Neoplasms of the exocrine pancreas
Robert A. Wolff, MD Christopher H. Crane, MD
Christopher H. Crane, MD  Donghui Li, PhD
Donghui Li, PhD  Douglas B. Evans, MD
Douglas B. Evans, MD  Anirban Maitra, MD
Anirban Maitra, MD  Susan Tsai, MD
Susan Tsai, MD
Overview
Adenocarcinoma of the pancreas is one of the most morbid and lethal malignancies. Worldwide, it accounts for 330,000 deaths annually. Smoking and obesity are both risk factors for pancreatic cancer and current estimates suggest half of all cases may be preventable. Genetic predisposition to pancreatic cancer is associated with some genetic syndromes to include BRCA1 or BRCA2 germline mutations, Peutz–Jeghers syndrome, and hereditary nonpolyposis colon cancer. Somatic mutations in KRAS, CDKN2A/INK4A, TP53, and SMAD4/DPC4 are common. Pancreatic cancer is often diagnosed when the primary tumor is inoperable or there is metastatic spread rendering the cancer incurable. At present, clinical staging defines the malignancy as resectable (and potentially curable), borderline resectable, locally advanced, or metastatic. Surgery with curative intent is possible for patients with resectable disease and for a subset of patients with borderline resectable disease. While chemotherapy has been proved to prolong survival as a component of adjuvant therapy, and as therapy for locally advanced or metastatic disease, the role of radiation therapy in treatment is less-well defined and not universally accepted. Thus far, molecular targeted therapy has shown no clinically meaningful benefit. Adjuvant and neoadjuvant approaches are employed in patients with resectable and borderline resectable disease. Intense research in the molecular biology of pancreatic cancer continues with increasing interest in the role of the tumor microenvironment in invasion, metastatic potential, and resistance. Novel therapeutic strategies that focus on stromal modification and immunologic manipulation are at the forefront of clinical and translational research efforts.
Introduction
In common parlance, the moniker of “pancreatic cancer” most often refers to the entity known as pancreatic ductal adenocarcinoma (PDAC). This is one of the most lethal and morbid of all solid tumors, notorious for local invasiveness, early spread to regional lymph nodes, and vascular dissemination to distant sites. Cure is possible only in the setting of localized, resectable disease with relapse after surgery occurring in about 80% of patients. PDAC is currently the fourth leading cause of cancer-related death in the United States.1 In 2015, PDAC will lead to about 40,500 deaths.1 Worldwide, it is blamed for over 330,000 deaths annually,2 and by 2020, PDAC is predicted to become the second leading cause of cancer death by 2020.3
In this chapter, we will summarize the current literature relevant to the epidemiology, pathology, molecular and cellular biology, and diagnosis and treatment of pancreatic cancer. Future directions in prevention, early detection, clinical management and translational research in genomics, epigenetics, tumor metabolism, and the microenvironment will also be discussed.
Epidemiology
Worldwide, pancreatic cancer is the thirteenth most common type of cancer and the eighth leading cause of cancer-related death.2 The incidence rates of pancreatic cancer are the highest in industrialized societies and western countries.4 The age-adjusted incidence rates range from 10 to 15 per 100,000 people in parts of Northern, Central, and Eastern Europe to <1 per 100,000 in areas of Africa and Asia.5 In the United States, the age-adjusted incidence rate for all races was 13.0 per 100,000 men and 10.3 per 100,000 women per year based on SEER’s (Surveillance Epidemiology and End Results) 2001–2005 data.6 The age-adjusted mortality rate per 100,000 person-years was 15.4 for black men, 12.4 for black women, 12.1 for white men, and 9.0 for white women. The black/white disparity in incidence rate is likely related to environmental influences as the rates are considerably higher in African-Americans than in native Africans.
Most patients are diagnosed between the ages of 60 and 80 years.4, 6 Historically, pancreatic cancer had a male: female ratio of 1.2 : 1, now the incidence is about the same for both sexes.4
Etiologic factors
As most other types of human cancers, pancreatic cancer etiology involves both genetic and nongenetic factors (Figure 1).
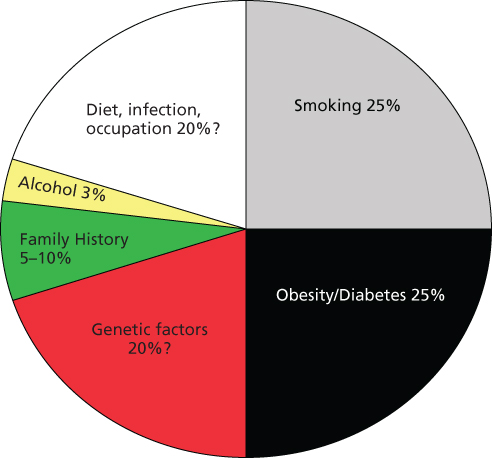
Figure 1 Schematic presentation of known and suspected risk factors for pancreatic cancer.
Hereditary risk factors
Inherited or familial pancreatic cancers represent approximately 5–10% of all pancreatic cancers.7 At-risk patients for familial pancreatic cancer include those with a minimum of two first-degree relatives with pancreatic cancer.8 Although the genetic mutations responsible for the majority of clustering of pancreatic cancer in families have not been identified, several pancreatic cancer genes that are associated with either a known cancer syndrome or chronic inflammatory diseases have been identified (Table 1).7
Table 1 Susceptibility genes for pancreatic cancer
| Gene | Syndrome | Chromosome location | Function |
| PRSS1 SPINK1 | Hereditary pancreatitis | 7q35 5q32 | Trypsinogen, serine protease Serine peptide (trypsin) inhibitor |
| CFTR | Cystic fibrosis | 7q31.2 | ABC transporter chloride channel |
| BRCA1 BRCA2 | Hereditary breast ovarian cancer | 17q21 13q12.3 | DNA damage repair |
| CDKN2A/P16 | Familial atypical multiple mole melanoma | 9p21 | Cyclin-dependent kinase inhibitor |
| STK11/LKB1 | Peutz–Jeghers syndrome | 19p13.3 | Serine/threonine kinase |
| MSH2, MLH1 PMS1/2, MGH6 | Hereditary nonpolyposis colorectal cancer | 2p21 3p21.3 | Mismatch repair |
| APC | Familial adenomatous polyposis | 5q21 | Negative regulator of β-catenin |
| PALB2 | Breast and pancreatic cancer | 16p12.2 | Partner and localizer of BRCA2 |
| ATM | Ataxia-telangiectasia | 11q22–q23 | DNA damage response |
PRSS1 and SPINK1
Germ line mutations in PRSS1 and SPINK1 genes are associated with hereditary pancreatitis. PRSS1 gene encodes the cationic trypsinogen protein and premature activation of the trypsinogen results in acute pancreatitis. SPINK1 gene encodes a trypsin inhibitor and mutation of this gene has been associated with chronic pancreatitis. Individuals with hereditary pancreatitis have been shown to have a 53-fold increased risk for developing pancreatic cancer and a lifetime risk (age 70) of pancreatic cancer of 30–40%.7
BRCA1 and BRCA2
Hereditary breast–ovarian cancer syndrome is associated with mutations in BRCA1 or BRCA2. The risk of pancreatic cancer among BRCA1 mutation carriers varied between studies.7 Germ line mutations in the BRCA2 gene have been found in 10% of patients with familial pancreatic cancer and in 7% of patients with sporadic pancreatic cancer.9–11 Germ line BRCA2 mutations represent the most common inherited predisposition to pancreatic cancer and are associated with an up to a 10-fold greater risk of pancreatic cancer than in the general population.12
LKB1/STK11
Peutz–Jeghers syndrome, an autosomal dominant trait, is caused by mutations in the LKB1/STK11 tumor suppressor gene. Individuals with Peutz–Jeghers syndrome have a highly increased risk of pancreatic cancer ranging from 96- to 132-fold higher risk than average risk individuals; the lifetime risk is 5–36%.13, 14 Biallelic inactivation of the LKB1/STK11 gene has also been found in 4% of patients with resected sporadic PDAC.15
CDKN2A (p16)
Germ line mutations of the CDKN2A (p16) tumor suppressor gene are mostly related to familial atypical multiple-mole melanoma (FAMMM) syndrome. It has been reported that individuals from FAMMM kindreds have a 13- to 22-fold increased risk of developing PDAC16 and individuals who carry p16/CDKN2A mutations have a 38-fold increased risk compared with the general population.17 CDKN2A gene is also frequently inactivated in sporadic pancreatic cancer.
