Neoplasms of the endocrine glands: pituitary neoplasms
Chirag D. Gandhi, MD, FACS, FAANS  Margaret Pain, MD
Margaret Pain, MD  Kalmon D. Post, MD, FACS, FAANS
Kalmon D. Post, MD, FACS, FAANS
Overview
Pituitary neoplasms represent a phenotypically and pathologically diverse family of tumors. Treatment and classification are both patient and tumor specific as symptoms may derive from abnormal hormone production, compression of adjacent nervous system structures, and, in rare cases, metastases. Transsphenoidal surgery represents a common and well-tolerated treatment for most pituitary adenomas; however, advances in radiation and chemotherapy have increased treatment options, enhancing treatment efficacy, and eligibility.
Pituitary adenomas are epithelial tumors arising from the adenohypophysis that can manifest with neurological symptoms from local mass effect such as headaches, visual disturbances, increased intracranial pressure, and cranial nerve palsies or as a variety of clinical entities depending on the hormones they secrete. In rare cases (<1%), patients can also present with diabetes insipidus. Pituitary adenomas constitute 10–15% of intracranial tumors, although the incidence is as high as 24% in autopsy series. They are most common in the third and fourth decades of life and overall affect both sexes equally. However, there are differences in frequency of certain subtypes between the sexes. Cushing’s disease, for example, is more common in women; Prolactin (PRL)-secreting adenomas are more common in young women; and null-cell adenomas, oncocytomas, and gonadotropin-secreting adenomas are more common in men.
Distinction between pituitary neoplasm subtypes has been refined progressively as we have come to understand more about the pathology and biochemistry of the pituitary gland. Early on, tumors were classified based on the light microscopy characteristics of the predominant cell cytoplasm and were therefore divided into categories as acidophilic, basophilic, or chromophobic tumors. Acidophils were thought to produce excess amounts of growth hormone (GH) and were linked to acromegaly and gigantism; basophils were thought to secrete adrenocorticotrophic hormone (ACTH) and to cause Cushing’s disease; and chromophobes were regarded as hormonally inactive. This system has become too simplistic for modern treatment algorithms as hormone secretion is only loosely associated with hematoxylin and eosin staining. It has been well established that both acidophilic and basophilic adenomas may secrete other hormones and that chromophobes can be hormonally active capable of secreting GH, PRL, ACTH, thyroid-secreting hormone (TSH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), or α-subunit.1
From a medical perspective, tumors are frequently categorized based on the predominant hormone that they secrete.2, 3 Tumors are further categorized as hormonally active or inactive and are considered active only if the amount of hormone they secrete exceeds normal levels in the blood and is clinically evident. Inactive adenomas contain secretory and cellular components necessary for hormone production, but they are not associated with clinical or biochemical evidence of hormone excess. There are several theories about the etiology of inactive adenomas. One theory is that the gland cells become neoplastic but inherently have low hormone production. Another proposed theory is that the cells produce abnormal hormone that is not recognized by antibodies in the standard radioimmunoassays, and finally that the cells have lost the ability to produce hormone through some acquired genetic defect.4 This functional classification is now being further modified to incorporate recent advances in molecular and immunohistochemical advances in pituitary research.
Anatomically and radiologically, tumors can also be classified based on their relationship to and involvement within the cavernous sinus. Magnetic resonance imaging (MRI) is the modality of choice and should be requested with a special consideration given toward imaging the pituitary. This can usually be achieved by obtaining thin slices through the pituitary in the coronal and sagittal planes (Figure 1). With high sensitivity for intracranial pathology, coronal T2-weighted images give greater clarity to possible optic chiasm displacement as well as hemorrhagic and cystic changes within the tumor. Gadolinium contrast enhancement is essential for increasing the diagnostic yield for microadenomas (<10 mm) and is also useful in delineating normal from abnormal tissue in macroadenomas (T1-weighted images pre- and postgadolinium).5 In general, pituitary adenomas appear as a hypointense lesion on a T1 precontrast image and will enhance less than normal pituitary tissue on T1 postcontrast series. Radiographic appearance has been used to further categorize parasellar invasive tumors according to the schema derived by Jules Hardy and/or Engelbert Knosp (Figure 2). The Knosp classification scheme has emerged as a relevant strategy for preoperative prediction of cavernous sinus invasion. Briefly, the MRI appearance of the tumor and its influence on the medial wall of the sinus relative to the position of the intracavernous internal carotid artery are used to estimate the likelihood of invasion. Increasing mass effect of the tumor on the medial wall correlates with increased likelihood of invasiveness. When compared with noninvasive tumors, invasive tumors are more likely to demonstrate aggressive behavior.7
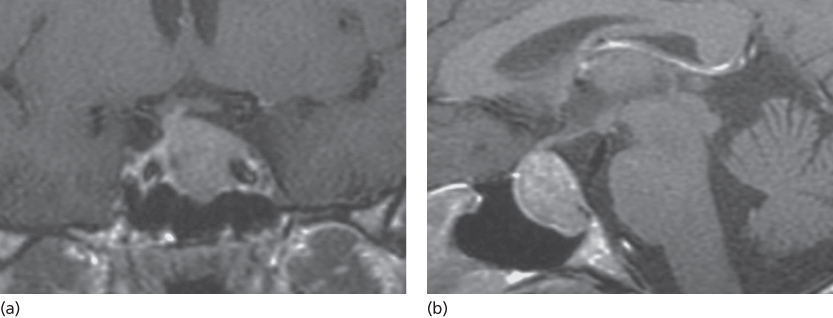
Figure 1 A 38-year-old woman with an ACTH-secreting adenoma. (a) Coronal T1-weighted magnetic resonance with contrast demonstrates a hypointense macroadenoma within the sella that extends into the left cavernous sinus and encases part of the carotid artery. (b) Sagittal T1-weighted magnetic resonance with contrast demonstrates a hyperintense macroadenoma within the sella.
