The Central Brain Tumor Registry of the United States (CBTRUS) estimated 51,410 new cases of malignant and nonmalignant brain tumors in 2007. Among children the incidence is 4.5 cases per 100,000 person-years. Among adults malignant tumors are estimated at 7.3 per 100,000 person-years and nonmalignant tumors at 9.2 cases per 100,000 person-years. An early peak in incidence starts at birth and extends to 4 years of age; after age 24 a gradual rise in incidence occurs, leading to a second peak at 50–79 years. For 2008 the SEER Cancer Statistics Review estimates that cancers of the brain and nervous system will account for 1.5% of all new cancer cases and 2.3% of cancer deaths annually (SEER 1975–2005). The relative risk of central nervous system (CNS) malignancy is 1.38 male to female, 3.18 elderly to young adult, and 1.86 Caucasian to African-American. In children, CNS tumors are the most common solid neoplasms and are the second leading cause of cancer deaths in patients younger than 15 years of age (SEER 1975–2005). The American Cancer Society estimates that in 2009 there will be 12,010 new cases of men with brain cancers with 7330 deaths and 10,060 new cases of women with brain cancer with 5590 deaths ( ).
The fourth edition of the World Health Organization (WHO) classification of primary brain tumors ( ) is presented in Figure 14.1 . All but the least common primary and secondary neoplasms of the CNS are reviewed in this chapter. Gliomas account for 36% of all primary brain tumors and 81% of malignant tumors (CBTRUS). Among these glioblastoma is the most common, accounting for at least 50% of cases. Meningiomas account for 32.1%, and pituitary tumors, nerve sheath tumors, lymphomas, medulloblastomas, and craniopharyngiomas range from 0.8% to 9%. Spinal cord neoplasms account for fewer than 15% of CNS tumors, and 10% of these represent spinal metastases from a primary intracranial tumor. Of all primary tumors of the spinal cord, schwannomas and meningiomas each account for 30%, ependymomas 13%, sarcomas 12%, astrocytomas 7%, and chordomas 4%. The distribution of CNS tumors varies with age: 90% of adult brain tumors are supratentorial, whereas 70% of childhood brain tumors arise in the posterior fossa. The distribution and differential diagnoses of CNS tumors are given in Figure 14.2 and Table 14.1 . Pituitary tumors, which represent between 5% and 15% of all brain tumors, are discussed in Chapter 7 .

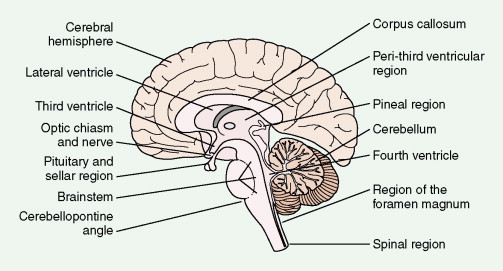
| Region | Adult Tumors | Childhood and Adolescent Tumors | ||
|---|---|---|---|---|
| Cerebral hemisphere | Astrocytoma Anaplastic astrocytoma Glioblastoma Meningioma | Metastatic carcinoma Oligodendroglioma Ependymoma Lymphoma Sarcoma | Astrocytoma Anaplastic astrocytoma Ependymoma | Oligodendroglioma Embryonal tumor Ganglion cell tumor |
| Lateral ventricle | Ependymoma Meningioma Subependymoma | Choroid plexus papilloma | Ependymoma Choroid plexus papilloma | Subependymal giant cell astrocytoma |
| Third ventricle | Colloid cyst | Ependymoma | Ependydoma | Choroid plexus papilloma |
| Peri-third ventricular region | Astrocytoma Anaplastic astrocytoma | Oligodendroglioma Ependymoma Pilocytic astrocytoma Glioblastoma | Pilocytic astrocytoma Astrocytoma | |
| Pineal region | Germ cell tumor Pineal parenchymal tumor | Glioma | Germ cell tumor | Pineal parenchymal tumor |
| Optic chiasm and nerve | Meningioma | Astrocytoma | Astrocytoma | |
| Pituitary and sellar region | Pituitary adenoma Craniopharyngioma | Meningioma Germ cell neoplasms | Craniopharyngioma Germ cell neoplasms | Pituitary adenoma |
| Corpus callosum | Astrocytoma Anaplastic astrocytoma | Glioblastoma Oligodendroglioma Lipoma | Astrocytoma Anaplastic astrocytoma | Oligodendroglioma Lipoma |
| Brain stem | Astrocytoma Anaplastic astrocytoma | Glioblastoma | Astrocytoma Anaplastic astrocytoma | Glioblastoma |
| Cerebellopontine angle | Schwannoma Meningioma Epidermoid cyst | Choroid plexus papilloma | Ependymoma | |
| Cerebellum | Hemangioblastoma Metastatic carcinomas | Astrocytoma Medulloblastoma | Medulloblastoma | Dermoid cyst Astrocytoma |
| Fourth ventricle | Ependymoma Subependymoma | Choroid plexus papilloma | Ependymoma | Choroid plexus papilloma |
| Region of foramen magnum | Meningioma | Schwannoma | ||
| Spinal region | Ependymoma Astrocytoma Hemangioblastoma Meningioma | Schwannoma Neurofibroma Paraganglioma | Ependymoma | Astrocytoma |
The biologic potential of CNS neoplasms depends largely on three factors: (1) the histology and degree of malignancy (grade) of the tumor; (2) the anatomic compartments involved (cerebral hemisphere, basal ganglia, posterior fossa, brain stem, third ventricle, visual system, spinal cord, etc.); and (3) the spatial delimitation of the tumor (e.g., diffuse, circumscribed, multifocal). CNS tumors of low histologic grade may have as poor a prognosis as high-grade malignancies if they are considered surgically unresectable—because they show a diffusely infiltrating growth pattern, because they involve a critical anatomic structure, or because they are technically unapproachable by surgery.