HNPCC and FAP
The hereditary nonpolyposis colon cancer (HNPCC, also known as Lynch syndrome), is caused by germ line mutations in several DNA mismatch repair genes.18 Families with Lynch syndrome have an 8.6-fold increased risk of developing pancreatic cancer.19 Families with familial adenomatous polyposis (FAP), which is caused by genetic defect in APC tumor suppressor gene, have also been reported to have increased risk of PDAC.20
PALB2 and ATM
Mutations of the PALB2 and ATM genes have been discovered in familial pancreatic cancer cases.21–23 PALB2 protein binds with BRCA2 protein stabilizing it in the nucleus, and this complex acts in double stranded DNA repair. ATM is an important regulator of cellular response to DNA damage. It is estimated that PALB2 mutations may account for 1–3% of the familial clustering of pancreatic cancer.21, 22
A recent study conducted in 727 unrelated familial pancreatic cancer probands has found that the prevalence of deleterious mutations was BRCA1, 1.2%; BRCA2, 3.7%; PALB2, 0.6%; and CDKN2A, 2.5%. BRCA2 and CDKN2A account for the majority of mutations in familial pancreatic cancer.24
Susceptibility variants
Three genome-wide association studies (GWAS) have been conducted in the United States with a majority of the study population of European ancestry.25–27 Polymorphic variants of the ABO, NR5A2, LINC-PINT, PDX1, BCAR1/CTRB1/CTRB2, ZNRF3, and TERT genes and a nongene chromosome region 13q22.1 have been identified as susceptibility factors for pancreatic cancer with genome-wide significance (p < 5 × 10−8). Two additional GWAS have also been conducted in Chinese28 and Japanese populations.29 Post GWAS analyses suggest that genes involved in the pancreas development pathway may play an important role in modifying the risk of pancreatic cancer.30–32 Initial examination on the interactions of genes with known etiological factors, such as smoking,33 obesity, and diabetes,34, 35 has generated interesting clues to the genetic factors that predispose individuals with such exposure to pancreatic cancer. Identification of the causal alleles and functional characterization of the genes or variants may involve many genes with modest effect on the phenotype.36, 37
Tobacco and alcohol
The risk factor most firmly associated with pancreatic cancer is cigarette smoking. It is estimated that approximately 25% of cases of pancreatic cancer are due to cigarette smoking with a 1.5- to 2.0-fold increased risk.38–41 The risk increases as the amount and duration of smoking increase. Long-term smoking cessation (>10 years) reduces the risk by approximately 30% relative to current smokers.38, 42 Whether smokeless tobacco products, pipe or cigar smoking increases risk remains controversial.43, 44
The mechanisms of smoking-induced pancreatic cancer involve tobacco carcinogen-induced DNA damage and pancreatic tissue injury.45 Tobacco carcinogens can be metabolically activated to cause DNA damage and gene mutations.46, 47 Nicotine also plays an important role in tumor promotion and progression via nicotinic acetylcholine receptor and adrenergic receptor-mediated signaling in multifactorial events that lead to pancreatic injury.45, 46, 48
The association between alcohol consumption and PDAC has also been investigated. In general, heavy alcohol consumption (30–40 g alcohol or ≥3 drinks/day) but not light or moderate consumption has been associated with increased risk.49–52 It appears that alcohol may account for 2–5% of all pancreatic cancer.
Obesity
Similar to tobacco, it has been estimated that obesity accounts for another 25% of the pancreatic cancer cases in the US population.53 Positive associations between obesity and pancreatic cancer risk have been reported in several large prospective studies and meta-analyses.54–62 In addition to BMI (body mass index), abdominal fat, a measured by waist to hip ratio, has been related to increased risk of pancreatic cancer, especially in women.60, 61 Obesity at younger age has been associated with a higher risk of pancreatic cancer than weight gain at older age. Furthermore, obesity has been associated with a younger age of disease onset and reduced overall survival (OS).63
Biological mechanisms linking obesity to pancreatic cancer are insulin resistance and inflammation.64 Studies using prediagnostic blood samples have shown positive associations of PDAC risk and markers for hyperglycemia and insulin resistance.65–68 Insulin and insulin-like growth factors are key regulators for cellular metabolism and growth and their signaling transduction networks play an important role in tumor development and progression.69–71 The mechanisms driving pancreatic carcinogenesis may have similarities to other inflammatory conditions associated with an increased risk of cancer. Fatty infiltration of the pancreas may lead to steatopancreatitis, suggesting a potential link among obesity, nonalcoholic fatty pancreatic disease, nonalcoholic steatopancreatitis, and PDAC.72, 73
Diabetes mellitus and antidiabetic medication
Diabetes mellitus has been implicated as both an early manifestation of pancreatic cancer and a predisposing factor.74 A consistent association of long-term diabetes and risk of pancreatic cancer has been reported from three large-scale meta-analyses75–77 and three pooled data analyses68, 78, 79 with each involved more than 1500 PDAC cases. Of note, PDAC can induce peripheral insulin resistance80 and type 3c (pancreatogenic) diabetes.81 It was observed that hyperglycemia could occur 2 years before clinical diagnosis of pancreatic cancer, and one in 125 new-onset diabetics would develop PDAC in 3 years.82, 83 Biomarkers that could help to distinguish type II diabetes mellitus from type 3c diabetes are being pursued.84, 85
Since the discovery that biguanides, the most commonly used antidiabetic medications, inhibited pancreatic tumor development in experimental animals,86 the role of antidiabetic therapy in PDAC risk reduction has been intensely studied. Other experimental evidence supports the antitumor effect of metformin.87, 88 Metformin has diverse regulatory effects and the major molecular targets of metformin are the liver kinase B1 (LKB1)-AMP-activated protein kinase (AMPK) signaling and mammalian target of rapamycin (mTOR) pathways, which are central in the regulation of cellular energy homeostasis, cell division, and cell proliferation.89 Metformin also inhibits the inflammatory response associated with cellular transformation and selectively targets the cancer stem cell.90, 91 In humans, eight cohort studies92–98 and three case–control studies99–101 have examined the impact of metformin use on risk of PDAC. Meta-analysis of these studies estimates RR of 0.63.102 Two additional studies have reported that metformin use in patients with pancreatic cancer and concurrent diabetes is associated with longer survival and reduced risk of death.103, 104
Pancreatitis and other medical conditions
Chronic pancreatitis is a risk factor for pancreatic cancer, and new onset pancreatitis could be a sign of occult pancreatic cancer.105 Data pooled from 10 case–control studies with adjustment for obesity and alcohol consumption, demonstrated that the risk of pancreatic cancer was nearly threefold for patients with an interval of >2 years between the pancreatitis and pancreatic cancer diagnoses.106
Infectious diseases
Helicobacter pylori may cause subclinical pancreatitis or increased gastrin levels, with resultant trophic effects on the pancreas. Several studies have examined the association between H. pylori and pancreatic cancer; with large meta-analyses being showing a weak yet statistically significant association between prior H. pylori infection and pancreatic cancer.107 It has been hypothesized that ABO genotype/phenotype status influences the behavior of H. pylori that in turn affects gastric and pancreatic secretory function, and these ultimately influence the pancreatic carcinogenicity of dietary- and smoking-related N-nitrosamine exposures, and thus risk of PDAC.108
Hepatitis B may also be a risk factor for pancreatic cancer.109 Subsequent studies were conducted mostly in Asian and a meta-analysis of nine such studies reported that the pooled RR was 1.39 (95% CI: 1.22–1.59) in chronic HBV (hepatitis B virus) carriers and 1.41 (95% CI: 0.06–1.87) in past exposure to HBV.110 Hepatitis B surface antigen (HBsAg) has been detected in pancreatic and bile juice,111 and there is evidence of HBV replication in pancreatic cells. HBV results in damage to pancreatic exocrine and endocrine epithelial cells via an inflammatory response.112, 113
Dietary factors
Various dietary factors have been suspected to play a role in the development of pancreatic cancer. Generally, high intakes of fat or meat increase the risk; whereas high intakes of fruits and vegetables reduce the risk.5 Less is known about which type of fatty acids pose the greatest risk.114 Several studies suggest that the method of meat preparation (deep fried, grilled, or barbequed), and subsequent intake of food mutagens may contribute to the development of pancreatic cancer.115–117
