The increased utilization of cranial imaging for headaches, seizures, and trauma has led to an increase in the diagnosis of benign tumors. SEER suggests that between 1975 and 1987, there was a significant increase in the incidence of CNS tumors, which leveled off between 1991 and 2006. Because many patients with CNS tumors survive for several years, the prevalence exceeds the incidence. Overall prevalence rate of individuals with a brain tumor was estimated to be 209 per 100,000 in 2004 and 221.8 per 100,000 in 2010. The female prevalence rate (264.8 per 100,000) was higher than that in males (158.7 per 100,000). The averaged prevalence rate for malignant tumors (42.5 per 100,000) was lower than the prevalence for nonmalignant tumors (166.5 per 100,000). Estimates of the expected number of individuals living with primary brain tumor diagnoses in the United States was 612,770 in 2004 and 688,096 in 2010.5
Etiologic Factors
No agent has been definitively implicated in the causation of CNS tumors, and risk factors can be identified only in a minority. Commonly implicated associations described with other malignancies, such as diet, exercise, alcohol, tobacco, and viruses, are generally not considered to be significant for CNS tumors.6
Environmental Factors
Farmers and petrochemical workers have been shown to have a higher incidence of primary brain tumors. A variety of chemical exposures have been linked.7 Ionizing and nonionizing radiation has been implicated, with the clearest association coming from the occurrence of superficial meningiomas in individuals receiving cranial or scalp irradiation, with the association being stronger for young children receiving low doses of irradiation for benign conditions.8 Exposure to ionizing radiation is a known risk factor for a small percentage of astrocytomas, sarcomas, and other tumors.9 There is a 2.3% incidence of primary brain tumors in long-term survivors among children given prophylactic cranial irradiation for acute leukemia, a fourfold increase over the expected rate.10
In addition, a retrospective study suggests an increased risk for developing gliomas in children undergoing computed tomography (CT) scans.11 Exposure to dental x-rays performed at a time when radiation exposure was greater than currently used appears to be associated with an increased risk of intracranial meningioma.12
There are conflicting reports regarding nonionizing radiation emitted by cellular telephones.13–19 Several investigators have reported meta-analyses of case control studies evaluating cell phone use and the development of a brain tumor. Kan et al.16 reviewed nine studies (5,259 cases and 12,074 controls) and showed an overall odds ratio (OR) of 0.90 for cellular phone use and brain tumor development; the OR was 1.25 for long-term users. An OR of 0.98 for developing malignant and benign tumors of the brain as well as the head and neck was reported by Myung et al.17 when collating 23 case control studies (12,544 cases and 25,572 controls). The International Commission for Non-Ionizing Radiation Protection Standing Committee on Epidemiology reviewed the epidemiologic evidence, and they concluded that there was not a causal association between mobile phone use and malignant gliomas, but for slow-growing tumors, the observation period was too short for conclusive statements.18 A recent report of the INTERPHONE study, an international, population-based case control study, also did not find an increased risk of gliomas or meningiomas.19 Glioma incidence has not followed the increase in cell phone use, but because of the potential for a lag in trends, continued surveillance on children who are exposed from an early age is warranted.15
Viral Associations
Although certain canine and feline CNS tumors may have a viral association, the human evidence remains weak. Specifically, no increase in the risk of developing a brain tumor has been associated with previous polio vaccination, which discredits claims that simian virus 40, which contaminated older polio vaccine preparations, caused brain tumors.20 The exception to this is primary CNS lymphoma, which has been shown to be associated with Epstein-Barr virus.21 An increase in incidence of primary CNS lymphoma is most likely due to the increasing numbers of immunosuppressed patients in the setting of HIV and posttransplant use of immunosuppressants.21–22
The association between human cytomegalovirus (HCMV) infection and glioblastoma was first described by Cobbs et al.23 in 2002. The presence of HCMV was also demonstrated in glioblastoma and in other gliomas.24–25 HCMV may have tropism for microglia and CD133-positive glioma cancer stem cells and further work is needed to evaluate the role of this virus.26
Hereditary Syndromes
Neurofibromatosis type 1 (NF1) is an autosomal-dominant disorder associated with intra- and extracranial Schwann cell tumors. Optic gliomas, astrocytomas, and meningiomas also occur at higher frequency in NF1. NF2 is characterized by bilateral vestibular schwannomas and meningiomas. Systemic schwannomas also occur in NF2. Subependymal giant cell astrocytoma commonly occur in children with tuberous sclerosis, an autosomal-dominant disorder caused by mutation in the TSC1 and TSC2 genes. Other hereditary tumor syndromes affecting the CNS include Li-Fraumeni syndrome (germline mutation in one p53 allele; malignant gliomas); von Hippel-Lindau syndrome (germline mutation of the VHL gene; hemangioblastomas), and Turcot syndrome (germline mutations of the adenomatous polyposis gene; medulloblastoma).27,28 The nevoid basal cell carcinoma syndrome (Gorlin syndrome) is associated with medulloblastomas (and possibly meningiomas) and represents mutations in the PTCH suppressor gene or other members of the Sonic hedgehog pathway.29,30
Meningiomas and schwannomas are more common in females; gliomas, medulloblastomas, and most other CNS tumors are more common in males. Meningiomas are more common in African Americans and gliomas and medulloblastomas are more common in Caucasians. It has been suggested that there is a lower incidence of meningiomas and a higher incidence of gliomas and vestibular schwannomas in higher socioeconomic groups.31–34
Primary CNS tumors are of ecto- and mesodermal origin and arise from the brain, cranial nerves, meninges, pituitary, pineal, and vascular elements. The WHO classification lists approximately 100 subtypes of CNS malignancies in seven broad categories (Table 97.1).3,4,35 In spite of the low proliferation rate within the meninges, meningiomas are among the most common CNS tumors. Astrocytes are among the most mitogenically competent cells, and astrocytomas, also referred to interchangeably as gliomas, are among the more common primary CNS tumors. The precise cell of origin of gliomas, however, remains unclear.

The WHO classification can be reduced to a simpler working formulation, categorizing the neoplasms into tumors presumably derived from glia, neurons, or from cells that surround the CNS or form specialized anatomic structures. Glial cells are believed to give rise to astrocytomas, oligodendrogliomas, and ependymomas. Neuronal cells are involved in the development of medulloblastoma and primitive neuroectodermal tumors (PNET). In PNETs, anatomic location is pivotal; the transformation of cortical neuroblasts leads to cortical PNETs, retinal neuroblasts form retinoblastoma, and pineal neuroblasts form pineoblastomas. Specialized anatomic structures within the CNS give rise to pituitary adenomas, pineocytomas, chordomas, hemangioblastomas, germ cell tumors, and choroid plexus papillomas and carcinomas.
This working formulation is speculative, based on scant phenotypical and immunohistochemical evidence. For example, oligodendrogliomas are diagnosed based on cellular morphology, including prominent nuclei surrounded by a cytoplasmic halo with a characteristic “fried egg” appearance, and many have codeletions of 1p and 19q. However, no definitive markers for oligodendrogliomas currently exist; these tumors can stain both for glial fibrillary acidic protein, an astrocytic marker, and for synaptophysin, a presumptive neuronal marker.36 A third of all gliomas have morphologic characteristics of both astrocytoma and oligodendroglioma, leading some to separate gliomas based on their molecular and genetic characteristics.37 Evidence that suggests that some oligodendrocytes derive from a neuronal lineage, whereas some neuron-derived tumors (embryonal tumors) can show significant areas of glial differentiation, highlights the uncertainty.38,39 An alternative hypothesis is that all neuroepithelial cells are derived from a common precursor cell (i.e., a multipotent neural stem cell), and hence all neuroepithelial tumors are derived from neural stem cells or their committed progeny.40 The recent discovery, isolation, and characterization of cancer stem cells from human brain tumors provides supportive evidence.41 However, more recently it was shown in an animal experiment that gliomas can originate from differentiated cells in the CNS, including cortical neurons.42
Approximately 15% of all primary CNS tumors arise in the spinal cord, where the distribution of tumor types is significantly different from that in the brain. Tumors of the lining of the spinal cord and nerve roots predominate (50% to 80% of all spinal tumors); schwannomas and meningiomas are most common, followed by ependymomas. Primary gliomas of the spinal cord are uncommon.43 In children, three-quarters of tumors are comprised of ependymomas, pilocytic astrocytomas, and other neuroepithelial neoplasms.2
ANATOMIC LOCATION AND CLINICAL CONSIDERATIONS
Intracranial Tumors
Intracranial tumors produce five categories of symptoms: those arising from increased intracranial pressure (ICP), seizures, physiologic deficits specific to location, higher order neurocognitive deficits, and endocrinologic dysfunction. A headache arises from irritation of the dura or intracranial vessels or due to elevated ICP from tumor bulk, edema, or obstruction of a cerebrospinal fluid (CSF) pathway. Slow-growing tumors may grow to a remarkably large size without producing headaches, whereas rapidly growing tumors can cause headaches early in their course. Small tumors can cause headaches by growing in an enclosed space that is richly innervated with pain fibers, such as the cavernous sinus, or by causing obstructive hydrocephalus. Nausea and vomiting, gait and balance alterations, personality changes, and slowing of psychomotor function or even somnolence may be present with increased ICP. Because ICP increases with recumbency and hypoventilation during sleep, early-morning headaches that awaken the patient are typical. Sometimes the only presenting symptoms are changes in personality, mood, or mental capacity or slowing of psychomotor activity. Such changes may be confused with depression, especially in older patients. Although fewer than 6% of first seizures result from brain tumors, almost one-half of patients with supratentorial brain tumors present with seizures. An adult with a first seizure that occurs without an obvious precipitating event should undergo magnetic resonance imaging (MRI).