There has been an increase in the incidence of primary malignant brain tumors over the past 25 years, with rates increasing at approximately 1.2% per year, particularly among the elderly. This increase does not seem to be related to an increase in lifespan over this same period of analysis. Although there have been significant improvements in diagnostic capabilities over the past 25 years, there is growing concern that the increase in incidence reflects exposure to an unrecognized environmental toxin. The only known environmental risk for malignant brain tumors is irradiation to the brain in childhood, usually as part of treatment for leukemia ( ) or fungal infection of the scalp ( ). Large epidemiologic studies have not identified absolute environmental risks, but there have been trends toward increased risks from vinyl chloride, pesticides, or fungicides, chemicals used in the rubber industry, and electronic and electrical equipment ( ). In addition, it was reported in 2005 that Gulf War veterans exposed to sarin nerve gas have a 2.5-fold increase in fatal brain tumors when compared with unexposed veterans in the same theater of operations ( ).
Despite the recent heightened concern that the low-level radiation associated with cellular telephone use poses an increased risk for the development of brain tumors, a meta-analysis of nine case-control studies containing 5259 cases of primary brain tumors and 12,074 controls did not detect an overall risk (OR 0.90, 95% confidence interval [CI] 0.81–0.99). However, more than 10 years of use had an OR of 1.25 (95% CI 1.01–1.54) ( ), suggesting that longer-term follow-up may be necessary to adequately evaluate risk.
There are well-recognized associations between malignant brain tumors and familial syndromes of germline mutations, although these account for only a small proportion of total cases ( ). Patients with Li-Fraumeni syndrome carry a germline mutation in TP53 and develop a variety of tumors, including those of bone, breast, blood, adrenal cortex, and brain. The majority of brain tumors are gliomas, predominantly low grade, and occasional glioblastomas. Less common familial syndromes include neurofibromatosis type 1 (NF1), linked to a gene on chromosome 17, which is associated with nerve sheath tumors, astrocytomas, and meningiomas in 5% to 10% of patients. Patients with neurofibromatosis type 2 (NF2) carry a genetic mutation on chromosome 22 that predisposes to schwannomas and meningiomas of the cranial nerves and spinal nerve roots, as well as astrocytomas in rare cases ( ). Tuberous sclerosis, associated with two distinct inherited loci, 9q34 ( TSC1 ) and 16p13 ( TSC2 ), predisposes to subependymal giant cell astrocytomas and subcortical glioneuronal hamartomas in addition to a wide variety of non-CNS tumors. Turcot’s syndrome, familial intestinal polyposis, results from a mutation of 5q21 ( ANAPC1 ) and predisposes to medulloblastoma. Other patients with this syndrome have lesions in 3p21 ( MLH1 ) or 7p22 ( GPSM2 ), both associated with glioblastoma at low frequency. Medulloblastoma is also associated with Gorlin syndrome, resulting from a mutation of 9q31 ( PTCH1 ). In some instances primary brain neoplasms constitute an essential feature of the familial syndrome, as for example cerebellar hemangioblastoma in von Hippel-Lindau syndrome, which results from a lesion in the VHL gene (3p25).
Sporadic mutations seem to play a major role in the genesis and maintenance of brain tumors, although how the genetic pathways govern the biologic behavior of the tumors is largely unknown. The data are perhaps strongest for gliomas, wherein mutations in cell cycle control and receptor tyrosine kinase pathways are common (see below).
Primary Neoplasms of the Central Nervous System
TUMORS OF NEUROEPITHELIAL TISSUE
In the adult, over 60% of all primary CNS tumors are gliomas. In children gliomas constitute 80% to 90% of all CNS neoplasms. Gliomas have been defined pathologically as tumors that display histologic, immunohistochemical, and ultrastructural evidence of glial differentiation. They are classified according to their differentiation lineage (i.e., astrocytic, oligodendroglial, or ependymal cells) and further subdivided by tumor grade ( ); see below.
Astrocytic Tumors
Astrocytoma
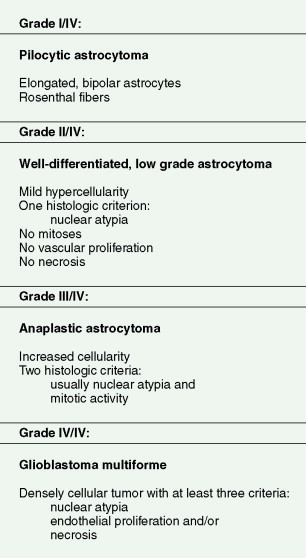
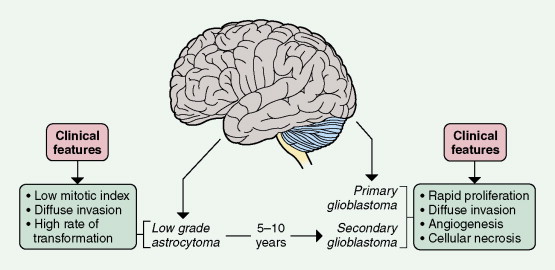
Astrocytomas range in incidence from five to seven new cases per 100,000 population per year and are predominantly diffusely infiltrating tumors. Although they can arise anywhere in the CNS, they preferentially develop in the cerebral hemispheres. Three histologic types are recognized: fibrillary, gemis- tocytic, and protoplasmic. Of these, fibrillary astrocytoma is by far the most common and protoplasmic astrocytoma the most unusual. Astrocytomas are graded on a scale of I–IV according to their degree of malignancy as judged by various histologic features (see below). Unlike other solid tumors, gliomas do not metastasize outside the CNS, and thus tumor grade is the primary determinant of clinical outcome. Grade I tumors are biologically benign and can be surgically cured if deemed resectable at the time of diagnosis. Grades II–IV tumors are diffusely infiltrating tumors and are incurable with current therapies. They differ in their aggressiveness, with grade II tumors, referred to as low-grade gliomas, often following long clinical courses (see below) and grade III tumors initially responding well to chemotherapy and radiation therapy but usually progressing to death within 3 years. Grade IV tumors (glioblastoma) have a median survival of 14.6 months when treated with the standard regimen of concurrent temozolomide and radiation therapy followed by 6–12 months of adjuvant temozolomide ( ). A subset of patients seems to have prolonged survival with this regimen, although the determinants of the response have not yet been elucidated. Seventy percent of grade II gliomas in adults transform into grade III and IV tumors within 5–10 years of diagnosis and then behave clinically like the higher- grade tumors.