The associations of dietary carbohydrates, refined sugars, and glycemic index or load with pancreatic cancer have been investigated in many studies but the results are inconsistent. A recent meta-analysis of data from eight prospective cohort studies concluded that there was no association among diets high in glycemic index, glycemic load, total carbohydrates or sucrose, and pancreatic cancer risk.118
Decreased pancreatic cancer rates have been associated with the high consumption of vegetables, citrus fruits, fiber, and vitamin C.5 It remains uncertain whether vitamin D has a protective effect on PDAC.114
Occupational exposures
The role of occupational or industrial factors in pancreatic cancer has been investigated extensively. Increased risk of pancreatic cancer has been associated with exposures to some chemicals (e.g., organochlorines, chlorinated hydrocarbons, and formaldehyde) or some specific occupations (e.g., stone miners, cement workers, gardeners, and textile workers).4, 88 The strongest and most consistent occupational risk factors associated with pancreatic cancer to date are chlorinated hydrocarbons and polycyclic aromatic hydrocarbons.88
In summary, cigarette smoking and obesity may each be responsible for as many as 25% of the cases of pancreatic cancer implying that half of pancreatic cancer cases may be preventable.119, 120 Conversely, the proportion of pancreatic cancer cases attributable to known inherited pancreatic cancer syndromes is small. Identification of individuals with excessively high risk of pancreatic cancer may allow for focused screening.121–123 Thus far, proved benefit to screening remains elusive, except in highly selective populations such as high-risk familial pancreatic cancer kindreds.124 Currently, for those individuals determined to have structural,124 or blood-based abnormalities,125 suggesting premalignancy or early stage neoplasm, management remains extremely controversial. Current options range from close observation to aggressive surgical intervention with a variety of approaches in evolution.126–128
Prevention of pancreatic cancer
The epidemiology of PDAC described earlier provides strong evidence for the prevention of pancreatic cancer; possibly resulting in a substantial reduction in global pancreatic cancer mortality. Primary prevention strategies expected to significantly reduce the risk of developing pancreatic cancer likely require lifestyle and dietary modifications. In addition, a number of available over the counter agents and widely prescribed medications have potential to reduce the risk of pancreatic cancer (Table 2).129–132
Table 2 Interventions that may reduce pancreatic cancer risk
| Lifestyle and nutritional factors |
| Cessation/avoidance of tobacco smoking and smokeless tobacco Regular exercise Weight control and weight reduction if needed to BMI < 25 kg/m2 Healthy diet high in fruits and vegetables |
| Potential chemopreventive agents |
| Metformin Aspirin Curcumin Celecoxib Atorvastatin and other HMG-coenzyme reductase inhibitors β-Carotene Vitamins C, D, and E |
Pathology
Histopathology of exocrine pancreatic neoplasms
PDAC accounts for approximately 95% of all neoplasms arising in the pancreas.133 On histological examination of surgically resected tumors, it is not uncommon to find neoplastic cells infiltrating into lymphatic structures or into the many nerve endings enervating the pancreatic bed, which may explain the pain associated with PDAC. Another defining histological feature of PDAC is an exuberant host stromal reaction to the cancer (desmoplasia), wherein smooth muscle actin expressing myofibroblasts generate abundant extracellular collagen that surrounds infiltrating neoplastic glands (Figure 2).134 Other components of this host desmoplastic response include secreted glycosaminoglycans (e.g., hyaluronan) that contribute to high interstitial pressures and impede the passive efflux of chemotherapeutics from the circulation into the peritumoral milieu.135
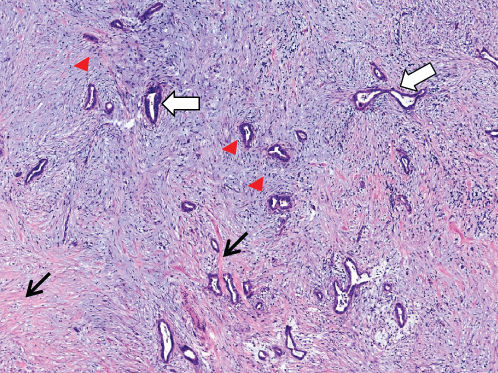
Figure 2 Stromal desmoplasia in pancreatic ductal adenocarcinoma. Infiltrating neoplastic glands (white arrows) are surrounded by a florid host response, comprised spindle-shaped cancer-associated fibroblasts and myofibroblasts (red arrowheads) and extracellular matrix, including collagen 1 (black arrow).
PDAC arises through a stepwise histological progression of well-recognized precursor lesions. The most common precursor lesions are microscopic, and known as pancreatic intraepithelial neoplasia or PanIN, which are almost always present in the pancreatic parenchyma surrounding an invasive cancer. PanINs vary from innocuous low-grade PanIN-1 lesions to the ominous highest grade PanIN-3 (previously known as carcinoma in situ), wherein the constituent cells morphologically resemble PDAC, except for their containment within a basement membrane. PanIN-3 lesions share many of the genetic aberrations found in infiltrating cancer. Of note, while PanIN-1 and -2 lesions can occur incidentally (with autopsy studies reporting as many as 50% of elderly individuals with low-grade PanINs in their pancreas),136 PanIN-3 is essentially never found in the absence of a synchronous PDAC.137 Thus, the emergence of high-grade PanIN-3 lesions likely represents the critical inflection point in the multistep progression of PDAC. By definition, and in contrast to cystic precursor lesions of PDAC (see below), isolated PanINs are not seen on abdominal imaging, although individuals with germ line mutations or a familial predisposition may have structural pancreatic changes discernible on endoscopic ultrasound (EUS).128
Cystic precursor lesions of PDAC comprised two distinct entities—intraductal papillary mucinous neoplasms (IPMNs) and mucinous cystic neoplasms (MCNs).138 IPMNs arise in either the main duct or one of the side branches of the pancreas and are characterized by broad “finger-like” papillae and secretion of abundant extracellular mucin. MCNs are less common and occur in women with significantly greater propensity than men (9 : 1 ratio). In contrast to IPMNs, MCNs have no communication with the ductal system. Akin to PanINs, the epithelial lining of cystic precursor lesions also demonstrates histological progression from low-grade (adenoma) through high-grade (carcinoma in situ) to invasive cancer. Approximately 25–33% of surgically resected cystic lesions are associated with an invasive adenocarcinoma, and the onset of invasion is associated with a significant reduction in median OS.139, 140 The precise timeline of progression from noninvasive cyst to invasive carcinoma is not known, although the median age of presentation for cystic lesions is about a decade earlier than the median age for PDAC, suggesting that this might reflect the period required for progression. Of note, the explosion in the use of abdominal imaging for symptoms not related to the pancreas has led to a veritable “man-made” epidemic of incidentally discovered cystic lesions of the pancreas. As the overwhelming majority of these asymptomatic cysts are not likely to progress to invasive cancer, radiological criteria have been proposed that help distinguish “high risk” from “indolent” cysts.141, 142 Intensive efforts are underway to identify molecular markers from cyst fluid or blood that would further refine this distinction and help prevent unnecessary surgeries and frequent monitoring.123
Other uncommon exocrine tumors include (1) acinar cell carcinoma, which bears morphological resemblance to pancreatic acini, wherein the neoplastic cells express acinar enzymes, such as trypsin and chymotrypsin;143 (2) solid pseudopapillary neoplasm, a cystic tumor of uncertain histogenesis that often arises in the tail of the pancreas in young adults, and typically has a favorable prognosis;122 (3) serous cystic neoplasm, a cystic tumor characterized by serous (watery) secretion, in contrast to the mucinous contents of IPMN and MCN, and which does not harbor a risk of progression to PDAC;144 and (4) pancreatoblastoma, the most common pancreatic tumor in children.145
Approximately 2% of primary tumors arise from the endocrine compartment (islets of Langerhans) and are known as pancreatic neuroendocrine tumors or PanNETs or “islet cell tumors.”146 PanNETs can be nonfunctional in up to a quarter of cases, or they can express a variety of islet cell hormones such as insulin or glucagon, which may lead to characteristic syndromes owing to systemic hormonal excess. PanNETs do not form glandular structures and are typically not associated with an exuberant host desmoplastic reaction. PanNETs can be readily identified based on expression of neuroendocrine markers such as chromogranin or synaptophysin in tissue sections.