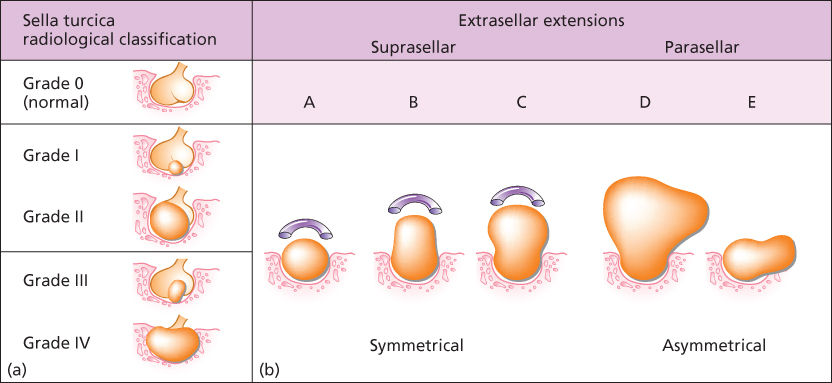
Figure 2 (a) Hardy’s classification of pituitary adenomas. Grade I and II are enclosed within the sella. Grades III and IV are invasive. Extrasellar classifications A, B, and C are increasing amounts of direct suprasellar adenomas. D is asymmetric extension, and E is lateral extension into the cavernous sinus. Source: Adapted from Hardy J and Somma M.6 (b) Knosp classification of cavernous sinus invasion.
Source: Woodworth, GF et al. J. Neurosurgery 120:1086–1094, 2014.
Molecular biological investigation into pituitary tumorigenesis has demonstrated that adenomas are a result of monoclonal proliferation8–10 and that neoplastic progression is related to both oncogenes and dysfunctional tumor suppression. Although several mutations have been implicated in pituitary tumorigenesis, only one mutation has been identified with significant prevalence in affected patients. Approximately 10–40% of GH-secreting adenomas result from a point mutation affecting the α-chain of the GTP-binding protein, leading to constitutive cAMP activation. It is postulated that the elevated cAMP formation leads to GH hypersecretion and adenoma proliferation.11 Although it has been theorized that adenomas with this gsp oncogene mutation are smaller and more sensitive to medical therapy, recent studies have failed to demonstrate any phenotypic differences between patient with the mutation and those without it.12 Many other additional factors have been identified to influence tumor progression. Oncogenes linked to adenoma progression include cAMP-responsive nuclear transcription factor, CREB, which is thought to be promoted by the overexpression of Gsα12; ras oncogene mutations that have been detected in aggressive prolactinomas13; and the pituitary tumor transforming gene (PTTG) thought to be a marker for invasiveness in secretory adenomas.14, 15 Tumor suppressor genes such as Rb, menin, TP53, p27, p16,16, 17 hormone-promoting factors such as hypothalamic neurohormones, and locally produced growth factors and cytokines are also being studied for their contribution to adenoma growth.18 With research into galectin-3 (Gal-3), a therapeutic drug target found only in PRL- and ACTH-secreting tumors, as well as nuclear receptor, peroxisome proliferator-activated receptor gamma (PPAR-γ), isolated from a number of studied adenomas, there is promise for novel drug development.19–21
Evaluation of a patient with suspected or known pituitary adenoma requires both complete radiographic and endocrinological and potentially genetic assessment before initiation of therapy.22 Endocrine assessment should include a complete metabolic panel, pregnancy test, serum GH, insulin-like growth factor (IGF-1), TSH, free T4, T3, PRL, ACTH, cortisol, LH, FSH, and testosterone. Following these tests, the determination of further medical or surgical treatments can be made.
Surgery is the preferred and definitive treatment of all pituitary adenomas with the exception of prolactinomas and asymptomatic, hormonally inactive tumors (incidentalomas). On the basis of the innovative work of Cushing, Guiot, and Hardy, the transsphenoidal approach is the preferred surgical technique for most adenomas and is associated with minimal morbidity and rapid recovery.23, 24 The goals of surgery include the reduction of tumor-related mass effect on adjacent structures, the cessation of endocrine hyperactivity, and the preservation or restoration of normal pituitary function. Adjuvant treatment can then be initiated based on the histopathology and immunochemistry, postoperative hormone secretory status, and the degree of extrasellar extension. In contrast, prolactinomas frequently respond well to medical therapy and incidental tumors require only observation until they are associated with symptoms.
Finally, a small percentage of pituitary neoplasms demonstrate aggressive behavior or carcinomatous change. Hormonally active tumors usually require hormone-specific therapy but this may not be sufficient for disease control. Temozolomide, an oral alkylating agent, has emerged as an effective adjunctive therapy for these patients.25 Its efficacy is explained by the phenotype of low O6-methylguanine-DNA methyltransferase (MGMT) expression frequently found in aggressive pituitary neoplasms.26
Prolactin-secreting pituitary adenomas
Prolactinomas are the most commonly diagnosed pituitary adenoma, representing approximately 30% of cases.27 Although rarely life threatening, when symptomatic, they can present with clinical findings relating to hyperprolactinemia (abnormal reproductive or sexual function) or mass effect on adjacent structures owing to size. Because of their reliable responsiveness to medical management, chemotherapeutics are often the first-line treatment for these tumors (Figure 3). However, surgical treatment has been shown to be equally effective and may be indicated at a primary intervention for certain cases.