The diffuse gliomas are classified histologically as astrocytomas, oligodendrogliomas, or tumors with morphologic features of both astrocytes and oligodendrocytes, termed oligoastrocytomas. Astrocytic tumors are subsequently graded as pilocytic astrocytoma, grade I; astrocytoma, grade II; anaplastic astrocytoma, grade III; and glioblastoma, grade IV. Oligodendrogliomas and oligoastrocytomas are subsequently graded as grade II or anaplastic, grade III. Such grading is related to the presence of histologic features of malignancy, such as high cellularity, cellular pleomorphism, mitotic activity, microvascular proliferation, and necrosis ( Fig. 14.3 ).
Diffuse, Low-Grade Astrocytoma (WHO Grade II/IV)
The clinical hallmarks of low-grade astrocytomas are low mitotic rate, ability to migrate long distances away from the original site of tumor development, and high propensity to progress to a higher-grade tumor after a long latency. These are tumors primarily of young adults, with peak age of incidence at 34 years, and often present initially with seizures. The tumor cells are well differentiated, show robust glial marker immunoreactivity, and are not associated with neovascularization or cellular necrosis. Magnetic resonance imaging (MRI) often demonstrates a diffuse large mass that is hypointense on T 1 -weighted imaging and does not enhance following administration of gadolinium. Whereas the reported median survival approaches 10 years, approximately 70% of patients transform to high-grade astrocytomas within 5 years of initial diagnosis (see Fig. 14.5A and B ), the remaining 30% die of infiltrating low-grade tumor. Surgical resection is the primary modality of treatment. Although radiation therapy is associated with prolongation of progression-free survival, there is no increase in overall survival when compared to surgery alone. Chemotherapy, utilizing BCNU (carmustine) or temozolomide, has not been shown to prolong either progression-free survival or overall survival. The basic strategy is to follow patients with serial MRI scans and start radiation therapy, with or without chemotherapy, at the time of progression or transformation to high-grade tumor. Mutational analysis of these tumors has identified two common genetic lesions: p53 loss-of-function mutations ( ) and platelet-derived growth factor ligand and receptor overexpression ( ). Whole-genome high-resolution-array comparative hybridization has identified additional copy number gains and losses (E. Maher, unpublished observations) and may ultimately yield insights into genes and pathways governing tumor maintenance and the transition to high-grade astrocytoma.
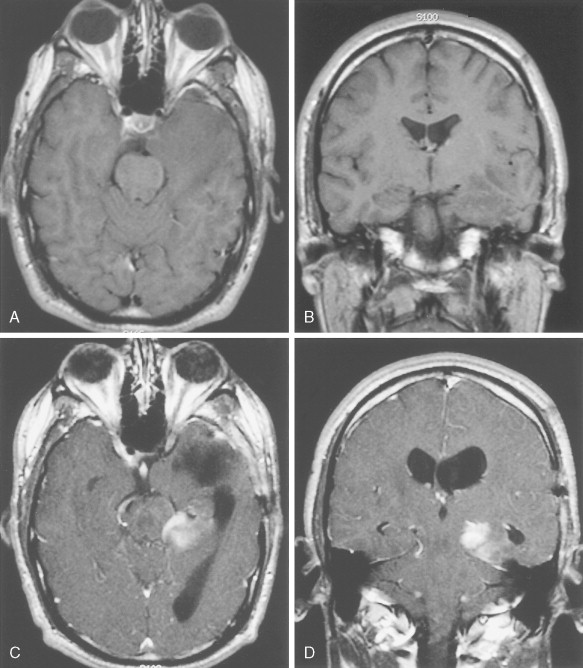
Anaplastic Astrocytoma (WHO Grade III/IV)
Anaplastic astrocytomas, also referred to as intermediate-grade astrocytomas, may arise de novo or develop from low-grade lesions. They are characterized histologically by nuclear atypia, increased cellularity, and a significant increase in mitotic rate over that seen in low-grade lesions without induction of neovascularization. MRI demonstrates enhancement of tumor following administration of gadolinium in approximately 80% of cases ( Fig. 14.8 ). The median age at diagnosis is 41 years. Patients present with symptoms similar to those described above for patients with low-grade astrocytomas. Survival is significantly shorter than with low-grade astrocytomas, ranging from 3 to 4 years. Treatment consists of surgery, external-beam irradiation, and chemotherapy using temozolomide. Genetic mutations associated with anaplastic astrocytomas include allelic losses on chromosome 9p or 13q, and, less frequently, by 12q amplification. Notably, these mutations are mutually exclusive events ( ) and are key components of the retinoblastoma pathway governing cell cycle progression.