Molecular pathology of exocrine pancreatic neoplasms
The advent of next-generation sequencing technology has enabled considerable advances over the last 5 years in elucidating the genomic landscape of PDAC,147 as well as that of other variant neoplasms of the exocrine and endocrine pancreas.148, 149 The International Cancer Genome Consortium (ICGC) has sequenced hundreds of PDAC, where the sequencing data is publicly available.150
By far, the most common genetic abnormality in the multistep pathogenesis of PDAC happens to be oncogenic mutation of KRAS, located on chromosome 12p, which encodes for Ras, a membrane-bound small GTPase.151 Somatic mutations of KRAS are reported in >90% of PDAC cell lines and patient-derived xenografts, although the frequency is somewhat lower (75–90%) in primary tumor samples. The majority of KRAS mutations cluster at codon 12, with mutations also found at codons 13 or 61, respectively. Somatic KRAS mutations result in constitutive activation of the GTPase activity of the encoded Ras protein, which, in turn, activates multiple downstream effector pathways.152 Somatic KRAS mutations are present even in the lowest grade PanIN lesions, underscoring its status as an early genetic “hit.”153 In genetically engineered mouse models (GEMMs), expression of a mutant KRAS in the pancreas results in preinvasive neoplasia closely recapitulating human PanINs, with a minority of animals progressing to full-fledged invasive cancer.154 Additional genetic events, such as expression of mutant p53 or biallelic deletion of the Ink4a/Arf locus, significantly accelerate the natural history of Ras-induced pancreatic neoplasia.155, 156 Conversely, in GEMMs with an inducible mutant KRAS allele, turning off Ras expression in established PDAC, leads to variable tumor regression.157, 158 Not surprisingly, a multitude of avenues have been explored toward pharmacological inhibition of aberrant Ras activity in PDAC and other KRAS-mutant tumors,159–161 but the clinical results overall have been disappointing. Nonetheless, Ras remains an attractive target, and a large federally funded “RAS Initiative” is currently underway at the National Cancer Institute to identify strategies to block Ras in PDAC and other cancers (http://www.cancer.gov/researchandfunding/priorities/ras).
Somatic inactivation of the CDKN2A/INK4A gene, which encodes for the cell cycle regulator p16, is another early genetic event in the multistep progression of PDAC. The p16 protein inhibits the cyclin D1/Cdk-4 complex that normally acts to constrain the retinoblastoma (Rb) protein via phosphorylation.162 In approximately 95% of PDAC, the CDKN2A/INK4A gene is inactivated by one of several mechanisms, including homozygous deletion, the loss of one allele combined with an intragenic mutation in the other allele, or epigenetic inactivation via promoter methylation. A potential therapeutic vulnerability that emerges in the setting of p16 loss pertains to the dependence on Cdk4 activity for unregulated proliferation, which can be targeted using small molecule inhibitors of Cdk4 function.163
In the multistep progression model of PDAC, loss of the TP53, SMAD4/DPC4, and BRCA2 tumor suppressor genes are considered later genetic events.164 Somatic mutations of TP53 are found in approximately 75% of human PDAC, while nuclear accumulation of p53 protein on immunohistochemistry, indicative of an underlying mutation, occurs in the majority of PanIN-3 lesions.165 The SMAD4/DPC4 tumor suppressor gene on chromosome 18q21 encodes for an intracellular transducer of the transforming growth factor-β signaling pathway that inhibits cell growth under physiological conditions. The SMAD4/DPC4 gene is inactivated in approximately 55% of human PDAC through either homozygous deletions or the loss of one allele coupled with an intragenic mutation in the second allele.166 Epithelial Dpc4 expression is nearly always retained in low-grade PanIN lesions and in noninvasive cystic precursor lesions of PDAC, reiterating that SMAD4/DPC4 abnormalities generally occur concomitant with progression to invasive cancer.167, 168 Overall, KRAS, CDKN2A/INK4A, TP53, and SMAD4/DPC4 are the “big four” peaks in the genomic landscape of PDAC, each mutated in over 50% of cases. Patients that harbor in all four genes tend to have a significantly worse OS than those with mutations in 1 or 2 of these genes, underscoring their role in prognosis.169
The BRCA2 gene, which has been discussed in the context of familial predisposition to PDAC, is also somatically mutated in approximately 5% of cancers. The BRCA2 protein plays a critical role in homologous recombination-mediated repair (HRR), and neoplastic cells with BRCA2 or related gene defects in the HRR pathway are exquisitely sensitive to double-strand breaks induced by agents such as cisplatin or mitomycin C.170 Another newer class of agents that is being selectively evaluated in tumors with BRCA2 mutations is PARP ([poly (ADP-ribose) polymerase]) inhibitors, which inhibits a second mechanism of DNA repair in cells already compromised in HRR. The concurrent PARP inhibition in BRCA2-mutant PDAC is expected to create a “synthetic lethality” vis-à-vis DNA repair that can be exploited toward a therapeutic advantage.171
Although a comprehensive tabulation of genetic abnormalities in PDAC is beyond the scope of this book, significant molecular aberrations are observed within every interrogated cellular component, including DNA methylation, histone marks, coding and noncoding RNAs (latter incorporating both long noncoding RNA and microRNAs), and protein. Many of these abnormalities, such as DNA mutations, aberrant DNA methylation, or deregulated microRNAs, are considered candidate biomarkers for the diagnosis of PDAC in surrogate biospecimens such as blood or pancreatic juice.172–175 Newly emerging areas of biomarker discovery for early diagnosis of PDAC, and in monitoring for recurrence postresection, include circulating cell-free mutant DNA (cfDNA) and DNA contained within extracellular microvesicles (exosomes),176, 177 which can be detected using ultra-sensitive PCR techniques.178
Molecular biology of exocrine pancreatic neoplasms: Translational implications
Below are a few selected examples of recent advances made in understanding the tumor biology of PDAC that have translational relevance within the field of cancer medicine.
A genetic timeline for PDAC progression: The overwhelming majority of PDAC patients present with advanced disease, and early detection remains one of the most promising avenues for improving OS. In a seminal report of PDAC autopsy cases that compared the genomic aberrations present in primary versus synchronous metastatic lesions, investigators calculated that almost two decades might be required for progression from the initial mutant clone(s) to terminal metastatic disease.179 No more than 2 years of this prolonged genetic timeline occurs during the symptomatic phase of disease, reiterating the importance of elucidating clinically applicable biomarkers that can identify emerging PDAC at a preclinical stage.