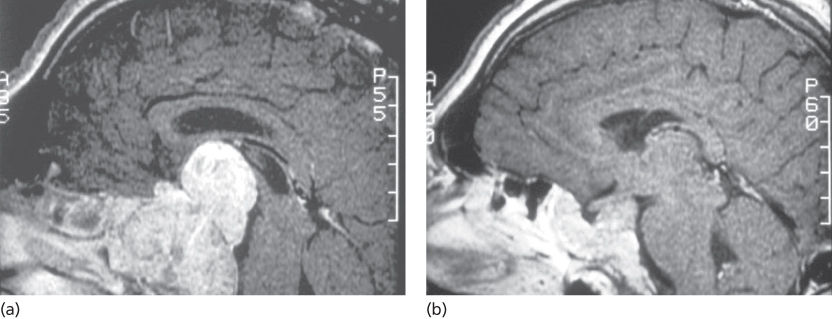
Figure 3 A 52-year-old male with diminished libido and a macroprolactinoma. (a) Sagittal T1-weighted magnetic resonance with contrast demonstrates a large sellar/suprasellar adenoma with compression of the optic chiasm. (b) Sagittal T1-weighted with contrast demonstrates a significant reduction in tumor volume after being treated medically with bromocriptine.
Clinical presentation
Although autopsy studies have demonstrated that prolactinomas have similar prevalence between the genders, women are four times more likely than men to become symptomatic. In women of reproductive age, hyperprolactinemia may cause oligomenorrhea, secondary amenorrhea, galactorrhea, and sterility. Less common symptoms in this population are decreased libido, dry vaginal mucosa caused by estrogen deficiency, weight gain, and psychological symptoms such as depression and anxiety. Prolactinomas account for approximately 5% of women with primary amenorrhea and 25% of women with secondary amenorrhea (excluding pregnancy).28 When the presentations of amenorrhea are accompanied by concomitant galactorrhea, the incidence of prolactinoma increases to 70–80%.29
In men and postmenopausal women, where symptomatic hyperprolactinemia is much more subtle, the tumors may grow to much larger sizes and are detected only when they begin to produce symptoms related to mass effect. Macroadenoma-related mass effect may result in headaches, visual disturbances (typically bitemporal hemianopsia as a result of chiasmatic compression), hypopituitarism, ophthalmoplegia, and, rarely, noncommunicating hydrocephalus from obstruction of the foramen of Monro. Men may also experience a diminished libido, impotence, gynecomastia, and infertility from decreased androgen production. The mechanism of hypogonadism caused by hyperprolactinemia is still controversial. One possibility is that PRL alters the hypothalamic release of gonadotropin-releasing hormone (GnRH) through dopamine inhibition, which in turn causes disruptions in the normal pulsatile secretion of LH.30 Prolonged periods of hyperprolactinemia have also been linked to bone demineralization by way of hypogonadism as bone density is normal in eumenorrheic hyperprolactinemic women.31
Diagnosis
Beside prolactinomas, hyperprolactinemia can be associated with a variety of causes such as pregnancy, hypothyroidism, PRL-stimulating drugs (e.g., phenothiazines, butyrophenones, and metoclopramide), and renal failure that need to be considered in the differential diagnosis. In addition, physiological hyperprolactinemia can occur from psychological and physical stresses, such as exercise, surgery, and hypoglycemia, but the PRL level rarely excessed 40 ng/mL in these cases.32 A definitive diagnosis of prolactinoma is difficult and in most cases requires radiographic evidence in addition to elevated PRL levels. In men, a basal PRL value greater than 100 ng/mL is almost always indicative of a prolactinoma. In women, PRL levels of 100–200 ng/mL should raise suspicion for prolactinoma, while a level of over 200 ng/mL is highly suggestive of prolactinoma.33, 34 The greatest diagnostic uncertainty lies in patients with PRL levels between 50 and 100 ng/mL. Within this range, hyperprolactinemia may also be related to “stalk effect,” where mass of a tumor interferes with the flow of prolactin-inhibitory factor (PRIF) from the hypothalamus. Although various endocrine stimulation tests to differentiate these two possibilities have been suggested, they have not been proven reliable.35
Serum PRL levels are an index of secretory activity and have been found to be associated with the size of the prolactinoma (excluding cystic or necrotic components). Eighty percent of tumors producing less than 200 ng/mL are microadenomas, while at levels above 200 ng/mL only 20% are microadenomas. This relationship becomes unreliable for very large tumors (>4 cm). While the total PRL level may usually correlate with size and may even be greater than 1000 ng/mL, the laboratory value may be falsely low (25–150 ng/mL) and not reflect the actual serum concentration because of a phenomenon known as the “Hook effect.”36, 37 Briefly, the Hook effect results from saturation of the antibody binding sites during the radioimmunoassay and therefore distorting the binding curve. This can be resolved by performing serial dilutions and should be considered for all giant pituitary adenomas.
Treatment
The treatment options available to a patient with a prolactinoma include observation, medical therapy, surgery, and radiotherapy. Treatment decisions should take into consideration the tumor size, PRL level, clinical manifestations, tolerance of medical therapy, and the desire for fertility. The vast majority of microprolactinomas (<10 mm) do not increase in size.34 Together with autopsy studies suggesting higher prevalence of undiagnosed tumors within the population, many clinicians and patient opt for observation and against treatment for microadenomas in the absence of clinical hyperprolactinemia, normal pituitary function, and no desire for pregnancy.