Tumors are sometimes associated with location-specific symptoms. Frontal tumors cause changes in personality, loss of initiative, and abulia (loss of ability to make independent decisions). Posterior frontal tumors can produce contralateral weakness by affecting the motor cortex and expressive aphasia if they involve the dominant (usually the left) frontal lobe. Bifrontal disease, seen with “butterfly” gliomas and lymphomas, may cause memory impairment, labile mood, gait imbalance, and urinary incontinence. These symptoms may be related to alteration of normal cortex and white matter by the tumor itself, or by surrounding tumor-related edema. Improvement of symptoms after a short course of high-dose glucocorticoids is often an indicator of whether the findings are related to tumor-associated edema. In the case of CNS lymphoma, corticosteroids can have a cytotoxic effect also with a reduction in the tumor mass.
Temporal tumors might cause symptoms detectable only on careful testing of perception and spatial judgment, but can also impair memory. Homonymous superior quadrantanopsia, auditory hallucinations, and abnormal behavior can occur with tumors in either temporal lobe. Nondominant temporal tumors can cause minor perceptual problems and spatial disorientation. Dominant temporal lobe tumors can present with dysnomia, impaired perception of verbal commands, and ultimately fluent (Wernicke-like) aphasia. Seizures are more common from tumors in this location.
Parietal tumors affect sensory and perceptual functions. Sensory disorders range from mild sensory extinction or stereognosis, which are observable only by testing, to a more severe sensory loss such as hemianesthesia. Poor proprioception in the affected limb is common and is sometimes associated with gait instability. Homonymous inferior quadrantanopsia, incongruent hemianopsia, or visual inattention may occur. Nondominant parietal tumors may cause contralateral neglect and, in severe cases, anosognosia and apraxia. Dominant parietal tumors lead to alexia, dysgraphia, and certain types of apraxia. Occipital tumors can produce contralateral homonymous hemianopsia or complex visual aberrations, affecting perception of color, size, or location. Bilateral occipital tumors can produce cortical blindness.
Classic corpus callosum disconnection syndromes are rare in brain tumor patients, even though infiltrative gliomas often cross the corpus callosum in the region of the genu or the splenium. Interruption of the anterior corpus callosum can cause a failure of the left hand to carry out spoken commands. Lesions in the posterior corpus callosum interrupt visual fibers that connect the right occipital lobe to the left angular gyrus, causing an inability to read or name colors.
Thalamic tumors can cause local effects and also obstructive hydrocephalus. Either sensory or motor syndromes or, on the dominant side, aphasia is possible. Thalamic pain disorders or motor syndromes from basal ganglia involvement may also occur. The most common brainstem tumor is the pontine glioma, which presents most frequently with cranial nerve VI and VII palsies. Long tract signs usually follow, with hemiplegia, unilateral limb ataxia, gait ataxia, paraplegia, hemisensory syndromes, gaze disorders, and occasionally, hiccups. Tectal involvement causes Parinaud syndrome, peduncular lesions cause contralateral motor impairment, and obstruction of the aqueduct causes hydrocephalus.
Tumors in the medulla can have a fulminant course, including dysphagia, dysarthria, and deficits in cranial nerves IX, X, and XII. Involvement of the medullary cardiac and respiratory centers can result in a rapidly fatal course. Fourth ventricular tumors, because of their location, cause symptomatic obstructive hydrocephalus at a relatively small size, with associated disturbances of gait and balance. In addition, nausea and vomiting can be a symptom of a fourth ventricular mass. Rapidly enlarging lesions may end in cerebellar herniation.
Cerebellar tumors have variable localizing presentations. Midline lesions in and around the vermis cause truncal and gait ataxia, whereas more lateral hemispheric lesions lead to unilateral appendicular ataxia, usually worst in the arm. Abnormal head position, with the head tilting back and away from the side of the tumor, is seen often in children but rarely in adults. Mass lesions within or abutting the brain or spinal cord can cause displacement of vital neurologic structures. This can lead, in the brain, to herniation syndromes with respiratory arrest and death and, in the spine, to paraplegia or quadriplegia. A hemorrhage into a tumor can also cause acute neurologic deterioration. This is often associated with iatrogenic coagulopathies such as thrombocytopenia due to chemotherapy or anticoagulation therapy for deep venous thrombosis. Primary tumors that most often bleed de novo are glioblastoma and oligodendrogliomas; of the metastatic tumors, lung cancer, melanoma, renal cell cancer, thyroid cancer, and choriocarcinoma most often show hemorrhage.
Lumbar puncture should not be performed in any of the acute herniation syndromes or when herniation is imminent. In fact, a lumbar puncture should be avoided in the setting of significantly elevated ICP that is directly related to a tumor’s mass effect or to obstructive hydrocephalus.
Spinal Axis Tumors
For the clinical presentation of tumors of the spinal axis to be understood, the local anatomy must be appreciated. A spinal tumor can produce local (focal) and distal (remote) symptoms, or both. Local effects indicate the tumor’s location along the spinal axis, and distal effects reflect involvement of motor and sensory long tracts within the cord.
Distal symptoms and signs are confined to structures innervated below the level of the tumor. Neurologic manifestations often begin unilaterally, with weakness and spasticity, if the tumor lies above the conus medullaris, or weakness and flaccidity if the tumor is at or below the conus. Impairment of sphincter and sexual function occurs later unless the tumor is in the conus. The upper level of impaired long-tract function usually is several segments below the tumor’s actual site. Local manifestations may reflect involvement of bone (with axial pain) or spinal roots, with radicular pain and loss of motor and sensory functions of the root or roots.
Magnetic Resonance Imaging
The imaging modality of choice for most CNS tumors is MRI, which can demonstrate anatomy and pathologic processes in detail.44 CT is generally reserved for those unable (e.g., because of an implanted pacemaker, metal fragment, or paramagnetic surgical clips) or unwilling (e.g., because of claustrophobia) to undergo MRI. Because of the link of nephrogenic systemic fibrosis to the infusion of gadolinium-based contrast agents, there are new preventative guidelines regarding the administration of gadolinium in patients who may be at high risk.45
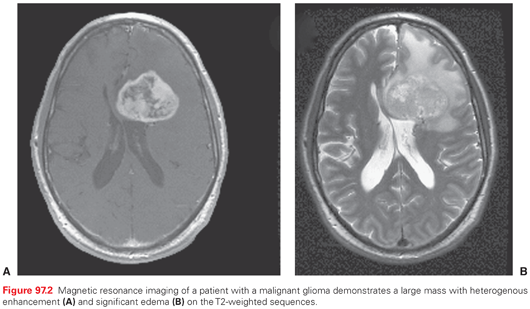
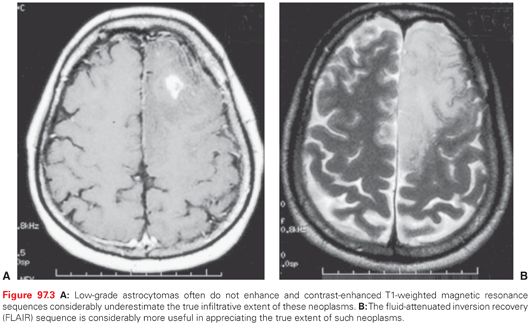
The most useful imaging studies are T1-weighted sagittal images, gadolinium (Gd)-enhanced and unenhanced T1 axial images, and T2-weighted axial images (Fig. 97.2). Contrast-enhanced MRI provides an improved ability to discern tumors from other pathologic entities, one tumor type from another, and putatively higher from lower grade malignancies. There are, however, limitations in anatomic MRI to definitively diagnose a mass lesion as a tumor.46 Other confounding diagnoses include bacterial abscesses, inflammatory disease such as sarcoidosis, tumefactive demyelination, and acute ischemic disease. It is conventionally believed that most low-grade gliomas (except pilocytic astrocytomas and pleomorphic xanthoastrocytoma) do not enhance, but in reviewing imaging studies of patients enrolled in several clinical trials, it is apparent that this may not be so categorical, in that even low-grade gliomas may frequently contain areas of enhancement, raising the concern that these areas might represent high-grade or malignant transformation (Fig. 97.3).47 In addition, some high-grade lesions may not have contrast enhancement on MRI. Imaging is also unable to discern different histologic subtypes; however, the presence of calcifications is typical of oligodendroglioma.
Neuraxis or Spinal Imaging
In the evaluation of spinal cord tumors, MRI is also the preferred modality, providing superb visualization of the spinal cord contour and (with gadolinium contrast) of most intrinsic tumors (such as ependymomas, astrocytomas, meningiomas, and schwannomas), as well as facilitating the diagnosis of leptomeningeal dissemination. Tumor cysts are readily identified on MRI, and spinal cord tumors can often be distinguished from syringomyelia. Ideally, neuraxis imaging should be performed before surgery. In the immediate postoperative period, spinal MRI scans may be difficult to interpret because arachnoiditis and blood products can mimic leptomeningeal metastasis. Delayed spinal MRI (more than 3 weeks after surgery) combined with an increased dose of gadolinium is a sensitive imaging study for leptomeningeal disease.