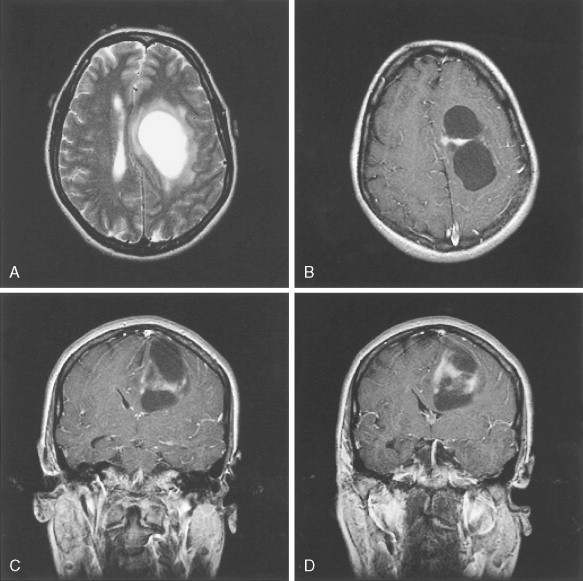
Glioblastoma Multiforme (WHO Grade IV/IV)
Two glioblastoma subtypes have been identified clinically ( ) ( Fig. 14.11 ). “Primary glioblastoma” typically presents in older patients as an aggressive, highly invasive tumor, usually without any evidence of prior clinical disease. “Secondary glioblastoma” has a very different clinical history. It is usually observed in younger patients who initially present with a low-grade astrocytoma that transforms into glioblastoma within 5–10 years of the initial diagnosis, regardless of prior therapy. Despite their distinctive clinical courses, they arrive at an indistinguishable clinical and pathologic endpoint characterized by widespread invasion and resistance to therapy. MRI is characterized by a diffuse enhancing mass, often with areas of necrosis. As such, tumors are managed as if they are one disease entity. However, global genomic analysis of these two glioblastoma subgroups showed wide-scale differences in their genomes that were previously unappreciated. Secondary glioblastoma was further classified into two distinct molecular subclasses, one characterized by multiple regions of loss and the other characterized by gain of chromosome 7 (without EGFR amplification) and several regions of gain and loss. Primary glioblastoma was characterized by the classic findings of EGFR amplification, and by chromosome 9p21 and chromosome 10 loss ( ). Ongoing studies are directed at functional characterization of the unique genes and pathways in the molecular subclasses.
The treatment of glioblastoma has evolved over the past several years with the demonstration that treatment with temozolomide, an oral alkylating agent, when given concurrently with radiation therapy as initial therapy after surgical resection or debulking and as adjuvant therapy for six cycles, improved overall survival from 12.1 to 14.6 months and 2-year survival from 10.4% to 26.5% when compared with surgery followed by radiation therapy alone ( ). Correlation of methylation status of MGMT , a gene that repairs DNA after alkylation, with survival in patients treated with combined temozolomide and radiation therapy demonstrated marked prolongation of survival in the patients with MGMT , with approximately 40% alive at 3 years ( ). The predictive potential of MGMT status is currently under evaluation in a large multicenter international study (RTOG 0525; http://www.rtog.org/members/protocols/0525/0525.pdf ). Assessment of tumor response has been improved by the addition of 2-[ 18 F]-fluoro-2-deoxy- d -glucose–positron emission tomography (FDG-PET) imaging for differentiation between true progression and treatment effect and/or radiation necrosis ( Fig. 14.16A and B ).
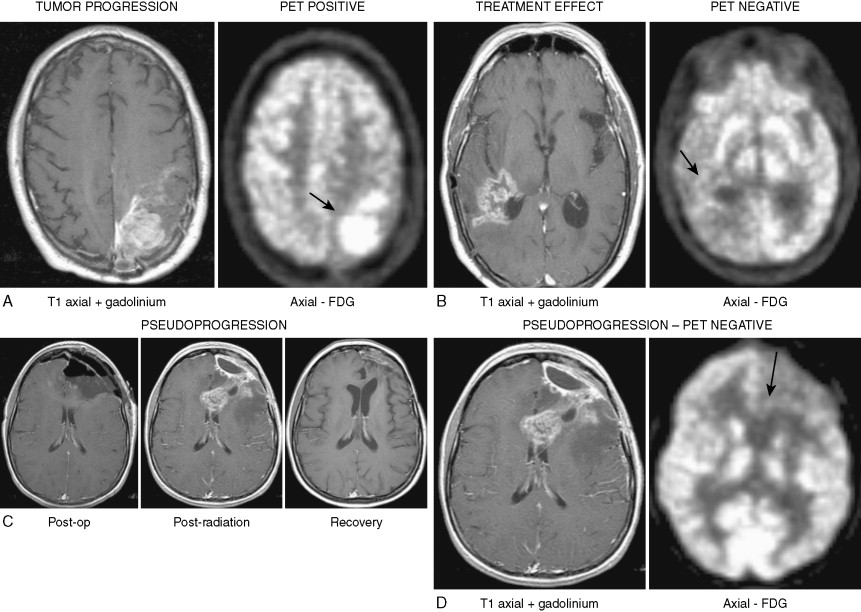
A well-recognized complication of the combined treatment is the development of “pseudoprogression,” which is the development of enhancement and T 2 /FLAIR abnormalities on MRI at the completion of concurrent temozolomide and radiation therapy ( Fig. 14.16C ), most often without clinical deterioration ( ). Despite MRI findings that are often indistinguishable from true progression, FDG-PET shows no uptake, the imaging abnormalities resolve over 2–6 months, and the patients may have long disease-free intervals ( Fig. 14.16D ). Patients who have undergone reoperation seem to consistently have necrosis without clear evidence of recurrent tumor. The pathobiology (reviewed in ) seems to be consistent with treatment-related exaggerated local tissue reaction with an inflammatory component, edema, and abnormal vessel permeability leading to increased contrast enhancement. In severe cases this can lead to treatment-related necrosis. The condition may be self-limiting or require prolonged steroid administration and, in severe cases, reoperation.