Dopamine agonists have been shown to be highly effective in suppressing tumor growth and PRL production because dopamine, when secreted by the hypothalamus, is an endogenous inhibitor of PRL production. Bromocriptine was the first dopamine agonist to be used for treatment of prolactinomas, but newer agents such as cabergoline, quinagolide, lisuride, and terguride have also gained widespread acceptance. On a molecular level, dopamine agonists selectively activate type D2 dopamine receptors, thereby blocking transcription of the PRL gene.38 The shrinkage of PRL cells results in amyloid deposition and fibrosis of the tumor.39 Dopamine agonists have been shown to have a high response rate with normalization of PRL levels in 70–80% of patients, tumor shrinkage in 80–90%, and restoration of ovulation in 80–90%.40 On the basis of these results, some argue that medical treatment should be used in all patients except the 10% who experience significant side effects.41 In addition, there is evidence to suggest that a significant percentage of patients who respond to cabergoline with normalization of PRL levels and tumor shrinkage will remain in remission following discontinuation of the drug after 2 years.42, 43 However, even in patients who have optimal response to cabergoline (normalization of PRL, tumor volume reduction, no invasion of critical structures, or optic chiasm compression), the majority do experience recurrence and so close follow-up after treatment discontinuation is critical.43
Dopamine agonists do have some significant disadvantages that may temper their use. For many patients, the effects are reversible and therefore lifelong compliance is needed to control the disease. In addition, significant side effects such as nausea, vomiting, postural hypotension, headaches, XX depression, and anxiety have been reported with medication use. In cases of pituitary apoplexy and in tumors with large cystic components, dopamine agonists are not effective in shrinkage as tumor size is not correlated with tumor cellularity. More harmful side effects of dopamine agonists have also been reported. At much higher doses of cabergoline than the maximum dose used for prolactinomas, cardiac toxicity has occurred. Although subclinical valvular fibrosis and trace mitral regurgitation with cabergoline therapy of prolactinomas has been reported, no patients experienced clinically significant change in cardiac function as a result.44, 45, 136 Finally, dopamine agonists are not always effective for the treatment of prolactinomas. Bromocriptine resistance may be an indicator of increased tumor aggressiveness, and there may be an association between gand medication resistance.46
Currently, there is controversy in the role of surgery for treatment of prolactinomas. With requirement for strict long-term compliance and frequent medical follow-up, some argue that surgery should be the initial treatment with medical therapy as an adjunct only in cases without remission.47 The rate of long-term remission after surgery varies significantly with preoperative PRL levels and tumor size. Remission rates are best, 50–80%, for microadenomas with preoperative PRL levels of less than 200 ng/mL.40, 47–49 Rates drop significantly for macroadenomas and in patients with PRL levels of 200–500 ng/mL. Surgery has not been shown to be useful in giant adenomas and tumors with PRL levels greater than 500 ng/mL, as a complete resection is rarely possible. Even in these patients, however, dopamine agonists appear to be beneficial. Meta-analysis has found that three-fourths of patients experienced significant reduction in tumor volume and normalization of PRL level in 60–70%.50 There is evidence to suggest that surgery is more effective when dopamine agonists are used as neo-adjuvant therapy.51 In addition, some have demonstrated that surgery to be beneficial in cases of dopamine resistance as partial tumor resection was associated with better hormonal control and lower doses of dopamine agonists.52, 53 Overall, there is an inverse correlation between PRL levels and remission rates.
Conventional radiotherapy is generally not considered a primary mode of treatment.54 The use of stereotactic radiosurgery as either a primary55 or adjuvant56 therapy is becoming increasingly common. Radiosurgery has been shown to be successful in decreasing PRL levels (and in one study, 30% of patients achieved an endocrinological cure.57 As radiotherapy is rarely used alone, these data may be skewed by confounding factors.
Medical therapy by means of dopamine agonists remains the first-line treatment for most prolactinomas58 This treatment should be considered for all patients, including those desiring pregnancy and those with primary amenorrhea.
For tumors larger than 2 cm or with PRL levels between 200 and 500 ng/mL, neo-adjuvant treatment with dopamine agonists appears to reduce tumor volume, followed by surgery. Any residual tumor can then be treated medically. For very large, invasive, or tumors associated with PRL levels of greater than 500 ng/mL, medical therapy should be the primary treatment modality.50
Special consideration should be given to patients with prolactinomas during pregnancy. There is a small but serious risk of rapid expansion of the tumor off medication. Complications related to this growth occur primarily from macroadenomas with suprasellar extension; they are much less frequent for microadenomas.59 If these patients become symptomatic, a dopamine agonist can be administered. Both bromocriptine and cabergolide are US FDA pregnancy category B, indicating that animal studies have not shown risk to the fetus but that randomized, controlled trials in humans are inadequate to assess the risk comprehensively.60, 61 During pregnancy, surgery should be undertaken only if the tumor does not respond to medical treatment or if there are progressive neurological symptoms. To reduce the risk of pregnancy-related complications, patients with macroadenomas who desire pregnancy should undergo transsphenoidal resection before conception or remain on bromocriptine throughout the pregnancy.
Growth hormone-secreting pituitary adenomas
GH secretion is under hypothalamic control through somatostatin and growth hormone-releasing hormone (GHRH). Upon release, GH causes the production of IGF-1 from the liver, which affects bone and tissue growth and provides negative-feedback inhibition to GH and GHRH secretion. Acromegaly, the clinical condition associated with GH-producing adenomas, results in autonomous GH secretion and loss of this negative-feedback inhibition. However, the clinical manifestations of GH-secreting tumors have been found to correlate better with IGF-1 levels than with GH levels.