Newer Imaging Modalities
Newer MRI techniques include magnetic resonance spectroscopy, dynamic contrast-enhanced MRI, diffusion-perfusion MRI, and functional MRI.48 In addition, metabolic imaging using positron-emission tomography using various tracers is being explored.49 These newer techniques remain to be validated as biomarkers of biological behavior or clinical outcome. Posttreatment metabolic scans may help distinguish recurrence from treatment-related changes, although most modalities have a relatively high false negative rate. A modification of the standard MRI is quick brain MRI, which uses single-shot, fast-spin echo imaging to allow for adequate demonstration of ventricular anatomy and appropriate evaluation of shunt function.50
Pseudoresponse and Pseudoprogression
In malignant gliomas treated with combined modality therapy, it is speculated that 25% to 40% or even more may experience imaging changes relatively early in the course of therapy, usually within a few months, which appears consistent with radiographic progression. However, with time and without any therapy, many of these changes actually improve or even resolve (pseudoprogression), and in patients operated on with a presumptive diagnosis of tumor, the histopathology often reveals large areas of tumor necrosis.51,52 With the advent of antiangiogenic therapies for malignant gliomas, rapid resolution of tumor enhancement is visualized on MRI, sometimes within days. This is consistent with the traditional definition of response, but in several instances, especially with time, even in the absence of contrast enhancement, tumor progression and clinical deterioration occurs, which is sometimes appreciated as T2 or fluid-attenuated inversion recovery (FLAIR) changes; this phenomenon is labeled as pseudoresponse.53
Cerebrospinal Fluid Examination
Typically, medulloblastoma, ependymoma, choroid plexus carcinoma, lymphoma, and some embryonal pineal and suprasellar region tumors have a high enough likelihood of spreading to justify CSF examinations to evaluate for malignant cells (cytology) and specific markers, such as human chorionic gonadotropin-β and α-fetoprotein.
CSF spread of a tumor may be associated with several possible findings, including CSF pressure above 150 mm H2O at the lumbar level in a laterally positioned patient; elevated protein, typically greater than 40 mg/dL; reduced glucose (below 50 mg/mL); and tumor cells by cytologic examination. A high protein concentration with normal glucose levels and normal cytology is also seen with base of skull tumors, such as vestibular schwannoma, and with spinal cord tumors that obstruct the subarachnoid space and produce stasis of the CSF in the caudal lumbar sac. Sampling of the CSF in the immediate postoperative period may lead to false-positive results, however, and is best done before surgery or more than 3 weeks after surgery, as long as there is no uncontrolled raised intracranial pressure.
Preoperative Considerations
The major objectives of surgery are to maximally remove bulk tumor, reduce tumor-associated mass effect and elevated ICP, and provide tissue for pathologic analysis in a manner that minimizes risk to neurologic functioning. For some tumors, a complete resection can be curative. However, most brain tumors are diffusely infiltrative; for these, surgical cure is rarely possible. Nonetheless, surgery can rapidly reduce tumor bulk with potential benefits in terms of mass effect, edema, and hydrocephalus. Furthermore, there is mostly retrospective evidence for both high-grade and low-grade infiltrative gliomas for which maximizing the extent of bulk tumor removal is associated with a better outcome, albeit so long as new, permanent neurologic deficits are avoided.54–57 The requirement for histopathologic confirmation of diagnosis is not necessary in certain well-defined situations, but a tissue diagnosis is still required to determine the appropriate treatment course in most circumstances. As molecularly targeted therapies become useful, tissue removal for molecular analysis will become more necessary to guide therapy. Pseudoprogression may make tissue-based confirmation necessary before changes in therapy are instituted.53 Technologic advances in surgical approaches, techniques, and instrumentation have rendered most tumors amenable to resection; however, for some tumor types or locations, the risk of open operation supports the choice of biopsy for obtaining diagnostic tissue. Biopsy techniques include stereotactic biopsy (with or without a stereotactic frame) using CT, MRI, or both, to choose the target. Metabolic or spectroscopic imaging can be coregistered with anatomic images to choose targets that may be of higher biologic aggressiveness within a tumor that appears homogeneous on standard imaging. In certain settings, an approach using simple ultrasonic guidance can also be considered for obtaining diagnostic tissue.
Unless a lymphoma is being considered, patients are given corticosteroids, usually dexamethasone, immediately preoperatively and often for several days before surgery to reduce cerebral edema and thus minimize secondary brain injury from cerebral retraction. Steroid administration is then continued in the immediate postoperative period and tapered off as quickly as possible. Antibiotics are given just before making the incision to decrease the risk of wound infection.
Anesthesia and Positioning
The routine use of prophylactic anticonvulsants in the perioperative period is a common practice despite recommendations that would seem to discourage that practice.58,59 Patients with a history of seizures need to have their anticonvulsants maintained at therapeutic dose levels. Under certain circumstances, such as for awake craniotomies with electrocorticography, the use of anticonvulsants for a short time might be warranted.
General Surgical Principles
In the past, localization of the surgical incision and craniotomy were most often performed by a neurosurgeon’s understanding of cranial anatomy and an interpretation of preoperative imaging. More recently, image-guided navigation systems have been employed to more effectively localize tumor margins as they project to the cranial surface and thus allow for smaller, precisely positioned craniotomies.59,60 For tumors not resectable because of their location or diffuseness, a biopsy can be performed stereotactically using frameless or frame-based techniques. Tumors that are limited to the cortical surface may be best sampled with an open biopsy, under direct vision, due to the risk of inadvertent injury to a cortical vessel with a more limited, needle-based approach.
Specialized technology can be used to help define the completeness of a resection. Often, preoperative mapping of functional areas and their connections with MRI-based techniques are used to delineate both cortical areas and important subcortical white matter tracts that subserve speech and motor function.61 Image-guided navigation systems are almost always employed, but the guidance may lose accuracy over the course of an operation due to brain shift or cyst decompression. Intraoperative imaging with ultrasound, CT, or MRI may be used to determine the extent of residual tumor and to further localize areas where additional tumor may be removed safely.62 There has been growing use of 5-aminoleuvilinic acid (5-ALA), a prodrug, which is converted by glioma cells into fluorescent porphyrins that can be visualized with an operating microscope equipped with a fluorescent imaging system. The impact of the use of 5-ALA to guide resection of glioblastoma (GBM) on completeness of surgical resection and progression-free survival (PFS) has been demonstrated in a phase III trial.63 Its use is limited to tumors that enhance with contrast on MRI (or CT), because the conversion of prodrug in low-grade tumors does not produce a sufficient amount of fluorescent porphyrin to be visualized intraoperatively. However, this conversion can be detected with the use of specialized optical instrumentation.57 Use of 5-ALA in this manner is approved by regulatory authorities in Europe; its use in the United States remains investigational at this time.
Intraoperative cortical-stimulation mapping facilitates the resection of tumors in or adjacent to functionally critical areas. Motor functions can be mapped even under general anesthesia; however, anesthetic agents may increase the threshold to response and hence decrease the sensitivity of mapping. Sensory and speech-associated cortex are typically mapped during an awake craniotomy. Patients are monitored in the specialized care unit overnight after surgery, and an MRI is done within 24 to 48 hours to evaluate the extent of any remaining tumor. It is important that this MRI is done before 72 hours to minimize the appearance of nonspecific contrast enhancement that is related to surgery and might be mistaken for residual tumor.64,65
Re-resection of recurrent cerebral astrocytomas can be modestly efficacious.66 When the initial tumor was low grade, histologic resampling may be necessary to guide further treatment at recurrence. Reoperation offers a chance to implant polymer wafers containing carmustine (bis-chloroethylnitrosourea [BCNU]) or to administer experimental agents, such as gene therapy agents or immunotoxins. A smaller volume of disease at initiation of chemotherapy predicts longer survival; thus, reoperation may improve the efficacy of adjuvant treatment as well as relieve mass effect in some patients.67 An increasingly important aspect of resection is the need for tumor sampling to allow for a molecular marker analysis, which might provide and aid in assessing the prognosis as well as the probability of benefit from both chemotherapeutic and targeted therapies.
General Concepts
Radiation therapy plays an integral role in the treatment of most malignant and many benign primary CNS tumors. It is often employed postoperatively as adjuvant treatment to decrease local failure, to delay recurrence, and to prolong survival in gliomas; as definitive treatment in more radiosensitive diseases such as PNET and germ cell tumors; or as therapy to halt further tumor growth in schwannomas, meningiomas, pituitary tumors, and craniopharyngiomas, and as ablative therapy to abrogate hormonal overproduction in secretory pituitary adenomas. Radiation therapy is also the primary modality in palliating brain metastases, and symptomatic spinal and osseous, as well as soft-tissue skull lesions.