Gliosarcoma
Gliosarcoma is a variant of glioblastoma characterized by the presence of both glial and sarcomatous elements. The origins of this tumor are unknown, although it has been speculated that it represents malignant transformation of a neural stem cell or glial progenitor that retained the ability to differentiate into both glial and mesenchymal lineages. Gliosarcomas carry the same prognosis as glioblastomas, and the general approach to treatment is the same as that described above for glioblastoma.
Pilocytic Astrocytoma (WHO Grade I/IV)
These tumors of childhood and adolescence differ from the diffuse astrocytomas previously discussed in that they are relatively well circumscribed and of low grade with little potential for malignant transformation. They are uncommon in the cerebral hemispheres and show geographic preferences for the region of the third ventricle, optic chiasm, and thalamus. Surgical resection is associated with long-term survival. Pilocytic astrocytomas are not associated with TP53 mutations, suggesting a different genetic basis for these low-grade tumors.
Pleomorphic Xanthoastrocytoma
These rare tumors occur most often in the temporal or parietal lobe of young people (third or fourth decade) with a history of epilepsy. Usually there is prominent leptomeningeal involvement; underlying cyst formation with mural nodules is also typical. These tumors are typically densely cellular and cytologically pleomorphic. However, mitoses are rare and necrosis is absent. The tumor is notable because it has a favorable prognosis yet bears superficial resemblance to a giant cell glioblastoma or malignant fibrous histiocytoma. Some tumors may eventually develop malignant transformation.
Subependymal Giant Cell Astrocytoma
Though characteristically associated with tuberous sclerosis, subependymal giant cell astrocytoma occasionally occurs in the absence of the disease. It usually arises from the wall of the lateral ventricle and presents as an intraventricular mass obstructing the foramen of Monro. The clinical signs are commonly those of obstructive hydrocephalus. Subependymal astrocytomas are low-grade tumors, with essentially no tendency for malignant transformation.
Astrocytoma: Sites of Preference
OPTIC NERVE AND CHIASMAL ASTROCYTOMA
Representing 1% of intracranial neoplasms in adults and 5% of intracranial tumors in children younger than 10 years old, optic nerve and chiasmal astrocytomas most commonly (≈70%) arise in the first decade. The most frequent symptom is visual loss, which may be pronounced. Bilateral optic astrocytomas may arise in association with von Recklinghausen’s neurofibromatosis, more often affecting the chiasm than the optic nerves. Although malignant transformation is rare, it occurs more frequently in adults with chiasmal lesions. Treatment is surgical; however, 20% of optic nerve tumors and 33% of optic chiasm tumors recur. The 20-year survival rate for optic nerve astrocytomas is 85%, as compared with 50% for optic chiasm tumors. The tumor grows by local extension, and chiasmal tumors frequently extend into the third ventricle or the optic tract. The histology is that of a pilocytic astrocytoma.
ASTROCYTOMA OF THE THIRD VENTRICULAR REGION
Both pilocytic astrocytomas and diffuse astrocytomas may be found in this site, most commonly in children. Although such tumors are benign and slow growing, their deep location limits surgical resection. The clinical signs are usually those of obstructive hydrocephalus.
BRAINSTEM ASTROCYTOMA
Most commonly occurring in children, this tumor usually presents as a diffuse astrocytoma originating in the pons. As with astrocytomas of the third ventricle, surgical resection is hindered by the deep location and infiltrating character of this tumor. Malignant transformation is frequent and may occur early in the disease course. The clinical signs include symptoms of brain stem dysfunction and cranial nerve palsies. Obstructive hydrocephalus occurs late in the course as a result of obstruction of the fourth ventricle. The prognosis depends on tumor grade; 30% of patients with well-differentiated astrocytomas survive for 15 years. Patients with high-grade astrocytomas have a typical survival time of less than 1 year. Occasionally brain stem astrocytomas are of the discrete pilocytic type, which is associated with prolonged survival.
CEREBELLAR ASTROCYTOMA
Accounting for 5% of all brain gliomas and 15% of all intracranial tumors of children and adolescents, cerebellar astrocytomas may be either diffuse (15%) or, more commonly, pilocytic (85%). The presenting signs are usually those of cerebellar dysfunction and hydrocephalus resulting from obstruction of the fourth ventricle. Surgical resection, even if partial, is associated with long-term survival. Malignant transformation and cerebrospinal dissemination are rare.
SPINAL CORD ASTROCYTOMA
Representing approximately 13% of all neoplasms affecting the spinal cord, these tumors commonly appear as fusiform enlargements affecting the thoracic and cervical segments. Diffuse low-grade fibrillary astrocytoma is the usual histologic type, although high-grade astrocytomas may occur. As many as 40% of these tumors are associated with proximal or distal syringomyelia. The prognosis is related to tumor grade. Mean survival time for patients with well-differentiated tumors may be as long as 8 years, whereas with high-grade lesions it may be as short as 6 months. Death is usually the result of intercurrent infection or medullary extension of the tumor.