Clinical presentation
GH-secreting pituitary adenomas account for about 30% of all endocrine-active pituitary tumors. Most patients present in their third to fifth decade with macroadenomas and a long history of subclinical symptoms that usually ranges between 4 and 10 years.62 GH excess classically presents as acromegaly or gigantism, both marked by an insidious coarsening of features with frontal bossing and prognathism, macroglossia, and exaggerated acral growth. Patients are also found to develop organomegaly, hypertension, cardiomyopathy, congestive heart failure, restrictive lung disease, sleep apnea, and arthropathies. In addition, a very high percentage of these patients develop impaired glucose metabolism and diabetes mellitus. Common presenting symptoms include fatigue, headaches, arthralgias, oily skin, and hyperhidrosis. For patients with variant tumors that also cosecrete PRL, amenorrhea, galactorrhea, and loss of libido may also be present. With an insidious onset and high rate of medical comorbidities at time of presentation, untreated patients have a high rate of early mortality- as high as 50% at 50 years old and 2–3 times that of the general population.63
Diagnosis
Acromegaly is frequently suspected based on change of appearance and sub-clinical history. Hormone hypersecretion can be assessed through analysis of basal fasting GH levels or age- and gender-adjusted IGF-1. Elevated basal fasting GH levels greater than 2.5 ng/mL suggest acromegaly. However, because GH has both a short half-life and is secreted in a pulsatile manner, assessment of IGF-1 often generates a more accurate assessment of GH excess.64 Dynamic testing is typically reserved for monitoring therapeutic efficacy. The oral glucose tolerance test (OGTT) tracks GH response to a large oral glucose load (75–100g). Normal patients respond to the glucose load with GH suppression and levels fall below 1 ng/mL. Loss of glucose-induced suppression is diagnostic for a GH—secreting tumor. A study of 92 patients with acromegaly suggests that nadir levels of GH did not vary significantly based on either age or gender.65
Although rare, ectopic GHRH has been reported to account for approximately 1% of acromegaly cases. When responsible for GH excess, the ectopic GHRH production can usually be attributed to tumors in the chest or abdomen. To avoid unnecessary surgery, image confirmation of a pituitary tumor should be obtained after making a clinical and biochemical diagnosis.
Treatment
The clinical definition for cure of acromegaly has been refined several times since the introduction of the GH assay in the 1960s. Currently, the biochemical goals of therapy are to reduce basal GH levels to less than 2.5 ng/mL, normalize IGF-1 levels, and achieve a nadir GH level of less than 1 ng/mL during an OGTT.66 A complete cure should lead to recovery of normal pituitary function and, in most cases, a slow reversal of both cosmetic and physiological abnormalities.
Transsphenoidal surgery remains the first-line therapy for GH-secreting tumors.63 The advantages of surgery include a prompt decrease in GH levels and a tissue diagnosis. At our institution, a 14-year review of 115 patients demonstrated that surgery alone produced biochemical remission in 61% of cases.67 The rate was 88% for microadenomas and 53% for macroadenomas. There was a negative correlation of surgical outcome with tumor size and preoperative GH levels. Immediate postoperative GH levels were found to correlate with long-term outcome. In cases where the postoperative GH level was less than 3 ng/mL, the chance of a favorable long-term outcome was 89%. Postoperative radiotherapy was given to 32 patients and led to remission in 31% of these patients. An additional three patients achieved remission with a combination of surgery, radiotherapy, and medical treatment. The overall complication rate was 6.9% with no CSF leaks, meningitis, permanent diabetes insipidus, or new hypopituitarism. The recurrence rate was 5.4%. Other published studies reflect similar results.68, 69
Both radiotherapy and radiosurgery have been used for treatment of acromegaly. The use of conventional external beam radiation has been found to reduce GH levels to less than 5 ng/mL in 50–70% of cases, but the effects can take as long as a decade to occur.70 Hypopituitarism has been found in 50% of patients 10 years after irradiation, with an increasing incidence annually thereafter.71 Radiosurgery has demonstrated normalization of GH levels within 3–5 years.72 The likelihood of hypopituitarism is similar. In addition, radiation to sensitive adjacent structures such as the optic chiasm remains a concern and a 4-mm safety boundary is recommended for radiotherapy for all pituitary adenomas. In general, radiosurgery continues to be an adjunctive therapy after failed transsphenoidal surgery.68, 73, 74 These results are somewhat difficult to interpret as the definition of “normal” was inconsistent.57 Endocrine cure rates in a variety of small studies were found to range from 0% to 96% and improvement in 0–67%. Out of the 20 studies reviewed, 6 did not cite their criteria for cure, and 11 of 14 used different criteria to define cure. Unfortunately, the studies are further confounded by the inconsistent use of somatostatin analogs in patients undergoing radiosurgery. Current studies on the efficacy of radiosurgery also have limited long-term follow-up. Additional randomized studies are needed. A more recent retrospective study assessed endocrine cure rates by normal OGTT for patients with adjuvant radiosurgery and found that 65% of patients achieved remission at 61.5 months with a trend toward increased remission at later time points.75 In spite of normalization of the oral glucose tolerance, a subset of patients experience persistent elevation of IGF-1 after radiotherapy, demonstrating persistent pituitary axis dysfunction and subtle clinical symptoms of acromegaly.76
Medical management as the primary or adjuvant treatment of acromegaly remains a controversial topic. In patients with no visual loss, some endocrinologists have advocated treatment with somatostatin analogs as the first-line therapy.77 However, in the context of the significant long-term morbidity associated with acromegaly, most practitioners recommend surgery as the first-line treatment with adjuvant medical or radiotherapy, if needed. Three classes of drugs are used to treat acromegaly: dopamine agonists, somatostatin analogs, and GH-receptor blockers. Dopamine agonists such as bromocriptine and cabergoline have been shown to provide symptomatic relief in some acromegalic patients.78 Although they have limited efficacy, these medications remain popular treatments for acromegaly because of their low cost and oral administration. In comparison, somatostatin analogs have been shown to be more effective. Octreotide was found to normalize IGF-1 and reduce GH levels to less than 5 ng/mL in approximately 50% of patients.79 The long-acting analogs such as lanreotide and octreotide LAR are slightly more effective and reduce GH to less than 2.5 ng/mL for 70% of patients and normalize IGF-1 in 88% of patients.80–82 Another class of medication for treatment of acromegaly are GH-receptor antagonists. At this time, pegvisomant is the only medication in this class with FDA approval. This class of medications is unique in that the site of action is not within the pituitary gland but rather on the receptors for GH. By preventing dimerization of the GH receptor, pegvisomant inhibits production of IGF-1, the hormone responsible for most of the clinical sequelae of acromegaly. Because it does not affect tumor size or GH production, this medication is typically reserved for cases of acromegaly that are refractory to other treatments. The medication has notable side effects including hepatic transaminitis and an association with rare tumor growth.83 Because of this, it is recommended that patients prescribed pegvisomant be monitored with regular MRI exams and liver function tests.