Radiobiologic and Toxicity Considerations
Most neoplasms can potentially be cured if the correct radiation dose can be delivered to the entire tumor and its microscopic extensions. This is not always feasible because the maximum radiation dose deliverable is limited by the tolerance of the surrounding normal tissues, and the identification of regions of microscopic extension remains vague. Radiation tolerance of the CNS depends on several factors, including total dose, fraction size, volume irradiated, underlying comorbidities (particularly hypertension and diabetes), and innate sensitivity. Adverse reactions to cranial irradiation differ in pathogenesis and temporal presentation and are not discussed in detail here.
A major radiobiologic consideration revolves around the selection of total dose and the fractionation schedule. Late or long-term toxicities are generally a function of fraction size (i.e., dose per daily fraction of treatment), and therefore, as the fraction size is increased, such as with radiosurgery, higher late toxicity rates must be anticipated, assuming that normal tissue is encompassed within the high dose field. These late toxicities from larger fraction sizes can be minimized by minimizing the volume irradiated, as is done with radiosurgery, thereby drastically reducing the volume of normal tissues exposed to high doses. Proton and charged particle therapy, such as carbon, etc., are characterized by minimal to no exit dose beyond the target (where the so-called Bragg peak [i.e., the peak region of dose deposition] is placed), thereby sharply targeting the dose, and advantageously sparing tissue distal to the target. For radiosurgery, doses in the order of 12 to 21 Gy in single fractions are often utilized. In conventional radiotherapy, fraction sizes of 2 Gy are routinely utilized and may be lowered to 1.8 Gy per fraction in proximity to the visual apparatus or may be increased to 3 Gy or more per fraction in patients in whom shorter palliative schedules, with lesser concern regarding long-term morbidities, exist. In general, the entire target is treated with a relatively uniform dose, but with the advent of newer delivery methods, it is possible to create dose gradients or dose inhomogeneities within the tumor to match differential radiosensitivity. However, this concept of dose painting remains investigational.
Treatment Planning and Delivery Methods
High-resolution MR fusion with CT planning images has allowed for more precise delineation of targets, although a significant margin, particularly with gliomas, is still necessary to cover microscopic extension.68 Patient immobilization devices limit intrafraction motion and provide precision in positioning, decreasing the margin required for setup variability. Image-guided radiotherapy (IGRT), using biplanar orthogonal x-ray imaging systems, cone beam CT, megavoltage CT, surface tracking, fiducial monitoring, etc., further improves setup reproducibility and allows for decreased margins. Newer systems in development incorporate on-board MRI, but remain investigational.
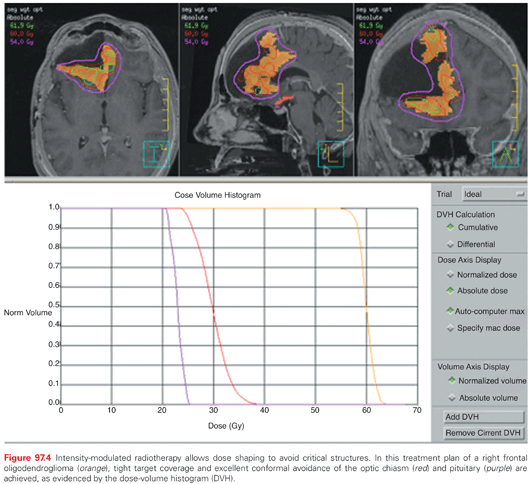
IGRT can be incorporated with any radiotherapy method, such as fractionated external-beam radiotherapy and stereotactic radiosurgery (SRS), and is practically mandatory for charged-particle therapy, frameless radiosurgery, fractionated stereotactic radiotherapy (FSRT), and intensity-modulated radiotherapy (IMRT). CT-based three-dimensional conformal radiation (3D-CRT) in which noncoplanar fields with unique entrance and exit pathways can be mapped on the target has improved normal tissue sparing. This allows for avoidance of critical structures, such as the brainstem, optic apparatus, and spinal cord. In IMRT, the photon flux of a beam is modulated in multiple directions during treatment, aimed at mimicking the shape of the target from various viewpoints, thereby producing improved conformality and nonuniform dose distribution. IMRT is increasingly being utilized for CNS tumors, based primarily on dosimetric studies, which suggest superior tumor coverage and reduction in the dose to critical structures (Fig. 97.4).69 This can be beneficial in specific instances, such as to preserve cochlear function, vision, or pituitary activity.70
In FSRT, the concepts of 3D-CRT or IMRT are merged with the accuracy and precision in delivery that characterizes SRS, and, typically, the radiation fraction size is considerably increased, so that the total course of therapy is reduced from the typical 20 to 30 or more fractions to 5 or fewer fractions. Various FSRT systems have been developed, with reported precision between 1 to 3 mm.71,72 FSRT is often used for larger lesions (e.g., 4 cm or more) and for lesions located in critical regions where single-fraction SRS is disadvantageous because of a higher risk of toxicity, such as larger vestibular schwannomas or meningiomas.
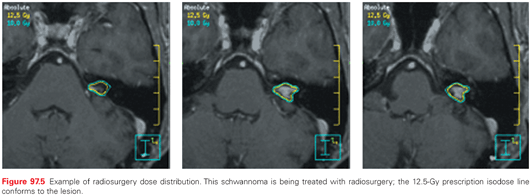
SRS is used to treat a diverse group of lesions. Treatment can be carried out using either a modified or dedicated linear accelerator, cobalt-60 units, or charged particle devices. Several commercial devices have now been developed, each with slightly unique features, including robots that position the linear accelerator at various angles, collimation systems that provide prefixed circular collimators of various sizes or shaped collimated beams, and even intensity-modulated delivery from one or multiple directions, delivered serially, helically, or volumetrically.73 Radiosurgery plays a dominant role in the treatment of oligometastases to the brain, arteriovenous malformations, schwannomas, and meningiomas and is occasionally used to treat malignant recurrences (Fig. 97.5).
Charged-particle beams, including protons (but not electrons), deposit the majority of their dose at a depth dependent on the initial energy, avoiding the exit dose of photon therapy. This localized dose is known as the Bragg peak. Historically, in order to cover larger volumes, proton beams have been modified by passive range modulators that disperse the Bragg peak and broaden the dose deposition, resulting in decreased proximal sparing, while still maintaining distal sparing. Charged-particle radiotherapy has been particularly utilized to treat tumors of the skull base to doses higher than can be achieved conventionally, and in reirradiation settings where conventional techniques are too unsafe. In particular, chordomas and chondrosarcomas require high radiation doses for local control. Proton beams have also been advocated for childhood tumors and tumors in young adults, because they decrease integral radiation dose, thereby decreasing the risk of second malignancies, although concern about incidental neutron production exists.74,75 The neutron contamination issue is almost nonexistent with approaches similar to the photon technique of intensity modulation, often also referred to as intensity modulated proton therapy (IMPT) and allows for significantly superior dose sculpting. Increasingly, this approach is being utilized in patients with lower grade neoplasms of the CNS, where survival is anticipated to be in years, and where reduction in the volume of normal brain irradiated is likely to produce benefits in cognitive, functional, and endocrine domains.
Brachytherapy has historically been widely used, but currently has a limited role in the CNS, although it has enjoyed some resurgence and is occasionally used for recurrent gliomas. A liquid colloid of organically bound iodine-125 (125I) in a spherical balloon continues to be used to treat both recurrent and newly diagnosed malignant gliomas and brain metastases in the postoperative context.76 At least two randomized trials using seed implants have failed to demonstrate a survival advantage in malignant gliomas. The injection of radioisotopes within the cystic portion of craniopharyngiomas allows ablation of the secretory lining. A select group of patients with cystic tumors may benefit from the direct instillation of colloidal phosphorus-32 (32P), yttrium-90 (90Y), or gold-198 (198Au).77,78 This technique will deliver between 200 to 400 Gy to the cyst wall.
Radiolabeled therapy is in the developmental phase. The most commonly used antigenic targets for CNS malignancies are the epidermal growth factor receptor (EGFR), neural cell adhesion molecule (NCAM), tenascin, placental alkaline phosphatase (PLAP), and phosphatidylinositide. Institutions using this technique have utilized murine, chimeric, or humanized monoclonal antibodies attached to 131I, 90Y, rhenium-188 (188Re), and astatine-211 (211At). The evolution of these trials has seen the delivery route move from systemic (intra-arterial or intravenous) to local instillation of the agent into a surgically created resection cavity. Even though the blood–brain barrier is often disrupted by a rapidly growing CNS malignancy, 150 kDa antibodies would still not likely cross to a significant degree.79 Most of the trials to date are of dose searching pilot or phase I design. Using 131I-81C6 (antitenascin monoclonal antibody), a trend toward significant improvement in median survival was shown for patients receiving 40 to 48 Gy versus less than 40 Gy.80 Unlike seed brachytherapy, there appears to be a very low rate of CNS toxicity with targeted isotope therapy, and a minimal need for surgical intervention for the removal of necrotic regions.
CHEMOTHERAPY AND TARGETED AGENTS
Drug therapies alone are effective for only a few types of CNS tumors (e.g., primary CNS lymphoma) but are useful as adjunctive therapy for many CNS tumors. Among the reasons for the poor efficacy of chemotherapeutic and targeted agents is the low concentration of drug penetration to the tumor because of the difficulty of agents to cross the blood–brain barrier, active transport mechanisms of drug efflux, and high plasma protein binding of agents, thereby lowering the volume of distribution of agents in the brain parenchyma.81 Intrinsic and acquired resistance remains an important reason for the lowered efficacy of chemotherapy. Although targeted agents are in early testing, multiplicity and alternate signaling pathways limit their efficacy.