Oligodendroglial Tumors
Oligodendroglioma
Constituting 4% of all CNS neoplasms and 5% to 19% of all gliomas, oligodendroglioma is predominantly a tumor of the middle decades, with a peak incidence between 35 and 40 years, although it occasionally arises in younger persons. Considered to be tumors of the white matter, they have geographic predilections based largely on the amount of white matter in a given location. Sites of preference include the frontal, parietal, and temporal lobes of the cerebral hemispheres, as well as the thalamus, particularly in the younger age groups. They occur rarely in the spinal cord and extremely rarely in the cerebellum. The clinical evolution may be prolonged and is frequently characterized by a long history of seizures. Calcification in these tumors is common, detectable radiographically in 40% of cases and histologically in 90%. Although previously graded like astrocytomas, the most recent WHO classification no longer recognizes glioblastoma as a grade of oligodendrogliomas. Thus, these tumors are grade II or maximum III, even when necrosis and neovascularization are present. This change reflects the clear difference in biologic behavior of the highest-grade tumor when compared to glioblastomas of astrocytic origin. The high-grade oligodendrogliomas are often exquisitely sensitive to the standard glioma treatments, PCV (combination therapy with procarbazine, CCNU [lomustine], and vincristine) or temozolamide (see response demonstrated in Fig. 14.22 after five cycles of chemotherapy), and median survival is often significantly longer than in patients with anaplastic astrocytomas. Genetic analysis of these tumors demonstrates a high incidence of mutations in 1p and 19q. Although the specific genes mutated in these tumors have not yet been identified, they are likely to be involved in conferring the chemosensitivity of these tumors.
Mixed Glioma (Oligoastrocytoma)
Mixed gliomas are tumors that clearly demonstrate both malignant oligodendrocytes and astrocytes. Similar to gliosarcoma, the origin of these tumors is unknown. They may represent malignant transformation of a neural stem cell or early glial progenitor. The molecular genetics are less clear than for pure oligodendrocytes; some have the characteristic 1p and 19q deletions, whereas most have a genetic profile similar to anaplastic astrocytomas. Treatment is similar to anaplastic astrocytomas, although prognosis may vary depending on the genetic profile of the tumor.
EPENDYMAL TUMORS
Ependymoma
Ependymomas represent approximately 3% to 9% of all neuroepithelial tumors. They are primarily tumors of childhood and adolescence, with peak incidence occurring between 10 and 15 years. They represent 6% to 12% of all intracranial tumors in childhood and a striking 30% in children under 3 years of age. The tumors can occur at any site along the ventricular system and spinal canal but are predominantly found in the fourth ventricle and spinal cord. Embryologically the ependyma is related to astrocytes and oligodendroglia, a glial heritage that is often expressed when the cells are neoplastically transformed. Characteristically, ependymomas are benign, slow-growing neoplasms; anaplastic transformation may occur, especially focally, but transformation to overt glioblastoma is rare. Because of its predominantly intraventricular location, symptoms are most often secondary to obstruction of cerebrospinal fluid (CSF) flow and resultant hydrocephalus. Tumors of the spinal cord are associated with symptoms related to the site of disease occurrence. The prognosis of ependymoma depends largely on the anatomic site of origin and the histologic grade. Long-term survivals tend to be the exception. Even benign-appearing tumors show a tendency to recur locally and metastasize via the subarachnoid space. Treatment is surgical resection, most often only partial, and radiation therapy.
Myxopapillary Ependymoma
These tumors represent a special variant of ependymoma found almost exclusively in the region of the filum terminale, although occasionally they have been found higher in the spinal cord or, rarely, in the brain. They may occur at any age, but most arise in the fourth decade. Myxopapillary ependymomas characteristically form a sausage-shaped mass in the lumbosacral region, displacing spinal nerve roots of the cauda equina. Their biologic behavior is usually benign, but because of their location they are often associated with significant compression-induced paralysis. Treatment consists of local excision, which must often be only partial because of the tumor’s location; approximately 20% recur even after complete initial resection. Metastases infiltrating the CSF and extradural space may occur, but transformation to anaplastic variants is extremely rare.
Subependymoma
This slow-growing, benign variant of ependymoma consists of proliferating ependyma and astrocytes. Seventy-five percent of these tumors are infratentorial, arising on ependymal surfaces. They are commonly found along the fourth ventricle, the walls of the lateral ventricles, the septum pellucidum, and the cerebellopontine angle. They are often an incidental finding at autopsy, particularly in the middle-aged and elderly. Symptomatic tumors may arise at any age, most commonly in the fourth decade, and show a male predominance. The clinical signs are usually those of hydrocephalus resulting from blockage of CSF flow through the ventricles. Treatment is surgical, and the prognosis depends entirely on the tumor’s location and resectability.
Choroid Plexus Tumors
Choroid Plexus Papilloma
Choroid plexus papillomas occur most frequently in the first decade of life, accounting for 10% to 20% of intracranial neoplasms in children; they are occasionally congenital. The lateral ventricle and third ventricle are the favored sites in children; the rare adult neoplasm favors the fourth ventricle. Symptoms are usually caused by hydrocephalus, which may result from mechanical obstruction to CSF flow or overproduction of CSF by the tumor. Although they are benign neoplasms and can be cured by surgery, they have a tendency to disseminate widely via the CSF, particularly after surgical intervention.
Choroid Plexus Carcinoma
This malignant tumor is distinguishable from choroid plexus papilloma on the basis of local brain invasion, a solid pattern of growth and cytologic features of anaplasia, including necrosis and mitoses. Choroid plexus carcinoma almost always occurs in patients under the age of 10, grows more rapidly than choroid plexus papillomas, and has a 5-year survival rate of approximately 40%. In older individuals it should be distinguished from the much more common metastatic papillary adenocarcinoma.
NEUROEPITHELIAL TUMORS OF UNCERTAIN ORIGIN
Gliomatosis Cerebri
This extreme form of diffuse astrocytoma in adults is characterized by widely infiltrating anaplastic glia, although the cell of origin is unknown. It typically presents in the second or third decade and diffusely enlarges the cerebral hemispheres, brain stem, and/or cerebellum. There is often expansion of compact fiber pathways, such as the optic nerves, corpus callosum, fornices, or cerebral peduncles. Its distinct clinical behavior is probably related to the overall very poor prognosis.