PDAC stroma: friend or foe? Traditionally, the cancer-associated myofibroblasts within the exuberant desmoplastic stroma in PDAC have been considered an ally of the invasive cancer.180, 181 One pathway that emerged as particularly relevant in facilitating tumor-stroma crosstalk is the Hedgehog signaling pathway,182 and depletion of stromal myofibroblasts in PDAC GEMMs using short-term therapy with Hedgehog inhibitors was shown to enhance chemotherapy delivery in preclinical studies.183 This prompted a randomized clinical trial of an inhibitor of Hedgehog signaling in combination with gemcitabine, but rather unexpectedly, the inhibitor to paradoxical disease progression (www.businesswire.com/news/home/20120127005146/en/Infinity-Reports-Update-Phase-2-Study-Saridegib). A more recent series of studies using both genetic and pharmacological ablation of myofibroblasts in PDAC GEMMs have been instrumental in implicating a tumor-constraining role for stroma.184–186 Specifically, stromal depletion led to tumor dedifferentiation and increased metastases, with reduced median survival of mice, mimicking the clinical findings. These paradigm-shifting reports have led to a reassessment of the role of stroma in PDAC. One approach that has been proposed in preclinical models is a shift away from stromal ablation to stromal “reprogramming,” wherein myofibroblasts are still retained in situ, but pharmacological strategies are used to revert their transcriptional profiles to a basal, resting state.187
Overcoming immune tolerance in PDAC: Cancer immunotherapy has emerged as one of the most promising therapeutic strategies over the last 5 years with agents known as checkpoint inhibitors resulting in dramatic improvements in survival for patients with melanoma.188 To date, these successes have not translated to PDAC.189, 190 Some of these failures have been attributed to the unique tumor microenvironment of PDAC. In contrast to certain “immunogenic” cancers such as melanoma, the immunological infiltrate in the PDAC microenvironment is sparse in effector T cells, and populated by cells that dampen tumor rejection, including regulatory T cells, macrophages, and suppressor cells of myeloid origin.191–193 This immunosuppressive milieu contributes to a state of immune tolerance and facilitates tumor progression. As immunotherapy of PDAC continues to evolve, there is increasing recognition of the importance of combination regimens, such as vaccines to induce an antigen-specific T-cell response in conjunction with blockade of checkpoints,194 or adoptive therapy using antigen-specific engineered T cells in conjunction with checkpoint inhibitors.195
Metabolic vulnerabilities in PDAC: As proposed by Otto Warburg nearly a century ago,196 cancer cells are known to mostly rely on aerobic glycolysis of exogenous glucose for their energy needs. In addition to glucose, exogenous glutamine is a key elemental fuel for PDAC survival and proliferation.197 Upon uptake, glutamine is converted by the enzyme glutaminase into glutamate. Not surprisingly, mutant Ras appears to be a critical orchestrator of the intracellular reprogramming. Mutant Ras rewires the metabolic machinery such that glutamate is oxidized via a noncanonical pathway distinct from normal cells, without increasing deleterious intracellular reactive oxygen species levels.198 In preclinical models, this unique glutamine utilization mechanism is essential for cancer cell survival and has generated enthusiasm about targeting glutaminase or other enzymes of this pathway in PDAC. Second, mutant Ras has been shown to promote dependence on autophagy, a nutrient recycling mechanism wherein lysosomal degradation of damaged proteins, macromolecules, and organelles, generates key intermediates for bioenergetics.199 Autophagy can be blocked using agents such as chloroquine and has shown promising results in preclinical settings.200 Finally, mutant Ras leads to profound increase in macropinocytosis, a primitive nutrient uptake mechanism that functions via endocytosis of extracellular fluid and nutrients via the so-called macropinocytic vesicles originating from the cell membrane.201 Macropinocytosis in PDAC cells enhances uptake of albumin and other extracellular proteins that are catabolized into essential amino acids channeled into various anabolic pathways. Thus, inhibition of macropinocytosis can be leveraged as potential therapeutic avenue. Alternatively, the propensity of PDAC cells to uptake extracellular albumin can be used as a “Trojan horse” strategy, by conjugating therapeutic agents with albumin. In fact, there is some evidence to suggest that the enhanced efficacy of nano-albumin-conjugated paclitaxel might be, at least in part, due to the macropinocytosis-induced uptake of the drug within KRAS-mutant cancer cells.
Taken together, these results provide greater understanding of the complexity of PDAC’s underlying biology with protracted genomic evolution, a dynamic microenvironment, and distinct differences in metabolism between normal and malignant cells. These insights suggest strategies to modulate the tumor’s cellular and extracellular defense mechanisms. A few specific examples translating these recent laboratory-based discoveries to the clinical arena will be discussed in the section titled “Future Directions” at the end of this chapter.
Diagnostic evaluation
The diagnostic work-up of patients ultimately found to have pancreatic cancer can be fragmented and inefficient. Surgeons or subspecialty oncologists may be asked to evaluate a patient at any point in the diagnostic process to include evaluation of abdominal pain, suspicion of a pancreatic mass, or biopsy-confirmed pancreatic cancer with or without complete staging evaluation. Essential clinical information which may have important therapeutic implications can be obtained from a thorough history and physical examination. Some of this information cannot be obtained from imaging studies and includes the assessment of performance status (PS), cardiopulmonary function, evaluation for left supraclavicular or periumbilical adenopathy, or evidence for venous thromboembolism (VTE).
Classification of clinical stage
It is critically important to use standardized, objective radiologic criteria for clinical staging. Modern imaging techniques have revolutionized the clinical staging of pancreatic cancer. Precise and objective anatomic radiographic criteria are used to determine the extent of the tumor–vascular relationship and to categorize clinical staging. The clinical stage of pancreatic cancer can be broadly divided into patients with inoperable disease (metastatic or locally advanced) and localized disease (borderline resectable or resectable); see Table 3 for specific radiographic criteria. The majority of patients will present with metastatic disease, as evidenced by ascites/peritoneal implants, liver, or lung metastases. Confirmation of metastatic disease can be achieved with percutaneous biopsy of the metastatic lesion (preferred) or at the time of endoscopy (EUS-FNA (fine needle aspiration)). In the absence of metastatic disease, the clinical stage is determined by the relationship of the primary tumor to adjacent vasculature. Radiographic findings of a potentially resectable pancreatic cancer (AJCC stages I or II) are (1) the absence of tumor-arterial abutment or encasement and (2) <50% involvement of the superior mesenteric vein (SMV)/portal vein (PV) (Figure 3). As a general rule, any tumor abutment (≤180° tumor-vessel interface) or encasement (>180°) of the celiac axis, common hepatic artery, or superior mesenteric artery (SMA) should be considered a contraindication to immediate surgery. A patient is deemed to have locally advanced, unresectable disease (AJCC stage III) when (1) the tumor encases the SMA or celiac axis, as defined by >180° of the circumference of the vessel (Figure 4), or (2) there is occlusion of the SMPV (superior mesenteric-portal vein) confluence without the possibility for venous reconstruction. Patients who have tumor abutment, without encasement, of the SMA or celiac axis, or short segment encasement of the hepatic artery are considered to have borderline resectable pancreatic cancer. In addition, patients with tumors that cause >50% narrowing or short segment occlusion of the SMV/PV that may be amenable to reconstruction are also considered to be borderline resectable. There is emerging consensus that even more subtle tumor-vein abutment may be best considered borderline resectable (especially as neoadjuvant therapy becomes more widely accepted).