The Blood–Brain Barrier
Central to treating CNS tumors is the issue of drug delivery, due to the blood–brain barrier (BBB), a physiologic and functional barrier. The CNS microvasculature has several unique features, including the lack of fenestrations between adjacent endothelial cells and relatively fewer pinocytotic and endocytotic endothelial vesicles. Additionally, adjacent BBB endothelial cells are connected by a continuous extension of tight junctions, which limit passive diffusion between endothelial cells and through capillary structures. Tight junctions within the BBB are also enveloped by astrocytic foot processes, which increase the barrier to passive diffusion across the BBB.
Brain microvasculature selectively transports nutrients through 20 or more active or facilitated carrier transport systems expressed on the endothelial surface.82 The endothelium is rich with efflux pumps, including the multidrug resistance (MDR) gene–encoded P-glycoprotein that actively removes substrate molecules that may have passed the BBB.83 There are several methods to disrupt or circumvent the BBB, including the intra-arterial administration of mannitol,84 which has resulted in significant toxicities and thus have limited its universal use.85 Noninvasive delivery systems using specialized carriers such as nanosystems (colloidal carriers) with favorable pharmacokinetic and pharmacodynamic properties are being explored.86,87 Other methods include local administration (Gliadel wafer)88 or local drug delivery such as convection-enhanced delivery (CED). CED requires the implantation of catheters directly into the brain, followed by continuous infusion of the drug under a constant pressure gradient. Proof of principle for CED has been demonstrated in several studies, but unfortunately, phase III results have been disappointing.89,90 Another approach is the direct administration of the agent into the CSF. With a few exceptions (e.g., methotrexate, cytarabine, thioTEPA), most compounds cause unacceptable neurologic toxicity, including death, when given into the CSF. Because of this, intrathecal chemotherapy is principally used to treat leptomeningeal metastases and for CNS prophylaxis for high-risk leukemia.
Challenges Specific for Targeted Agents
Despite the availability of targeted agents specific to aberrant signaling pathways in high-grade gliomas, the results of phase II studies of many agents have been disappointing. In addition to the difficulty of delivery of agents across the BBB, there are other challenges that limit the efficacy of these agents. These include accounting for the heterogeneity of tumors, redundancy of pathway interactions, a lack of accurate and reproducible biomarkers to select patients for specific therapies, and difficulty in assessing target modulation.91–93 Bayesian adaptive randomized designs in clinical trials may allow for more efficient trials compared to those with balanced randomization.94
Other Systemic Therapy Considerations
Many antiepileptic agents, including phenytoin, carbamazepine, and phenobarbital, induce the hepatic cytochrome P-450 isoenzyme and glucuronidation drug-elimination systems. The specific isoenzymes induced by these drugs are often capable of metabolizing many agents. For example, standard paclitaxel doses commonly result in subtherapeutic serum levels in patients also using phenytoin.95 In fact, the maximally tolerated paclitaxel dose in patients using enzyme-inducing P-450 antiepileptics is nearly threefold higher than in patients not using such agents. Similar observations have been made with regard to 9-aminocampothecin, vincristine, teniposide, irinotecan, and targeted agents.96–99 In addition to different maximal tolerated dose (MTDs) being established depending on the use of enzyme-inducing antiepileptics, the side effect profile and dose-limiting toxicities can also differ.99,100 Most phase I clinical trials in brain tumor patients now use separate arms for patients who are or are not taking enzyme-inducing antiepileptic drugs or limit enrollment to patients not taking enzyme-inducing antiepileptic drugs. It may be preferable to change to a non–enzyme-inducing antiepileptic agent (e.g., levetiracetam [Keppra]), although it may take days to make the switch and some time for the P-450 enzyme induction to resolve.
SPECIFIC CENTRAL NERVOUS SYSTEM NEOPLASMS
Cerebral Glioma
Pathologic Classification
The histologic subtypes of gliomas include tumors of astrocytic, oligodendroglial, ependymal, and neuroepithelial origin (Table 97.2). Based on the WHO classification,3 noninfiltrative gliomas are classified as grade I, and infiltrating gliomas are subsequently categorized from grades II to IV. Infiltrative astrocytic tumors are divided into three categories: astrocytoma (including grade II fibrillary, gemistocytic, and protoplasmic), anaplastic astrocytoma (grade III), and glioblastoma (including grade IV giant cell glioblastoma and gliosarcoma). Oligodendrogliomas and ependymomas are either grade II or anaplastic (grade III).

WHO Grade I: Astrocytoma
Low-grade astrocytomas (WHO grade I) such as pilocytic astrocytoma, pleomorphic xanthoastrocytoma, and subependymal giant cell astrocytoma are typically circumscribed and indolent tumors. Missense mutations of the V600E type in the v-RAF murine sarcoma viral oncogene homolog B1 (BRAF) gene were identified in these noninfiltrative neoplasms.101 The highest frequencies were found in pleomorphic xanthoastrocytomas (66%; 65% in its anaplastic variant), gangliogliomas (18%), and pilocytic astrocytomas (9%, especially in tumors with extracerebellar location).101
Complete surgical resection, whenever feasible, is the curative mainstay therapy for such tumors. Despite aggressive near total resection, delayed recurrence and eventual malignant transformation are, unfortunately, common. The resection of a low-grade glioma can be difficult in locations such as the optic pathway, hypothalamus, and in those involving deep midline structures. In these instances, asymptomatic patients can be observed carefully for a prolonged period of time and undergo a maximally safe resection only at the time of progression.
In patients who have a recurrent tumor that are not amenable to further resection or who have a residual tumor causing significant morbidity, adjuvant chemotherapy or radiotherapy can improve recurrence-free survival, although the role of chemotherapy in adults remains controversial. Immediate postoperative adjuvant therapies may be appropriate in some cases depending on the location of the tumor, the extent of residual disease, the impracticability of repeated surgical excision, and the availability for follow-up. Generally, radiotherapy is the primary adjuvant treatment used in older children and adults with low-grade gliomas. In young children with unresectable, progressive low-grade gliomas, there is a desire to avoid or delay radiotherapy owing to the long-term radiation-related sequelae; chemotherapy is often utilized here as the initial therapeutic option.102–105 Some responses from chemotherapy can last for years, and nearly half of all children treated with chemotherapy ultimately require radiotherapy for tumor progression.
In terms of radiotherapy used with a curative intent, in children, the most common situation is with cerebellar and optic-pathway pilocytic astrocytoma, typically after progression on chemotherapy, whereas in adults, this tends to occur most commonly with hypothalamic pilocytic astrocytoma. The typical radiation dose used in this setting is 50.4 to 54.0 Gy, in 1.8 Gy fractions. There is evidence of improved PFS in this situation.106 Given the young age and long expected survival of these patients, proton beam therapy is often considered for these patients, with the desire to decrease the risk of a second neoplasm, and to treat less normal brain tissue with radiation.106
Subependymal giant cell astrocytomas can be effectively treated with everolimus. In a prospective randomized study, 35% of patients in the everolimus group had at least a 50% reduction in the volume of their tumor versus none in the placebo group, although complete responses still remain uncommon, even with this therapy.107
WHO Grade II: Low-Grade Glioma
Nonpilocytic or diffusely infiltrating low-grade gliomas are classified as WHO grade II tumors. They may arise from astrocytic, oligodendrocytic, or mixed lineage. Like astrocytomas, oligodendrogliomas display various degrees of clinical aggressiveness. Three common genetic alterations, inactivation of the TP53 tumor suppressor gene, heterozygous point mutations of the isocitrate dehydrogenase-1 (IDH1), and loss of chromosome 22q are involved in the formation of WHO grade II astrocytoma. TP53, located on chromosome 17p, encodes the p53 protein that has an important role in a number of cellular processes, including cell cycle arrest, apoptosis, and response to DNA damage.108
Somatic mutations at codon 132 in IDH1 are present in 50% to 80% of WHO grade II and III astrocytic tumors and oligodendroglial tumors, as well as in secondary grade IV glioblastomas.109,110 These IDH mutations promote the conversion of α-ketoglutarate into D-2-hydroxyglutarate, an oncometabolite that mediates the oncogenic activity of IDH mutations and can be measured by magnetic spectroscopy.111 Tumors that have IDH mutations carry a better prognosis than do IDH wild-type gliomas of the same histologic grade.112,113
An unbalanced t(1;19)(q10;p10) translocation results in a combined loss of chromosomal arms 1p and 19q, which leads to the loss of one hybrid chromosome, and thus, a loss of heterozygosity.114 This cytogenetic alteration is usually associated with oligodendroglial histology and is rarely found in other tumors. Patients with 1p- and 19q-codeleted tumors have a better prognosis than do histologically similar tumors of the same grade that do not harbor this codeletion.115
In addition to histology and molecular characteristics, several variables have been found to be of prognostic importance in low-grade gliomas. Pignatti et al.116 performed the most comprehensive of these analyses and developed a scoring system to identify patients at varying level of risk for mortality. A multivariate analysis showed that age 40 years or older, astrocytoma histology, maximum diameter 6 cm or greater, tumor crossing the midline, and presence of neurologic deficits negatively impacted survival. Patients with up to two factors were considered low risk (median survival, 7.7 years) and patients with three or more were considered high risk (median survival, 3.2 years). Recently, 339 European Organisation for Research and Treatment of Cancer (EORTC) patients with central-pathology confirmed LGGs were used to develop a new prognostic model for PFS and overall survival (OS).117 Data from 450 patients with centrally diagnosed LGGs recruited into two large studies conducted by North American cooperative groups were used to validate the models. Both PFS and OS were negatively influenced by the presence of baseline neurologic deficits, a shorter time since first symptoms, an astrocytic tumor type, and tumors larger than 5 cm in diameter.117
Surgery for Low-Grade Glioma
Retrospective analyses have suggested that the extent of resection is a significant prognostic variable. The Radiation Therapy Oncology Group (RTOG) performed a prospective evaluation of the natural history of completely resected low-grade gliomas (RTOG-9802), evaluating the recurrence risk in 111 patients with surgeon-defined gross total resections (GTR) and found that the extent of postoperative residual disease was an important variable for time to first relapse.118 Five-year recurrence rates were 26% versus 68% for patients with less than 1-cm residual tumors versus 1- to 2-cm residual tumors.