Medical treatment demonstrates good results with regard to management of the clinical effects of GH excess. It has been found to provide symptomatic relief of headaches and hyperhidrosis as well as to improve arthropathy and cardiac function. If the medication is able to normalize IGF-1 levels and GH levels in the OGTT, patients experience significant improvement in left ventricular ejection fraction. This is especially notable as patients with uncontrolled acromegaly tend to also have deterioration of LVEF.84, 85 Unlike prolactinomas, medication does not cure acromegaly. Although current medical therapies are well tolerated and demonstrate good efficacy, lifelong treatment is required in order to avoid recurrence. This presents a serious downside to medical therapy as its success is both expensive and dependent on long-term compliance. Because of this, many practitioners and patients use chemotherapy as an adjuvant to either surgery or radiotherapy. There has not been statistically significant benefit demonstrated for the use of neo-adjuvant somatostatin analog treatment.86
A minority (20–30%) of GH-secreting adenomas also secrete PRL. If symptomatic, patients may experience amenorrhea, galactorrhea, impotence, or loss of libido. Dopamine agonists are particularly effective in the treatment of mixed GH- and PRL-secreting tumors, but transsphenoidal surgery still remains the first-line treatment.87
Adrenocorticotrophic hormone-secreting adenomas
Cushing’s disease represents the most frequent cause of clinical hypercortisolism, or “Cushing’s syndrome.” ACTH-producing tumors stimulate adrenal hyperplasia and ultimately result in hypercortisolism. Many consider Cushing’s disease the most challenging pituitary endocrinopathy both to diagnose and to treat. Tumors are often small and evade detection by radiological imaging. Macroadenomas are often aggressive and have a high rate of recurrence. Demographically, Cushing’s disease is nine times more common in women with a peak incidence in the third and fourth decades of life.
Clinical presentation
Only about 10–20% of ACTH-secreting adenomas are large enough to produce mass effect with resultant visual field deficits, cranial neuropathies, or hypopituitarism. The majority of tumors are less than 5 mm and present with clinical hypercortisolism. Common presenting features of Cushing’s disease include central obesity, moon facies, buffalo hump, hirsuitism, purple abdominal striae, and acne. Other clinical findings include hypertension, osteopenia, proximal myopathy, diabetes mellitus, and, very frequently, psychiatric disorders. Affected patients experience significant morbidity from the condition with a 5-year mortality rate of 50% in those left untreated.88 The increased morbidity and mortality are unique to secretory Cushing’s disease patients, thought to be sequelae of increased cortisol exposure. Among the predictors of increased mortality in Cushing’s patients, duration of clinical hypercortisolism, preoperative ACTH concentration, and depression have been shown to be significant.89
Diagnosis
Accurate diagnosis of Cushing’s disease depends on identifying evidence of hypercortisolism and localizing the pathology to the pituitary gland. Accuracy in diagnosis approaches 100% with a complete workup;90 however, each individual test has a failure rate of 10–30% (Table 1).91
Table 1 Biochemical evaluation for Cushing syndrome
|
|
Abbreviations: DST, dexamethasone suppression test; UFC, urinary free cortisol; CRH, corticotrophin releasing hormone; IPSS, inferior petrosal sinus sampling.