NEURONAL AND MIXED NEURONAL-GLIAL TUMORS
Gangliocytoma and Ganglioglioma
Gangliogliomas are distinguished from gangliocytomas (ganglioneuromas) by the presence of glial elements in gangliogliomas. Both tumors show a geographic predilection for the temporal lobes in children and young adults, and seizures are thus the most common presenting symptoms. However, these tumors occur in all brain regions, including the frontal lobes, third ventricle, and hypothalamus. They carry an excellent prognosis following surgical resection, although transformation of the glial elements in gangliogliomas can occur that then carry a less favorable prognosis.
Central Neurocytoma
The central neurocytoma is typically a tumor of young adults, in whom a discrete, often partially calcified mass intrudes into the lateral ventricle near the foramen of Monro. Symptoms are often related to increased intracranial pressure rather than focal neurologic deficits. Surgery may be curative if complete resection is achieved.
Paraganglioma
Paragangliomas are tumors derived from neural crest cells, the most common type of which is the pheochromocytoma. The designation also includes tumors of the carotid body, glomus jugulare, glomus tympanicum, filum terminale, vagus nerve, orbit, and duodenum. Certain of these tumors show a predilection for middle-aged women, such as the jugulotympanic paraganglioma, which usually arises from the lateral portion of the temporal bone, and the vagal body paraganglioma, which often presents as a mass in the neck or at the skull base beneath the jugular foramen. Clinically, these tumors manifest with signs of cranial nerve palsies. In the case of paragangliomas involving the cauda equina, which tend to be sausage-shaped intradural tumors, symptoms include lower back pain, sensorimotor deficits, and incontinence. Carotid body tumors present as painless masses of the skull base, where they may produce cranial nerve palsies, a palpable thrill, and an audible bruit. The incidence of carotid body tumors is markedly increased in regions of high altitude, possibly as a result of hypoxia-induced hyperplasia. An autosomal-dominant pattern of inheritance for these tumors has been recognized, and familial tumors may be bilateral. Most paragangliomas are benign and carry a favorable postoperative prognosis, although recurrences are not uncommon. Approximately 5% of these tumors are malignant and may invade tissue locally or metastasize to lymph nodes, lung, or bone marrow.
Olfactory Neuroblastoma (Esthesioneuroblastoma)
This rare neoplasm arises high in the nasal cavity from neurosensory receptor cells or basilar cells in the olfactory mucosa. The age distribution is bimodal, one peak occurring in adolescence and young adulthood and the second peak occurring in late middle age. Olfactory neuroblastomas are slow-growing but aggressive, locally invasive tumors that may invade the nasal sinuses, nasopharynx, palate, orbit, cribriform plate, and brain. Metastases to the CSF, lymph nodes, and viscera may occur. There seem to be several types of esthesioneuroblastomas, one with classic features of neuroblastoma, the type most likely to occur in young patients, and the other with characteristics of neuroendocrine carcinoma, more common in older patients. The importance of initial gross total surgical excision has been emphasized. Because the tumor is highly radiosensitive, radiation therapy is often indicated. The 10-year survival rate has been reported as 77% for patients with neuroendocrine carcinoma and 67% for those with neuroblastoma.
PINEAL PARENCHYMAL TUMORS
Pineocytoma and Pineoblastoma
These uncommon tumors, derived from pineal parenchymal cells, are divided into two types: the pineocytoma, originating from mature cells, and the pineoblastoma, derived from more primitive pineal cells. Pineocytoma, which may occur at any age, is typically well circumscribed, slow growing, and noninvasive, and it rarely metastasizes via the CSF. Its highly malignant anaplastic counterpart, the pineoblastoma, occurs primarily in children and frequently metastasizes via the CSF. Because pineocytomas tend to be less radiosensitive than pineoblastomas, their treatment usually includes surgical resection. Mean survival time is approximately 5 years for pineocytoma, whereas it is less than 2 years for pineoblastoma.
EMBRYONAL TUMORS
Medulloepithelioma
Believed to arise from the primitive medullary plate and neural tube, these rare, highly malignant tumors occur early in life, most frequently between the ages of 6 months and 5 years. The preferred geographic location is periventricular in the cerebral hemispheres; tumors are often deeply situated and lie near the midline. These tumors can also arise in the cauda equina, presacral area, outside the CNS along nerve trunks, and in the eye. Radical surgical removal, followed by extensive neuraxial irradiation, is the treatment of choice, given the highly primitive and malignant character of these tumors. Mortality is high, and extracranial metastases may occur.
Neuroblastoma (Cerebral)
Derived from ganglion cell precursors, central neuroblastomas are rare tumors, occurring most frequently in children in the first decade of life. They are frequently situated deep in the cerebrum, forming a well-defined mass. Approximately 50% disseminate via CSF pathways, and distant metastases may occur. Treatment consists of radical surgical excision followed by radiation therapy, in that the primitive character of these lesions suggests some degree of sensitivity to radiation therapy. The 5-year postoperative survival rate is approximately 30%.
Ependymoblastoma
Although their histologic designation is somewhat controversial, ependymoblastomas are distinguished from ependymomas by their highly malignant biologic behavior and the frequency of focal microscopic invasion and leptomeningeal involvement. They are rare tumors affecting predominantly the cerebral hemispheres of neonates and young children. They are generally large and supratentorial, closely approximated to the ventricles. They have a propensity for CSF seeding, rapid local growth, and extraneural and extracranial metastases.