Radiation Therapy
The role of radiotherapy—particularly the timing—remains somewhat controversial. Early intervention is indicated for patients with increasing symptoms and radiographic progression. In younger patients (less than 40 years) who have undergone complete resection, observation with imaging is an option. In RTOG-9802, median time to progression in 111 good-risk patients defined as younger than 40 years and with a gross total tumor resection was 5 years.118 In those who have undergone a subtotal resection or those with high-risk features, postoperative radiotherapy may be recommended, typically 50.4 Gy in 1.8 Gy fractions.
Three phase III trials provide the best evidence with respect to the indications for radiotherapy as well as the dose. In a study by the EORTC (EORTC-22845), 314 patients were randomized to postoperative radiotherapy to 54 Gy (n = 157) or radiotherapy at progression (n = 157).119 A statistically significant improvement in PFS was associated with early radiotherapy, 5.3 versus 3.4 years (p <0.0001), without a difference in median survival, 7.4 versus 7.2 years.
Two other trials investigated the dose question. In EORTC-22844, 379 patients were randomized to 45 Gy versus 59.4 Gy.120 With a median follow-up of 74 months, OS (58% versus 59%) and PFS (47% versus 50%) were similar. In an Intergroup study, 203 patients were randomized to 50.4 Gy (n = 101) or 64.8 Gy.121 There was no significant difference in PFS or OS.
To assess the OS and cause-specific survival (CSS) impact of early adjuvant radiotherapy (EART) following the resection of supratentorial LGG in adults (16 to 65 years), 2,021 patients in the SEER database from 1988 to 2007 were evaluated.122 Of the 2,021 patients, 871 (43%) received EART, and 1,150 (57%) did not. In the multivariate Cox proportional hazards model, EART was associated with worse OS and CSS. Using a propensity score and instrumental variable analyses to account for known and unknown prognostic factors demonstrated unmeasured confounding variables that may affect this finding.
Consequently, low-dose radiotherapy, 50.4 to 54.0 Gy in 1.8 Gy fractions, has become an accepted practice for selected patients with low-grade gliomas. The target volume is local, with a margin of 2 cm beyond changes demonstrated on traditional MRI sequences. FLAIR images usually show considerable abnormality beyond any enhancing or nonenhancing tumor and whether a smaller margin may be used for planning if FLAIR sequences are utilized is unknown.
Posttreatment cognition remains an important consideration. Brown et al.123 reviewed the results of the Mini-Mental Status Examination for 203 adults irradiated for low-grade gliomas. Most patients maintained stable neurocognitive status after radiotherapy, and patients with abnormal baseline results were more likely to have improvement in cognitive abilities than to deteriorate after therapy; few patients showed cognitive decline. A more in-depth analysis of formal neurocognitive testing suggest that the tumor itself may have the most deleterious effect on cognitive function.124 Recognition has been gaining that long-term neurocognitive functional (NCF) impairment following radiotherapy for benign or low-grade adult brain tumors could be associated with hippocampal dose. A dose to 40% of the bilateral hippocampi greater than 7.3 Gy was recently shown to be associated with long-term impairment in list-learning delayed recall.125 Based on such data, the role of proton therapy as a potential approach to reduce cognitive deficits and other side effects is being explored.
Chemotherapy
Low-grade gliomas have historically been considered chemotherapy resistant. With the recent demonstration of the chemotherapy responsiveness of some low-grade astrocytomas and oligodendrogliomas has renewed interest in investigating chemotherapy for low-grade gliomas.126,127 It has been demonstrated that some low-grade gliomas, especially optic pathway and hypothalamic tumors, can be responsive to chemotherapy.31,128 In children, various single and multichemotherapeutic and biological agents are effective at controlling the growth of a low-grade glioma in a setting of a newly progressive lesion, multiply recurrent, or unresectable residual tumors.102–105,129,130 Platinum-containing regimens result in radiographic response rates greater than 60%.129 Vinblastine has also demonstrated substantial activity in recurrent low-grade gliomas and is a commonly used second-line agent after treatment failure with vincristine and carboplatin.131,132 Other second- and third-line therapies for multiply recurrent tumors include thioguanine, procarbazine, lomustine, and vincristine (TPCV) and temozolomide. Irinotecan and bevacizumab are currently being investigated in a multi-institutional phase II trial for the treatment of progressive low-grade gliomas. Rapamycin, an oral immunosuppressive agent, has been effective at reducing the growth of astrocytomas associated with tuberous sclerosis.133 Most of the chemotherapy responses seen in children with low-grade gliomas are for contrast-enhancing masses that probably represent pilocytic astrocytomas. Some of these responses can last for years, although nearly half of all children treated with chemotherapy ultimately require radiotherapy. Nonenhancing, diffusely infiltrating astrocytomas in children appear to be much less responsive to chemotherapy. Data on the use of chemotherapy for low-grade glioma in adults are sparse. In a small Southwest Oncology Group trial, adults with incompletely excised low-grade gliomas were randomly assigned to radiation therapy (RT) alone or RT and lomustine ([2-chloroethyl]-3-cyclohexyl]-1-nitrosourea [CCNU]). There was no difference in survival between the two arms.134 The role of adjuvant procarbazine, CCNU, and vincristine (PCV) for high-risk patients (e.g., less than total resection, age older than 40 years) with low-grade gliomas was evaluated in RTOG-9802. From 1998 to 2002, 251 patients were randomly assigned to RT alone or RT followed by six cycles of PCV. An initial report of this study showed that the 5-year OS rates for RT versus RT/PCV were 7.5 years versus not reached respectively (hazard ratio [HR] = 0.72, 95% confidence interval [CI], 0.47 to 1.10; p = 0.33).135 At the time of that report, however, 65% of the patients were still alive. A recent National Institute of Health press release on more mature results of this study reported significant improvement in OS in the PCV chemotherapy plus RT group (13.3 years) compared to those assigned to RT alone (7.8 years) at a median follow-up of 12 years.136 Molecular and cytogenetic analyses (isocitrate dehydrogenase mutations and loss of heterozygosity of 1p and 19q, as well as methylation of methylguanine methyltransferase status) and clinical outcome are pending to identify predictive factors for patients with LGG.
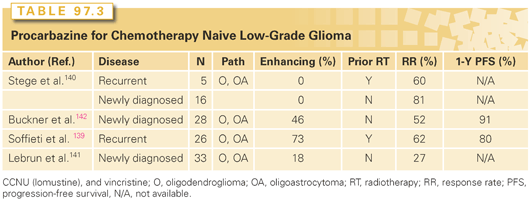
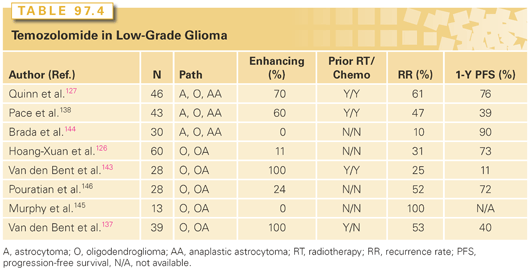
Several studies have evaluated PCV in the recurrent setting, and, more recently, temozolomide has also been evaluated (Tables 97.3 and 97.4).126,127,137–146 In general, approximately half of the patients treated with either temozolomide or PCV experienced imaging stability or improvement of neurologic symptoms. Although results are encouraging, the number of patients treated in these studies was small, and there are questions regarding the criteria used for radiographic response. In the first report of RTOG 0424, the primary endpoint was to compare the 3-year OS of a regimen of concurrent and adjuvant temozolomide and RT in a high-risk low-grade glioma population to the 3-year OS rate of the high-risk EORTC LGG patients reported by Pignatti et al.116 With a median follow-up time of 4.1 years and a minimum follow-up of 3 years, MST has not yet been reached. The 3-year OS rate was 73.1% (95% CI, 65.3 to 80.8%), significantly improved in comparison to the prespecified historical control with a p value <0.0001.147 An ongoing intergroup phase III trial is attempting to answer this issue more definitively.