Hypercortisolism can be demonstrated through elevation in 24-h urinary free cortisol and 17-OH corticosteroids. Assays for midnight salivary cortisol have also been shown to be both sensitive and specific for hypercortisolism.92 The etiology of hypercortisolism should be assessed first with a low-dose dexamethasone suppression test. This test assesses functioning of the hypothalamic–pituitary–adrenal negative-feedback loop for cortisol secretion. Patients with Cushing’s disease will have hypercortisolism that persists in spite of a low-dose bolus of dexamethasone. However, 95% of Cushing’s disease patients demonstrate a 50% decrease in plasma cortisol when a high dose of dexamethasone is used. Plasma ACTH levels should be assessed to differentiate a diagnosis of Cushing’s disease from adrenal causes of hypercortisolism.93 Diagnostic sensitivity for Cushing’s disease can be further increased if a corticotrophin-releasing hormone (CRH) stimulation test is added to the workup.94 Finally, inferior petrosal sinus sampling (IPSS) can be performed to improve tumor localization and diagnostic specificity if the previous workup is not definitive. While MRI is a sensitive study for tumor localization for tumors larger than 3 mm in diameter, conventional sequences fail to detect a high percentage of tumors.95 However, with optimization of T1 postcontrast sequences or utilizing spoiled gradient recalled acquisition (SPGR), significantly more tumors can be visualized.95, 96 IPSS and cavernous sinus sampling can be helpful in these situations as they can confirm a pituitary pathology but laterality is incorrect in 25–30% of exams.97
Treatment
Transsphenoidal surgery is the first-line treatment for Cushing’s disease. The overall remission rate following surgery ranges from 76% to 91%.98 Microadenomas tended to have slightly better outcomes with remission rates between 84% and 94%98 The major determining factor for success was the ability to localize the tumor on MRI.99 In contrast, remission rates for macroadenomas appear to depend chiefly on the degree of invasiveness. Surgery alone achieves remission for 64% of patients, and when combined with adjuvant radiotherapy or radiosurgery, this number improves to 83% and 70%, respectively.98, 100
While the postoperative course is unique for every patient, many patients with a successful tumor resection experience a rapid decline in their serum cortisol and ACTH levels. In some patients, this leads to life-threatening hypocortisolism (Addisonian crisis). Most patients require replacement therapy for up to 1 year as their hypothalamic–pituitary–adrenal axis recovers from the chronic ACTH overstimulation. Good prognostic factors for full postoperative remission include lower than normal serums levels of cortisol and ACTH. However, even with immediate improvement in serum cortisol, 15–25% of patients experience a recurrence.21, 101 In our experience, even normal cortisol and restoration of a normal response to dexamethasone suppression do not guarantee lasting remission. Because of this, long-term follow-up is essential in these patients.
Radiotherapy and radiosurgery are frequently used as adjuvant treatment when surgical results are inadequate. Conventional radiotherapy cure rates are as high as 90% at 5 years post-treatment.102 However, a large proportion of these patients began to experience hypopituitarism in later years and 5% developed Nelson’s syndrome.102 Conventional radiation is not used as a primary modality because the effects of treatment take several years to develop while the symptoms and morbidities of Cushing’s disease persist. In contrast, radiosurgery for Cushing’s disease produces results much sooner. Remission is achieved in 35–90% of patients with much lower rates of hypopituitarism.103, 104 Full interpretation of these data is complicated by the fact that there was discrepancy in what constituted a “cure” and long-term follow-up was limited.
While medical therapy is not recommended as a primary treatment of Cushing’s disease, it may be suitable for patients who do not experience improvement after surgery or who are otherwise not good candidates for surgery. Pasireotide was approved by the US FDA for these indications in 2012. A somatostatin analog with high specificity for the somatostatin receptor 5, it has been shown to be clinically effective in reduction of cortisol levels and should be considered the preferred medication to treat Cushing’s disease.105 In addition, chemotherapy can temporize patients with severe hypercortisolism as they await surgery. In these cases, ketoconazole is often the drug of choice as it works to block adrenal steroid synthesis. Up to 90% of patients experience normalization of serum cortisol and ACTH while taking ketoconazole. However, the effects of this medication end immediately after cessation and patients on this medication must be closely monitored for hepatotoxicity.106 Other medical treatments for Cushing’s disease include aminoglutethimide, metyrapone, mitotane, and cyproheptadine. Dopamine agonists such as cabergoline have also been shown to be successful in the treatment of ACTH-secreting tumors. After 3 months of cabergoline treatment, 60% of patients demonstrated cortisol inhibition and 40% had normalization of cortisol secretion.107 This finding could be explained by the observation that the majority of ACTH-secreting adenomas demonstrate positive dopamine receptor (D2) immunostaining.107
There are more radical surgical options for patients who do not respond to the above measures. Total adrenalectomy offers absolute control of cortisol secretion but requires lifelong dependence on exogenous glucocorticoid and mineralocorticoid replacement. Nelson’s syndrome, a rapid enlargement of the pituitary adenoma from loss of cortisol negative-feedback inhibition, results in 10–30% of patients.108 Nelson’s syndrome typically presents with hyperpigmentation caused by stimulation of melanocytes by elevated ACTH. Most patients with Nelson’s syndrome harbor large, invasive pituitary adenomas. Studies examining the effectiveness of neurosurgery for treatment of Nelson’s syndrome have demonstrated variable rates of success.109–111 There appears to be some prophylactic value in hypophyseal radiosurgery before adrenalectomy.112
In the event that ACTH and cortisol levels remain elevated after surgery, additional exploration of the pituitary is usually effective.113 To increase the likelihood of success during the second surgery, IPSS should be performed before the operation. A complete hypophysectomy can be considered if previous treatments were unsuccessful but it is rarely indicated.
Silent ACTH-secreting adenomas
There is a subset of pituitary adenomas where immunohistochemical staining suggests ACTH production but the patient does not have clinical evidence of hypercortisolism. The exact incidence of this phenomenon is unclear but has been reported to be between 6% and 43%.114 There is evidence that these tumors behave more aggressively than other ACTH-producing tumors and have a 37% rate of recurrence.115, 116 However, when compared to ACTH-negative nonfunctioning pituitary adenomas, these tumors demonstrated similar regrowth rates.117 A subset of patients with these tumors also goes on to develop HPA axis dysfunction postoperatively and require postoperative steroid replacement.114
Gonadotropin-secreting and nonsecretory pituitary adenomas
Approximately one-third of pituitary tumors are considered nonsecretory adenomas. The peak incidence is in the fifth decade and patients typically present with symptomatic hypopituitarism, visual field defects, and visual concerns. Despite their similar clinical presentation, these tumors comprise a heterogeneous group of pathologies. Ultrastructural examination frequently demonstrates secretory granules and staining to suggest production of FH, LSH, and the α-subunit. A smaller subset produces other anterior pituitary hormones without clinical manifestation.118 With such a heterogeneous group of tumors, there is no clear or consistent treatment algorithm or prognosis.