PRIMITIVE NEUROECTODERMAL TUMORS
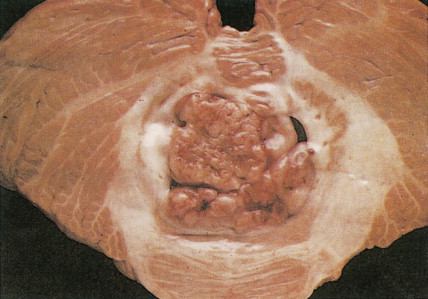
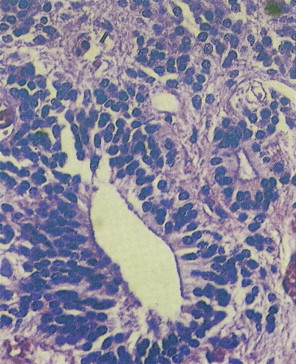
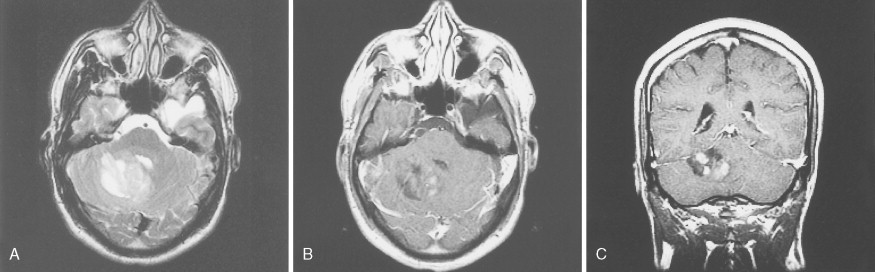
Medulloblastoma
These embryonic cerebellar tumors are believed to originate from remnants of the fetal external granular cell layer of the cerebellum. Overall, they account for less than 0.5% of intra- cranial primitive neuroectodermal tumors, but in children they represent 25% of intracranial tumors. Most arise in patients younger than 25 years of age, although occasionally they occur as late as the fifth decade and have a male predominance (65%).
Medulloblastomas arise in the cerebellum, particularly favoring the midline in early life, whereas in later life tumors tend to arise in the lateral hemispheres. The clinical signs are usually those of cerebellar dysfunction and increased intracranial pressure due to obstruction of the fourth ventricle. Medulloblastomas frequently infiltrate the subarachnoid space early and extensively and metastasize widely via CSF pathways. Systemic metastases to bone and lymph nodes may occur, although the lung characteristically remains free of metastatic deposits. Since these tumors are extremely radiosensitive, the treatment of choice is radiation therapy of the entire neuraxis, usually in combination with surgical extirpation. The 5-year survival rate ranges from 40% to 80%. Variants of medulloblastoma include medullomyoblastoma, containing myoblasts or myocytes, and melanotic medulloblastoma, containing melanosomes and premelanosomes.
Tumors of Cranial and Spinal Nerves
SCHWANNOMA
Constituting 5% to 10% of all intracranial tumors, schwannomas are usually solitary tumors discovered in the middle and later decades of life. Schwannomas presenting at an early age and/or bilaterally are seen in association with neurofibromatosis. The lesions are firm, encapsulated, slow growing, and benign. Schwannomas show a marked predilection for sensory nerves. In the cranial cavity they principally involve cranial nerve VIII (particularly the vestibular component) and, far less commonly, cranial nerves V, IX, and X. Clinical symptoms of auditory and/or cerebellar dysfunction are common. Intraspinal schwannomas, representing 30% of these tumors, most often involve the lumbar segment and give rise to signs of local root irritation and spinal cord compression. Both intradural and extradural growth is observed, and large lesions may traverse and expand the intervertebral foramina, resulting in a dumbbell-shaped lesion. Schwannomas are treated surgically and may recur if resection is not complete. Malignant transformation is rare.
NEUROFIBROMA
Variants: Circumscribed (Solitary) and Plexiform
Like schwannomas, neurofibromas are tumors of Schwann cells and can be distinguished by their morphology. Intraneural neurofibromas diffusely transform a nerve segment and its branches (the “plexiform” variant) and only infrequently produce an isolated lesion involving one nerve fascicle (the “circumscribed” or “solitary” variant). It is the plexiform variety that is pathognomonic of neurofibromatosis, whereas solitary neurofibromas infrequently share this association. Neurofibromas may be found along cranial or spinal nerve roots and ganglia, major nerves of the trunk and limbs, including the sympathetic system and subcutaneous branches, and along visceral sympathetic plexuses. Symptoms are related to compression of surrounding structures by tumor. Treatment is surgical, but resection almost invariably sacrifices the involved nerve because neurofibromas infiltrate the nerve directly. Partial resection may result in recurrence.
As discussed above, patients with neurofibromatosis type 2 (NF2) are predisposed to schwannomas and meningiomas of the cranial nerves and spinal nerve roots, whereas patients with neurofibromatosis type 1 (NF1) are susceptible to peripheral neurofibromas. The NF1 gene has been mapped to chromosome 17, and recently the NF2 gene was mapped to chromosome 22. Alterations in the NF1 gene may also be associated with pilocytic astrocytomas.
Malignant Peripheral Nerve Sheath Tumors
Malignant peripheral nerve sheath tumors arise by malignant transformation of a neurofibroma, usually plexiform, or arise de novo in a normal nerve sheath. Malignant peripheral nerve sheath tumors are highly malignant tumors that infiltrate locally and commonly metastasize to distant sites. The tumors occur primarily in adults, in whom they present as painful, rapidly enlarging masses that favor the trunk, neck, and proximal limbs, and only rarely affect cranial nerves. Treatment is usually surgical; the prognosis is directly related to tumor size. Fewer than 20% of patients survive 5 years.
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