Patients with low-grade oligodendroglial tumors with 1p/19q deletion or t(1p;19q) have longer PFS and OS than those without.114 Consequently, 1p/19q determination is important in patient counseling and in assessing the results of outcomes in future clinical trials. A randomized phase III EORTC trial stratified patients with low-grade glioma by 1p status prior to randomization to RT versus temozolomide.148 In the initial results of the trial presented, PFS was not significantly different, and median OS was not reached. 1p deletion was a positive prognostic factor irrespective of treatment (PFS: 0.0003; HR = 0.59; 95% CI, 0.45 to 0.78); OS: 0.002; HR = 0.49; 95% CI, 0.32 to 0.77). First-line treatment with temozolomide compared to radiotherapy did not improve PFS in high-risk LGG patients. A molecular and genetic analysis of LGG has revealed aberrant signaling in the phosphatidylinositol-3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) network; however, a defined role for the inhibition of this pathway in the treatment of LGG remains to be established.149,150 Targeting this pathway is a therapeutic approach that is being investigated in clinical trials in recurrent LGG patients.
WHO Grade III: Anaplastic Astrocytoma
Prospective and/or randomized evidence indicating that a complete resection of enhancing or an MRI-visible tumor improves survival is lacking, but retrospective analyses reaffirm that this relationship is likely to be present. Nonetheless, almost all of these tumors are characterized by postoperative residual microscopic disease, and radiotherapy is used adjunctively, resulting in a 3-year survival of approximately 55%.151
Radiation Therapy
Partial brain fields are used for the treatment of anaplastic astrocytoma; the initial gross tumor volume (GTV) is defined as the T2 or FLAIR abnormality; the boost GTV is defined as the contrast-enhancing volume or the surgical bed; for smaller, nonenhancing tumors, the initial and the boost GTV are often equivalent.152 The clinical tumor volume (CTV) is defined as an approximately 2-cm margin surrounding the GTV, but not expanding across natural barriers. The initial volume is typically treated to 46 Gy, with the boost volume to 60 Gy. No survival advantage for the use of bromodeoxyuridine as a radiosensitizer was demonstrated.153 Early versus delayed radiotherapy, utilizing chemotherapy as part of the regimen, was evaluated in a German phase III trial (NOA-04), which is described in the section that follows.
Chemotherapy
The role of chemotherapy remains controversial. Most phase III trials have demonstrated no benefit compared with radiation alone. Both single-agent carmustine and PCV are associated with minimal improvement in survival. Although, for a period of time, PCV was considered the “superior” regimen, database analyses have belied this claim.151 A meta-analysis by the Glioma Meta-Analysis Trialists’ group demonstrated an approximate 6% absolute increase in 1- and 2-year survival for patients who received chemotherapy (2-year survival of 37% versus 31%).154 A large randomized trial by the Medical Research Council found no benefit of adjuvant PCV compared with RT alone.155 Although temozolomide is effective for the treatment of recurrent anaplastic astrocytoma, its role as an adjuvant to RT has not been rigorously assessed. Based on these results in recurrent anaplastic astrocytoma, the RTOG initiated a phase III trial (RTOG 9813) to compare radiation with BCNU or CCNU to radiation with temozolomide, and the results are pending.
A comparison of the efficacy and safety of radiotherapy versus chemotherapy with either PCV or temozolomide as initial therapy on patients with newly diagnosed anaplastic glioma showed comparable results in terms of time to treatment failure.156 Because of the potential prognostic and predictive value of hypermethylation of the O6-methylguanine DNA-methyltransferase (MGMT) promoter and mutations of the IDH1 gene in malignant gliomas, analyses of these was a correlative part of the study.110,157,158 Hypermethylation of MGMT promoter and mutations of the IDH1 gene as well as oligodendroglioma histology reduced the risk of progression. Hypermethylation of MGMT promoter was associated with prolonged PFS in the chemotherapy and RT arms.
Difluoromethylornithine (DFMO), an inhibitor of ornithine decarboxylase, was evaluated in a phase III trial.159 Of 228 patients, the majority had anaplastic astrocytoma. Following RT, patients were randomized to PCV or PCV plus DFMO. There was a difference in survival during the first 2 years, but this did not continue after 2 years.
Chemotherapy for Recurrent Anaplastic Astrocytomas
Chemotherapy for anaplastic astrocytomas that recur following radiation is of benefit, and both nitrosourea-based regimens and temozolomide have efficacy. The U.S. Food and Drug Administration (FDA) granted accelerated approval for temozolomide on the basis of its activity in recurrent anaplastic astrocytoma; the response rate was 35% for patients who had not received chemotherapy and 20% for patients who had received nitrosourea-based therapy.160 Many patients are being treated with temozolomide early in the course of their illnesses; therefore, for recurrent anaplastic astrocytoma, nontemozolomide regimens used in glioblastoma are often considered.161 Several clinical trials to evaluate targeted agents in recurrent malignant glioma often include recurrent grade III histology. Based on documented activity of the antivascular endothelial growth factor antibody in recurrent glioblastoma, this agent has also been used in patients with recurrent anaplastic astrocytoma.162 A retrospective study reported a 64% radiographic response and a 6-month PFS rate of 60% in 25 patients.163 Prospective studies are pending.
The NOA-04 phase III trial compared efficacy and safety of RT followed by chemotherapy at progression with the reverse sequence in patients with newly diagnosed anaplastic gliomas.156 Patients received conventional RT: procarbazine, lomustine (CCNU), and vincristine, or temozolomide at diagnosis. At occurrence of unacceptable toxicity or disease progression, patients in the RT arm were treated with PCV or temozolomide, whereas patients in chemotherapy arms received RT. Median time to failure, PFS, and OS were similar for all arms. Hypermethylation of the MGMT promoter was associated with prolonged PFS in the chemotherapy and RT arm. This study showed that IDH1 mutations are a positive prognostic factor in anaplastic gliomas, with a favorable impact stronger than that of 1p/19q codeletion or MGMT promoter methylation.156
WHO Grade III: Anaplastic Oligodendroglioma
Surgery
Surgery retains its role as the principal modality of treatment, as with other glial neoplasms, and the maximum safe resection is considered the standard of care. However, the consideration of risks versus benefits of an aggressive surgical resection should take into account the 1p/19q deletional status of the tumor and the potential for a more favorable natural history and response to medical therapy.
Radiation Therapy
No randomized trials that focus only on these tumors comparing radiation versus no RT have been completed. In general, patients with pure and mixed anaplastic oligodendrogliomas receive postoperative irradiation to 60 Gy in conventional daily fractions of 1.8 to 2.0 Gy using an approach similar to that used for other malignant gliomas. Recent data show a categorical and very large survival benefit for both the 1p19q codeleted and the IDH-mutated anaplastic oligodendrogliomas treated with combination chemoradiotherapy, in comparison to RT alone. Therefore, up-front RT alone should not be the preferred treatment for these good prognosis patients. Conversely, no level 1 data exist to support treating these patients with up-front chemotherapy alone, either, and although this practice is sometimes adopted in practice, it should be subjected to rigorous clinical evaluation, because of the potential for the loss of long-term survivorship in these favorable patients, if either therapy is compromised. These results are described in greater detail in the section that follows.
Chemotherapy
Retrospective series and phase II trials first suggested that oligodendrogliomas are chemosensitive.137,164,165 In two phase III trials, RT alone was compared with RT plus PCV. In the North American trial (RTOG 9402) patients received PCV for four cycles prior to radiation or no up-front PCV. Survival in the two groups was the same. Patients with 1p and 19q deletions had significantly better outcomes, regardless of treatment.166 An unprespecified analysis of PFS demonstrated that the benefit from PCV was most notable in patients with 1p and 19q deletions. Long-term results of this study demonstrated that patients with codeleted tumors lived longer than those with non-codeleted tumors irrespective of therapy, and the median survival of those with codeleted tumors treated with PCV plus RT was twice that of patients receiving RT (14.7 versus 7.3 years).167 There was no difference in median survival for patients with tumors lacking 1p and 19q deletion.
In the European trial, patients received PCV or no immediate chemotherapy after radiation.168 PFS was better in the PCV group, but OS was not different. Patients with 1p and 19q deletion had superior survival, regardless of treatment. A further molecular analysis of this cohort demonstrated that MGMT promoter methylation was of prognostic value.169,170 Long-term follow-up showed that PFS and OS were better in the PCV group, but OS was not different between the two groups (OS in the RT/PCV arm, 42.3 months; in the RT arm, 30.6 months). In patients with a 1p/19q codeletion, there was a trend to more benefit from adjuvant PCV (OS not reached in the RT/PCV group versus 112 months in the RT group).171 Both trials confirmed the prognostic value of 1p and 19q.
Temozolomide has produced high response rates in patients with anaplastic oligodendroglioma. In 27 newly diagnosed patients treated with temozolomide prior to radiotherapy, the objective response rate was 33% and the 6-month progression rate was 10%.172
Chemotherapy for Recurrent Anaplastic Oligodendroglioma
Prospective trials have demonstrated that approximately 50% to 70% of patients with anaplastic oligodendrogliomas that recur after RT respond to chemotherapy.143 In a study of 48 patients with anaplastic oligodendroglioma/oligoastrocytoma who progressed on PCV, the objective response rate to temozolomide was 44%.173 Although there is no evidence that the sequence of temozolomide and PCV is superior in terms of efficacy, the absence of cumulative myelosuppression with temozolomide argues for its use initially in the setting of recurrent disease.