Clinical presentation
Because they are not associated with hypersecretion syndromes, patients with these tumors usually present with symptoms of mass effect. This can range from hypopituitarism to vision changes or headaches. The majority of cases are associated with a macroadenoma.
Treatment
Surgery remains the primary treatment of patients with inactive adenomas. The goals are to relieve the mass effect, restore pituitary function, and obtain a tissue diagnosis. As with other pituitary adenomas, a transsphenoidal approach is often preferred unless there is significant extrasellar extension. With no clear criteria for what is considered a cure, surgical efficacy is not as well described as with other tumors. Many patients do experience improvement in preoperative symptoms, and 70–80% experience significant improvement in visual function.119 Nearly 100% experience improvement in headaches120 and 15–57% with improvement in pituitary function.119 However, this improvement is largely due to decompression of mass effect and should not be taken to be indicative of complete remission. The extent of resection should be assessed routinely with an MRI 3–4 months postoperatively. Imaging acquired earlier than this is often confounded by edema and otherwise normal postoperative changes. Residual tumor is identified in 66–86% of patients but the rate of recurrence is highly variable.121, 122
Radiotherapy or radiosurgery is increasingly utilized either for primary or adjuvant treatment of nonsecretory adenomas. A recent multicenter study analyzing the safety and efficacy of radiosurgery for residual lesions found that overall tumor control was achieved in greater than 90% of patients at follow-up with low incidence of side effects.123 The same group later went on to study radiosurgery as a primary treatment modality in patients not well suited for surgery. They found tumor control in 85% of patients at 10 years with a 24% rate of new or worsened hypopituitarism.124 Because of the risks of radiation to the pituitary, many clinicians advocate reserving radiotherapy only for tumors that demonstrate growth in serial postoperative imaging.125, 126 Studies of the natural history of these tumors have demonstrated a difference in growth patterns and proliferation index (MIB-1) between patients younger patients and those 61 years of age or older.127 Time to tumor doubling was significantly longer for older patients, suggesting that adjuvant treatment may not always be necessary in this age group.
In addition, medical therapy has only limited efficacy for this class of tumors. In a study of nine patients with nonsecretory adenomas treated for 1 year with cabergoline, tumor dopamine reception expression was demonstrated in 67%, but only 56% demonstrated response to cabergoline.128 This response was clinically significant in only two patients. Other medical treatments such as bromocriptine, other dopamine agonists, octreotide, and GnRH agonists or antagonists have been used but their results have been inconsistent.
TSH-secreting pituitary adenomas
TSH-secreting adenomas are the least common variant of pituitary tumors, representing only 1–2% of cases.129
Clinical presentation
Patients present with signs and symptoms suggestive of hyperthyroidism, such as heat intolerance, diarrhea, visual changes, weight loss, tremulousness, fatigue, and exophthalmos. This frequently leads to a work up and presumptive diagnosis of Graves’ disease. As a result, patients with TSH-secreting pituitary adenomas are often diagnosed later in their course of illness, when their tumors are large and locally invasive. Thus, they may also suffer from symptoms related to tumor mass effect, such as headaches, vision changes, and hypopituitarism.130
Diagnosis
The majority of TSH-secreting adenomas is associated with elevated levels of TSH, in spite of elevated T3 and free T4. There is increased diagnostic sensitivity with an increased α-subunit/TSH ratio. Finally, ultrasensitive immunometric TSH assays allow for distinction between patients with primary hyperthyroidism from those with central hypothyroidism or resistance to thyroid hormone.
Treatment
Hyperthyroidism presents significant anesthesia and surgical risk owing to its effects on heart rate and cardiac function. Therefore, treatment of hyperthyroidism before surgery is essential. This is frequently accomplished through beta blockade. If surgery is nonemergent, an antithyroid drug (propylthiouracil or methimazole) may also be added. Transsphenoidal surgery is the primary treatment for this type of tumor but is associated with low rates of remission (35–62%). When adjuvant medical therapy or radiotherapy is added, the rate of remission increases to 55–81%.131–134 Remission is frequently characterized by resolution of clinical symptoms of hyperthyroidism. The criteria for remission are not well established. External radiation is used if clinical remission is not attained with surgery alone but is not used as first-line treatment.
Octreotide has demonstrated clinical efficacy in 92% of cases and is associated with a normalization of TSH and tumor shrinkage for the vast majority.135 While useful as an adjuvant therapy, significant long-term costs and continued compliance make octreotide unpopular for the primary treatment modality. Recent reports suggest that lanreotide, a long-acting octreotide analog, may have similar efficacy while avoiding some of the downsides of octreotide.
Conclusion
Pituitary adenomas are a heterogeneous group of tumors requiring treatments specific to their underlying pathology. Transsphenoidal surgery remains the most common intervention while medical and radiation therapies play important roles in long-term control. As our understanding of molecular biology, drug development, and radiosurgery evolves, there will no doubt be further improvements to clinical outcomes and patient satisfaction.
Summary
Pituitary neoplasms arise from the adenohypophysis and can be classified based on their cell type as well as their radiographic appearance. Transsphenoidal surgery is the treatment of choice for all adenomas except prolactinomas, for which medical therapy is the primary therapy. Hormone-specific chemotherapy and radiotherapy have demonstrated clinical benefit for many tumor subtypes. Prognosis depends heavily on hormonal control as the systemic effects from hormone overproduction are often more severe than mass effect from the neoplasm. Research developments into the genetics of these tumors as well as their tumor biology have led to a number of promising targets for future therapies.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