Table 3 Radiographic criteria for clinical staging of pancreatic cancer
| Resectable | |
| Tumor–artery relationship | No radiographic evidence of arterial abutment (celiac, SMA, or hepatic artery) |
| Tumor–vein relationship | Tumor-induced narrowing ≤50% of SMV, PV, or SMV-PV |
| Borderline resectable | |
| Artery | Tumor abutment (≤180°) of SMA or celiac artery. Tumor abutment or short segment encasement (>180°) of the hepatic artery |
| Vein | Tumor-induced narrowing of >50% of SMV, PV, or SMV-PV confluence. Short segment occlusion of SMV, PV, and SMV-PV with suitable PV (above) and SMV (below) to allow for safe vascular reconstruction |
| Extrapancreatic disease | CT scan findings suspicious, but not diagnostic of, metastatic disease (e.g., small indeterminate liver lesions which are too small to characterize) |
| Locally advanced | |
| Artery | Tumor encasement (>180°) of SMA or celiac artery |
| Vein | Occlusion of SMV, PV, or SMV-PV without suitable vessels above and below the tumor to allow for reconstruction (no distal or proximal target for vascular reconstruction) |
| Extrapancreatic disease | No evidence of peritoneal, hepatic, and extra-abdominal metastases |
| Metastatic | |
| Evidence of peritoneal or distant metastases | |
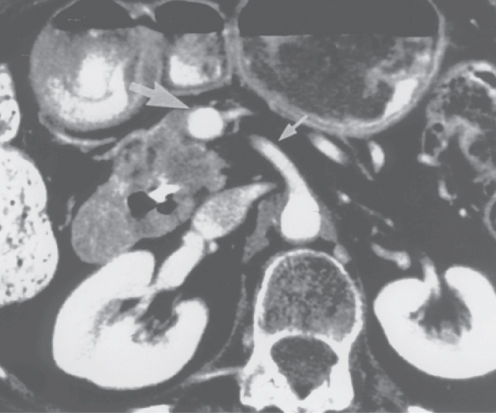
Figure 3 Contrast-enhanced computed tomography scan demonstrating a resectable adenocarcinoma of the pancreatic head. The low-density tumor is easily seen in the pancreatic head. Note the absence of tumor extension to the superior mesenteric artery (SMA) (small arrow); there is a normal fat plane between the low-density tumor and the SMA. However, the tumor (area of low density) does extend to the superior mesenteric vein (SMV) just inferior to the tip of the large arrow. This patient may require venous resection and reconstruction at the time of pancreaticoduodenectomy. This subtle finding would not be apparent on a lesser quality scan. The intrapancreatic portion of the common bile duct contains a stent that was endoscopically placed for biliary drainage.
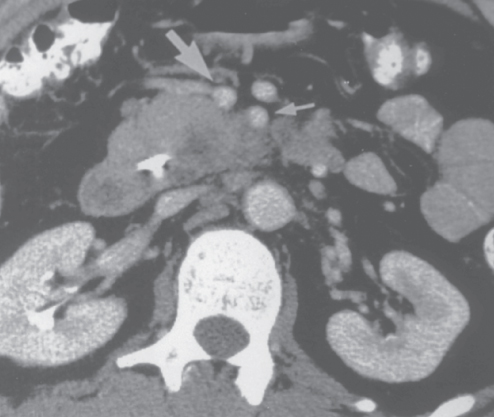
Figure 4 Contrast-enhanced computed tomography scan demonstrating an unresectable adenocarcinoma of the pancreatic head and uncinate process. The low-density tumor is inseparable from the posterior wall of the SMA (small arrow). Direct intraoperative assessment of the extent of retroperitoneal tumor growth in relation to the SMA is not possible until after gastric and pancreatic transection, at which point the surgeon has committed to resection; accurate preoperative imaging of this vital tumor–vessel relationship is thus critical. The large arrow identifies the SMV.
Imaging
As accurate characterization of tumor–vascular relationship is critical for clinical staging, interventions such as EUS and ERCP (endoscopic retrograde cholangiopancreatography, which can induce pancreatitis and interfere with accurate assessment of the extent of disease) should be deferred briefly until high-quality imaging is obtained. The single most important imaging tool for the detection and staging of pancreatic cancer is computed tomography (CT).202, 203 Current multidetector protocols utilize dual-phase technique. A rapid injection of intravenous contrast allows for the maximal enhancement of the pancreas and mesenteric vasculature.204 The first (arterial) phase is used for visualization of the primary tumor and optimal assessment of the tumor–arterial relationship. The second (venous) phase aids definition of the relationship of the tumor to the surrounding venous structures (SMV, PV, and splenic vein) and may uncover metastases to locoregional lymph nodes and distant organs (particularly to the liver).
If a low-density mass is not seen on CT, the presence of biliary or pancreatic duct obstruction remains concerning for malignancy. Additional imaging with magnetic resonance imaging (MRI) can be considered. Several studies have demonstrated that small pancreatic lesions (<2 cm) are more conspicuous on MRI relative to CT.205, 206 As in CT imaging, the hypovascular nature of pancreatic cancer may make tumors best seen on early phase (arterial) sequences.204 Ultimately, patients with suspicion of pancreatic cancer without evidence of a mass should undergo EUS. In addition, EUS combined with FNA may provide tissue confirmation of malignancy. Rarely, patients may present with radiographic features concerning for pancreatic cancer without an identifiable mass on CT, MRI, or EUS. If the patient is jaundiced, an ERCP may provide additional radiographic information to include evidence for a double duct sign or smooth tapering more suggestive of sequelae of pancreatitis (Figure 5). In these settings, biliary brushings can provide material for cytologic evaluation. Importantly, pancreatic cancer remains the major diagnostic consideration in patients (without a history of recurrent pancreatitis or alcohol abuse) who have a malignant-appearing stricture of the intrapancreatic portion of the common bile duct or the presence of pancreatic duct dilation without radiographic evidence of a mass. In the presence of an elevated CA 19-9 level, repeat EUS should be considered and if a cytologic diagnosis cannot be confirmed, upfront surgery would be the obvious next step assuming that the patient is otherwise healthy with no contraindications for major abdominal surgery.

Figure 5 Endoscopic retrograde cholangiopancreatography demonstrating the smooth tapering common bile duct stricture (arrowhead) seen with biliary obstruction secondary to chronic pancreatitis.
Tissue acquisition
Confirmation of malignancy is required in all patients with locally advanced or metastatic disease before treatment with systemic therapy or radiotherapy. For patients with radiographic evidence of metastatic disease, percutaneous biopsies of the metastatic site can usually be obtained with either US- or CT guidance. For patients with localized disease that may be amenable to surgical resection, we prefer EUS-guided FNA biopsy to minimize the risk of tumor seeding. Furthermore, EUS-guided biopsy is much safer and more accurate than percutaneous CT-guided biopsy for tumors that are smaller or difficult to access using CT guidance.207 A negative cytology from EUS-guided FNA should not be considered proof that a malignancy does not exist, and repeat EUS-guided FNA may improve diagnostic accuracy.208 Of note, FNA biopsy should only be performed if a mass is visualized; blind biopsy of the pancreas is inappropriate.
Laparoscopic staging
In the past, laparoscopy was performed in patients who had radiologic evidence of localized pancreatic cancer.209 However, with the advent of high-quality contrast-enhanced CT, the role of laparoscopy in staging (when performed under a separate anesthesia induction) appears to be decreasing. Moreover, the yield of staging laparoscopy may be improved in those patients with very high levels of serum carbohydrate antigen (CA) 19-9, and acquisition of preoperative CA 19-9 levels is recommended.210, 211 If the suspicion of extra-pancreatic disease is high, or if the patient is a marginal surgical candidate based on medical comorbidities, laparoscopy as a stand-alone procedure may be appropriate.
Biliary drainage
In patients with localized pancreatic cancer, placement of endobiliary stents before surgery relieves the symptoms of biliary obstruction and facilitates the normalization of liver function. The latter is particularly important for patients who will be treated with neoadjuvant therapy, as hyperbilirubinemia prevents the safe delivery of systemic therapy. If neoadjuvant therapy is planned, biliary complications, including stent occlusion and cholangitis, can be minimized with insertion of a short metal stent after cytologic confirmation of cancer.212 For patients with locally advanced or metastatic pancreatic cancer, an endobiliary self-expandable metal stent has superior long-term patency compared with plastic stents.213, 214
Certain groups of patients with symptomatic distal bile duct obstruction should not undergo endobiliary metal stent placement. Although covered metal stents can also be removed endoscopically,215 they should rarely be used to provide temporary biliary decompression in a patient without a tissue diagnosis of malignancy.
