a primary treatment for small peripheral retinoblastomas or as adjuvant or secondary treatment for tumors treated by other modalities.1 Rapid freezing (−90°C per minute) results in intracellular ice crystal formation, protein denaturation, pH changes, and finally cell membrane rupture. All studies examining the use of cryotherapy alone have demonstrated 90–100% cure rates in tumors that are <3 mm in diameter and <1 mm in thickness with minimal complications.
Brachytherapy
Episcleral brachytherapy was pioneered in 1933 by the British ophthalmologist Henry Stallard. Radioactive plaques can be used as primary therapy and their relative indications include tumors that are classified as Reese–Ellsworth stage IVa or less and tumors that are between 4 and 10 disc diameters in size. Brachytherapy can also be used as a salvage technique in eyes that have failed other types of therapy including intra-arterial chemotherapy, external-beam radiation, photocoagulation, or cryotherapy. 125Iodine is currently the most commonly used isotope in brachytherapy for retinoblastoma. This isotope is advantageous because the radioactive seeds can be placed into a custom-built plaque designed to match the size of the lesion.
Systemic chemotherapy
Chemoreduction can be used to shrink these tumors so that focal treatments such as cryotherapy, thermotherapy, or radioactive plaques can be administered afterword.1 Most studies of chemoreduction for retinoblastoma since 1996 have utilized vincristine, carboplatin, and an epipodophyllotoxin, either etoposide or teniposide. The addition of cyclosporine as a P-glycoprotein inhibitor has also been suggested to decrease multidrug resistance.11 Choice of agents and number and frequency of cycles vary currently at different institutions.
For patients with Reese–Ellsworth groups I–III eyes patients, many authors have demonstrated that enucleation can be successfully avoided almost 100% of the time. Results for patients with Reese–Ellsworth groups IV and V eyes have been more discouraging. In a meta-analysis of patients treated with chemoreduction, 37% of eyes avoided both external-beam radiation and enucleation. Forty-one percent of eyes required radiation but avoided enucleation, and 40% of eyes required enucleation (with or without prior radiation).12 Owing to concerns of short- and long-term toxicity, many centers have abandoned systemic chemotherapy as primary treatment of retinoblastoma in favor of more focal chemotherapy techniques.
Intra-arterial chemotherapy
Clinicians in Japan were the first to postulate that another method of local drug delivery, intra-arterial infusions of chemotherapy, may increase penetration of the drug into small intraocular tumors and decrease systemic side effects.13 Our group introduced a technique in which the ophthalmic artery is selectively cannulated and a combination of up to three drugs (melphalan, topotecan, and carboplatin) are injected at the ostium.1 Other techniques to access the ophthalmic artery include catheterization of the middle meningeal artery or the use of a balloon. Treatment typically consists of monthly infusions given an average of three times. This technique has been adopted by many centers worldwide and is our primary method of treatment for both unilateral and bilateral retinoblastoma. It can be used to treat eyes of all classifications, whether naïve or prior treated, and bilateral cases can be infused in the same session with tandem therapy. In children <3 months of age or 6 kg, bridge therapy is employed: single-agent carboplatin is given until the child reaches an adequate age and weight for intra-arterial chemotherapy. Two-year Kaplan–Meier estimates of ocular event-free survival are estimated at 82% for naïve eyes and 60% for prior treated eyes,14 although with the recent adoption of intravitreal chemotherapy, these numbers are likely higher (Figure 4).
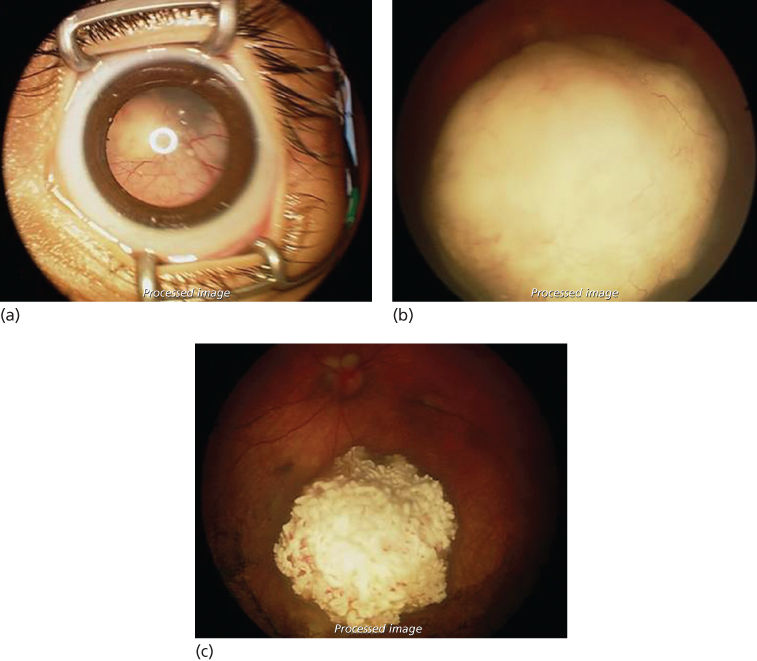
Figure 4 (a) An example of the typical patient undergoing intra-arterial chemotherapy. The globe is full of retinoblastoma with a total retinal detachment visible behind the lens. (b) Fundus photograph before intra-arterial chemotherapy. Note that the large tumor is obscuring the optic nerve head. (c) Fundus photograph after intra-arterial chemotherapy. The tumor has become calcified and there are no vitreous seeds visible. The optic nerve head can now be visualized above the tumor. The retina is flat.
Ocular side effects are minimal and may include periocular inflammation, medial forehead erythema, and chorioretinal changes.14 Electroretinogram monitoring of retinal function demonstrates that carboplatin and topotecan have no impact on retinal function; and while melphalan may have an effect, these changes are minimal and likely clinically irrelevant. Approximately 30% of children have at least one episode of grade 3 or 4 hematotoxicity during their treatment course (particularly when melphalan dose exceeds 0.4 mg/kg) and therefore monitoring of blood counts is advised. Vascular complication is a rare complication and no death has been reported from this procedure.
Periocular chemotherapy
Periocular injections of carboplatin or topotecan have been used, particularly as salvage or adjuvant treatment. However, side effects, particularly with carboplatin, include scarring and loss of vision. Furthermore, its long-term efficacy of this delivery method has been questioned.
Intravitreal chemotherapy
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