Treatment of localized, potentially resectable disease
Surgical considerations
If the primary tumor cannot be resected completely (gross complete resection), surgery for pancreatic cancer (pancreaticoduodenectomy) offers no survival advantage. Patients who undergo a gross incomplete resection have an expected median survival of <1 year.216 Even microscopically positive surgical margins have a negative impact on OS and the ability to obtain a margin negative resection should be viewed as necessary for long-term survival.217
Upfront surgery and adjuvant therapy
Currently, the standard of care for early-stage, resectable disease is surgery followed by adjuvant therapy. Owing to recent advances in operative technique, anesthesia, and critical care, the 30-day in-hospital mortality rate is 2% or less in patients who undergo pancreaticoduodenectomy performed by experienced surgeons.218–220 Data have also demonstrated that higher hospital volume is associated with lower surgery-related mortality.221 Upfront surgical resection followed by adjuvant therapy is potentially curative. However, median survival times of 22–24 months have remained stagnant for the last 30 years using this approach.222, 223
Pancreaticoduodenectomy
The standard surgical procedure for neoplasms of the pancreatic head and periampullary region is pancreaticoduodenectomy. The current technique of pancreaticoduodenectomy has evolved from the procedure first described by Whipple and colleagues in 1935.224 and incorporates selected aspects of the traditional Whipple procedure and emphasizes the importance of removing all soft tissue to the right of the SMA. The surgical resection is divided into six clearly defined steps (Figure 6); the most oncologically important and difficult part of the operation is step six, during which the pancreas is divided and the specimen is removed from the SMPV confluence and the right lateral border of the SMA.225
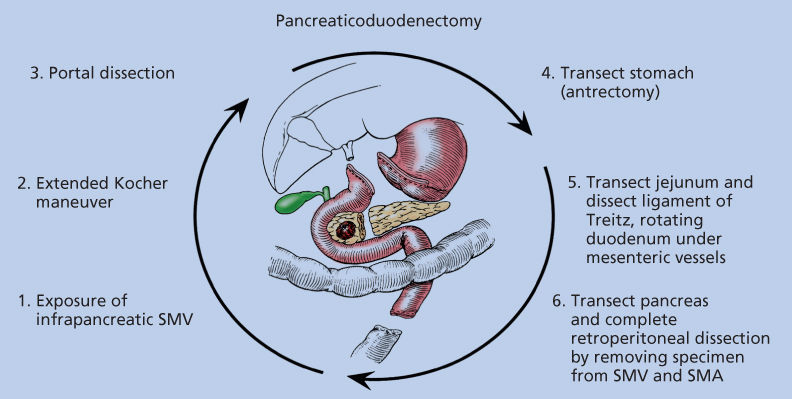
Figure 6 Six surgical steps of pancreaticoduodenectomy.
The high incidence of local recurrence after standard pancreaticoduodenectomy requires particular attention to the SMA margin, the soft tissue margin along the right lateral border of the proximal SMA (Figure 7a).226 It is critical that this margin be identified for the pathologist and assessed histologically; the residual disease status (termed “R” factor) cannot be determined if the retroperitoneal margin is not assessed histologically.220, 227
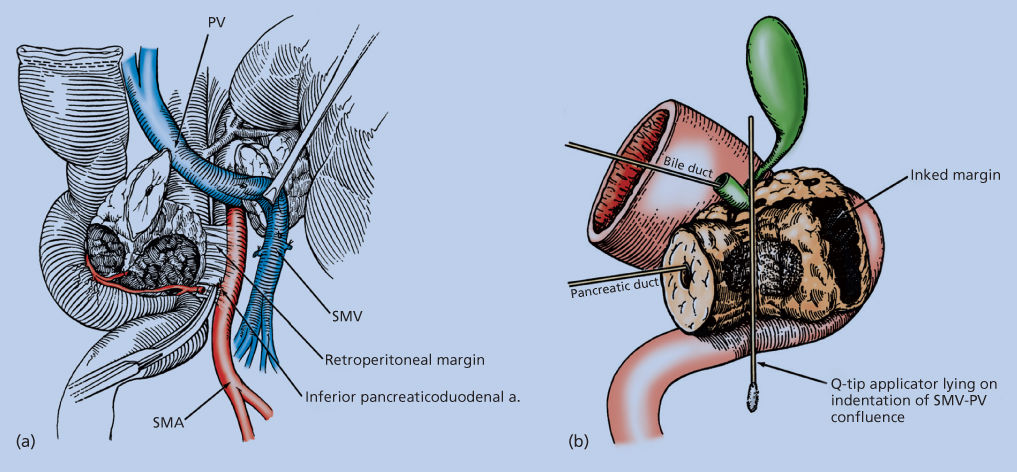
Figure 7 Illustration of the retroperitoneal margin as defined at the time of tumor resection (a). Medial retraction of the SMV and superior mesenteric-portal vein (SMPV) confluence facilitates dissection of the soft tissues adjacent to the lateral wall of the proximal SMA; this site represents the retroperitoneal margin. Complete permanent-section analysis of the pancreaticoduodenectomy specimen requires that it be oriented for the pathologist to enable accurate assessment of the retroperitoneal margin of excision and other standard pathologic variables. The retroperitoneal margin must be identified and inked with the pathologist (b); it cannot be assessed retrospectively. As shown here, a probe is in both the pancreatic duct and the bile duct, and a Q-tip applicator lies on the indentation of the SMPV confluence. One can then ink (for final pathologic assessment) the soft-tissue margin adjacent to the proximal SMA; this represents the retroperitoneal (or mesenteric) margin of excision.
Vascular resection
The goal of vascular resection is to obtain an R0 resection, which requires complete removal of the tumor from the SMV/PV, and exposure of the SMA to allow sharp dissection of the tumor off this artery. Venous resection and reconstruction should be performed for tumors adherent to the SMV or SMPV confluence (Figure 8) or in the presence of short segment occlusion of the SMV/PV with no tumor encasement of the SMA or celiac axis.220, 225
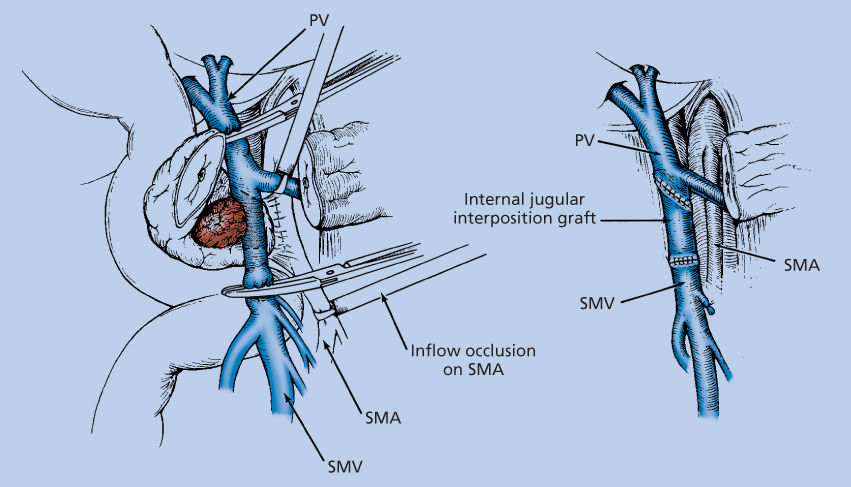
Figure 8 Venous resection/reconstruction. Illustration of resection of the SMV with splenic vein preservation. The intact splenic vein tethers the portal vein (PV), making a primary anastomosis impossible in most cases. Our preferred method of reconstruction of the SMV is to use an internal jugular vein interposition graft. With the splenic vein intact, exposure is inadequate to separate the specimen from the lateral aspect of the proximal SMA. Therefore, the graft can be placed before specimen removal, thereby allowing medial retraction of the reconstructed SMPV confluence, or after separation of the specimen from the SMA posteriorly. Segmental resection of the SMV with splenic vein preservation adds significant complexity to this operation.
A very select proportion of patients with locally advanced disease (as currently defined) may be considered for surgery (after neoadjuvant therapy) if (1) tumor encasement of the celiac axis or common hepatic artery can be removed with an Appleby procedure (extended distal pancreatectomy with celiac resection) and (2) there is evidence of radiographic, biochemical (CA 19-9), and physiologic response (improved PS) to preoperative therapy. In these patients, neoadjuvant chemoradiation is recommended and surgical candidacy is reassessed upon the completion of therapy. The most common operation performed for such patients has been a distal pancreatectomy, splenectomy, and en bloc resection of the celiac axis (Appleby procedure). In the small series of patients where resection and arterial reconstruction has been performed, results have been encouraging.228, 229
Pylorus preservation
Currently, available findings from randomized trials suggest no difference in perioperative factors or patient outcome between standard and pylorus-preserving pancreaticoduodenectomy.230–232 Most investigators would agree that pylorus preservation should not be performed in patients who have bulky tumors of the pancreatic head or duodenal tumors involving the first or second portions of the duodenum.
Minimally invasive pancreatectomy
Interest in applying minimally invasive techniques to pancreatic resection stems from the hypothesis that open surgical resection is associated with substantial physiologic stress and morbidity. While the advantages and appropriateness of minimally invasive approaches are still controversial, from 1998 to 2009, the rates of minimally invasive pancreatectomy have risen from 2.4% to 7.3%.233 Minimally invasive procedures include both laparoscopic and robotically assisted procedures.
Laparoscopic distal pancreatectomy is the most common minimally invasive pancreatic operation performed as there is no requirement for anatomic reconstruction after the tumor is removed. Although no prospective trials have been performed comparing open versus laparoscopic distal pancreatectomy, multiple retrospective studies have reported favorable perioperative outcomes, with decreased blood loss, complications rates, and shorter hospital stays associated with the laparoscopic approach.234, 235 The oncologic impact of open versus laparoscopic surgery has been less rigorously reported. Only three studies have described the oncologic outcomes of patients undergoing laparoscopic distal pancreatectomy.236–238 The largest study was a multicenter, retrospective matched cohort analysis; no differences in R0 resection status (74% vs 66%), lymph nodes examined (14 vs 12), or median OS (16 months vs 16 months) were observed between open and laparoscopic distal pancreatectomy.
In contrast to laparoscopic distal pancreatectomy, which is offered in most major centers, minimally invasive approaches to pancreaticoduodenectomy for pancreatic cancer have not yet achieved widespread acceptance. Few centers have a reported experience with 30 or more patients.239, 240 Among these studies, conversion to an open procedure occurred in approximately 6–10% of cases.
Robotic-assisted pancreaticoduodenectomy has also been reported with perioperative complication rates that appear comparable to the laparoscopic approach.241–243 Similar to distal pancreatectomy, the oncologic outcomes for minimally invasive pancreaticoduodenectomy are limited. In one of the largest series of minimally invasive pancreaticoduodenectomy for cancer, 108 patients were reported and their survival duration was no different than the larger cohort of patients who underwent an open pancreaticoduodenectomy. The authors suggested that with a minimally invasive approach, more patients may receive adjuvant therapy and in a more timely manner. However, time to the receipt of adjuvant therapy (duration of time from the date of surgery to the start of systemic therapy) has recently been shown to be less important than whether or not the patient receives all intended postoperative therapy.244
Pathologic (surgical) staging
The staging system of the AJCC and International Union Against Cancer is given in Table 4. The modifications to the TNM staging system in the sixth edition allow for the accurate staging of patients even if they do not undergo pancreatic resection. The T4 (and stage III) designation is reserved for locally advanced unresectable primary tumors in the absence of distant metastases.226
Table 4 TNM staging system
| Definitions | |||
| Primary tumor (T) | |||
| TX | Primary tumor cannot be assessed | ||
| T0 | No evidence of primary tumor | ||
| Tis | In situ carcinoma | ||
| T1 | Tumor limited to pancreas and 2 cm or less in greatest dimension | ||
| T2 | Tumor limited to pancreas and more than 2 cm in greatest dimension | ||
| T3 | Tumor extends beyond the pancreas but without involvement of the celiac axis or the superior mesenteric artery | ||
| T4 | Tumor involves the celiac axis or the superior mesenteric artery (unresectable primary tumor) | ||
| Regional lymph nodes (N) | |||
| NX | Regional lymph nodes cannot be assessed | ||
| N0 | No regional lymph node metastasis | ||
| N1 | Regional lymph node metastasis | ||
| Distant metastasis (M) | |||
| MX | Distant metastasis cannot be assessed | ||
| M0 | No distant metastasis | ||
| M1 | Distant metastasis | ||
| Stage grouping | |||
| Stage 0 | Tis | N0 | M0 |
| Stage IA | T1 | N0 | M0 |
| Stage IB | T2 | N0 | M0 |
| Stage IIA | T3 | N0 | M0 |
| Stage IIB | T1–3 | N1 | M0 |
| Stage III | T4 | N0/1 | M0 |
| Stage IV | T1–4 | N0/1 | M1 |
Source: Greene 2002.226 Reproduced with permission of Springer.
When the (pathologic) pancreaticoduodenectomy specimen is evaluated, the surgeon and pathologist should first evaluate frozen sections of the common bile duct transection margin and the pancreatic transection margin. The SMA margin (the soft tissue margin directly adjacent to the proximal 3–4 cm of the SMA—Figure 7b) is then evaluated on permanent sections by inking the margin and sectioning the tumor perpendicular to the margin.220 The R status of resection cannot be determined if the SMA margin is not assessed histologically. Most importantly, the surgeon and pathologist should classify this margin after integrating the operative findings and the histologic assessment of this margin. All pancreatic resections should be classified according to the R status: R0, no gross or microscopic residual disease; R1, microscopically positive surgical margins with no gross residual disease; and R2, grossly evident residual disease. The R designation should appear in the final operative report and should be consistent with the pathology report. The final pathologic evaluation of the permanent sections should include a description of the tumor histology and differentiation, gross and microscopic evaluation of the tissue of origin (pancreas, bile duct, ampulla of Vater, or duodenum), and a measurement of the maximum transverse tumor diameter, involved lymph nodes and total lymph nodes examined, and the presence or absence of perineural, lymphatic, and vascular invasion.
Prognostic factors
Investigators have examined pathologic factors of the resected tumor in an effort to establish reliable prognostic variables associated with decreased survival.218, 245 Regional lymph node metastases, poorly differentiated histology, and increased size of primary tumors have all been associated with decreased survival duration. Nevertheless, heterogeneity in patient outcomes remains. For example, in one study of 45 patients with pancreatic adenocarcinoma who underwent complete resection with both negative resection margins and negative regional lymph nodes, median survival was 32 months with a 5-year survival of 40%.246 However, another group reported on 12 five-year survivors (following complete resection); 4 had poorly differentiated histology, 5 had metastatic disease in regional lymph nodes, 9 had histologic evidence of malignant extrapancreatic soft tissue extension, and 10 had evidence of perineural invasion.247 Similarly, in a report of actual 5-year survivors, 36% were node positive, and 20% of all node positive patients survived 5 years; this last report utilized preoperative therapy before surgical resection.248
Adjuvant (postoperative) therapy
The first report of the beneficial adjuvant therapy was published in 1985 by the Gastrointestinal Tumor Study Group (GITSG).249 Since that time, several other prospective randomized trials have been conducted and published (Table 5).250–254
Table 5 Summary of randomized trials of adjuvant therapy in resected pancreatic cancer
| Study (year) | Number of enrolled patients | Patients with R1 resection (%) | Treatment assignment Median survival (months) | Treatment assignment Median survival (months) | p-Value | Local failure rate (%) |
| GITSG249 (1985) | 49 | 0 | 5-FU chemoradiation 21.0 | Observation 10.9 | 0.035 | NR |
| EORTC250 (1999) | 114 | 19 | 5-FU chemoradiation 17.1 | Observation 12.6 | 0.09 | 34 |
| ESPAC-1251 (2004) | 289 | 18 | 5-FU chemotherapy 20.1 | No chemotherapy 15.5 | 0.09 | 60 |
| 5-FU chemoradiation 15.9 | No chemoradiation 17.9 | 0.009 | ||||
| RTOG 9704252 (2011) | 388 (Head lesions) | ≥35 | Gemcitabinethen5-FU chemoradiationthengemcitabine 20.5 | 5-FUthen5-FU chemoradiationthen5-FU 16.9 | 0.09 | 26 |
| CONKO 001253 (2007) | 368 | 19 | Gemcitabine 22.8 | Observation 20.2 | 0.005 | >35% |
| ESPAC-3254 | 1088 | 35 | Gemcitabine 23.6 | 5-FU/Folinic acid 23.0 | 0.42 | NR |
Abbreviation: NR, not reported.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