Ongoing Clinical Trials for Newly Diagnosed Grade 3 Glioma
Two international trials are being conducted in patients with newly diagnosed grade 3 glioma stratified by 1p 19q status rather than histology. Nondeleted patients are randomized to radiation with or without temozolomide; following RT, there is a second randomization to adjuvant temozolomide or not. Codeleted patients are randomized to three arms, temozolomide alone (phase II group), RT with concomitant and adjuvant temozolomide, or RT with adjuvant PCV (phase III).
WHO Grade IV: Glioblastoma
Surgery
Gliomas are heterogeneous, and therapy is guided by the most aggressive grade in the specimen. Resection provides the best opportunity to obtain an accurate diagnosis. Studies have shown that more complete resections are more likely to provide a high-grade diagnosis and to detect an oligodendroglial component.174 Two randomized trials of resection of malignant gliomas have been published. In a study by Vuorinen et al.,175 survival was twice as long with resection compared to biopsy alone. Stummer et al.63 reported that patients without residual contrast-enhancing tumor had a higher overall median survival time than did those with residual enhancing tumor (17.9 months versus 12.9 months, respectively; p <0.001). Complete resection of an enhancing tumor enhances certain approved or investigational adjuvant therapies (e.g., carmustine wafers, immunotherapy). Resection also is superior to stereotactic biopsy alone for the provision of adequate tissue for the evaluation of molecular and cytogenetic classifications and certain prognostic markers (e.g., MGMT), which may be a requirement for entry into some clinical trials.
There has been extensive work in molecular subtypes of GBM in recent years that include a report of the Cancer Genome Atlas Research Network176 and follow-up transcriptome work of glioblastoma provided insights into the major structural and expression alterations that may drive disease pathogenesis and biology in glioblastoma.177,178 Verhaak et al.178 proposed a gene expression-based molecular classification of GBM into proneural, neural, classical, and mesenchymal subtypes. Aberrations and gene expression of EGFR, NF1, and platelet-derived growth factor receptor a (PDGFRA)/IDH1 were utilized to define the classical, mesenchymal, and proneural subtypes, respectively. These investigations into the genome and transcriptome reveal GBM as a heterogeneous collection of distinct diseases with multiple dependencies both within and across each particular subtype.179
Radiotherapy
Randomized trials have demonstrated a survival benefit with RT.180 Localized radiation volumes are recommended based on evidence from several sources that GBMs typically recur locally, and the bulk of the infiltrative disease is within a few centimeters of the enhancing rim. However, the wide and somewhat unpredictable degree and direction of dissemination, which is not visualized well with any imaging technique, renders an RT field definition difficult. Outside of clinical trials, consensus regarding the exact field design remains difficult to obtain. In the large randomized trial RTOG 0525 for newly diagnosed GBM, which allowed both a single field treatment or a separate boost field to be utilized, no survival differences were identified, although the number of patients treated with the single field was rather small, in comparison (>80% versus <20%).181
Standard therapy uses a total dose of 60 Gy in 30 to 33 fractions based on dose response studies showing a survival improvement for 60 Gy compared to lower doses and without a benefit for higher doses.182–215 For patients with poor prognostic factors and for those who are not able to tolerate conventional treatment, a shorter course may provide palliation. Older patients (older than 65 years), especially those with poor performance status, have been shown to have limited posttreatment improvement following conventional RT,183 and several studies have not shown a significant survival difference using shorter courses.184–218 A number of trials have evaluated the role of temozolomide versus RT in elderly patients with glioblastoma. The German phase III trial (NOA-08) randomized patients 65 years or older with anaplastic astrocytoma or glioblastoma with a minimum karnofsky performance status (KPS) of 60 to either temozolomide or radiotherapy. Of 412 patients who were randomized, 373 received at least one dose of treatment and were included in efficacy analyses. Median survival was 8.6 months in the temozolomide arm compared to 9.6 months with RT. These results met the criteria of “noninferiority” of temozolomide.185 In an unplanned post hoc analysis, MGMT promoter methylation status was evaluated in 209 patients; promoter methylation was associated with longer OS (11.9 versus 8.2 months). Event-free survival was longer in patients with MGMT methylation who received temozolomide alone versus RT, whereas the opposite was true for patients without MGMT promoter methylation. Therefore, MGMT methylation seems to be a useful predictive biomarker and could aid decision making in elderly patients not fit to receive concurrent chemoradiation.
The Nordic three-arm phase III trial randomized elderly (>60 years) patients with glioblastoma to two different radiotherapy schedules of 60 Gy in 2 Gy per fraction (fxs) over 6 weeks or a hypofractionated schedule of 34 Gy in 3.4 Gy fxs over 2 weeks, versus temozolomide alone.186 Median survival was significantly longer with temozolomide versus conventional RT (8.3 versus 6.0 months; HR, 0.70; p = 0.01), but not hypofractionated radiotherapy (7.5 months; HR, 0.85; p = 0.24). This trial suggests that both temozolomide alone and hypofractionated RT alone produce equivalent survival in elderly patients with glioblastoma, and both are superior to standard RT.
These trials did not address the issue of concomitant chemoradiotherapy for the elderly. An ongoing, phase III trial (EORTC 26062-22061 NCIC CTG CE6) compares the OS rates between short-course RT alone and short-course RT given together with temozolomide in newly diagnosed patients with glioblastoma who are older than 65 years of age and who are not fit for standard treatment. This study has completed accrual and results are pending.
Dose Escalation
In the pretemozolomide era, studies evaluating radiosurgery or brachytherapy boosts to conventional RT have not demonstrated a survival advantage.222–225
The feasibility and toxicity of dose-escalated photon radiotherapy concurrent with BCNU chemotherapy in patients with supratentorial GBM187 was the goal of RTOG 9803. There were 209 patients who were enrolled and stratified into two groups based on the size of planned target volume (<75 mL versus ≥75 mL). Within each stratum, four RT dose levels were evaluated: 64, 72, 78, and 84 Gy; all treatments were delivered with a fraction size of 2 Gy. Acute and late grade ≥3 radiotherapy-related toxicities were no more frequent at higher RT doses or with larger tumors. No dose-limiting toxicities were observed at any dose level in either stratum, and as a result, the dose was escalated to 84 Gy in both strata. Median time to RT-related necrosis was 8.8 months (range, 5.1 to 12.5 months). This study demonstrated the feasibility and tolerability of photon dose escalation with an acceptable risk of late CNS toxicity with doses as high as 84 Gy. However, this study was conducted with concurrent BCNU chemotherapy, not the current standard approach of concurrent and adjuvant temozolomide. This chemoradiotherapy regimen has become the backbone of standard postoperative treatment for patients with GBM but has never adequately been tested in a RT dose-escalation or -intensification context. With this standard postoperative chemoradiotherapy regimen, the predominant pattern of failure remains local, highlighting the importance of investigating more intensive local therapies.
Recently, the University of Michigan published results of a clinical trial that escalated dose and dose-per-fraction from 66 Gy to 81 Gy in 30 fractions during chemoradiotherapy with temozolomide for patients with GBM.188 The maximum tolerated dose with concurrent temozolomide was 75 Gy in 30 fractions (2.5 Gy per fraction). Median survival was 20.1 months, suggesting improved efficacy comparable to other contemporary studies. Interestingly, the probability of in-field failure decreased with increasing dose escalation, setting the stage for more definitive investigations of this approach. Small phase II studies of dose escalation using mixed photon/proton irradiation demonstrated median survival times of 20 to 22 months, and more formal comparative studies need to be performed.244,245 Alternate particle radiation modalities used in the treatment of gliomas include neutrons, helium ions, other heavy nuclei such as carbon, negative pi-mesons, and thermal neutrons in conjunction with boronated compounds (boron neutron capture therapy). To date, most studies have been designed to determine optimal dose scheduling, efficacy, and safety.
Radiosensitizers and Radioimmunotherapy
Studies using various radiation modifiers such as hyperbaric oxygen, nitroimidazoles and tirapazamine, RSR-13, or carbogen and nicotinamide to overcome the hypoxia present in malignant gliomas have generally yielded disappointing results with no survival advantage.228,232 Halogenated pyrimidines are incorporated into the DNA of dividing cells due to their biochemical similarity to thymidine. After being incorporated, cells are much more susceptible to single-strand breaks from radiation-induced free radicals and have impaired ability to repair DNA. Prospective clinical studies, however, have not demonstrated a survival advantage.169 Motexafin gadolinium (MGd) is a redox-active drug that selectively accumulates in tumor cells. It is thought to sensitize tumors through the production of reactive oxygen species that destabilize cellular metabolism. A phase II RTOG trial did not demonstrate superiority in survival.189 Studies of radiation synergistic cytotoxics such as the camptothecins or platinum agents also did not demonstrate a survival benefit.235 Radioimmunotherapy, using various monoclonal antibodies against EGFR or tenascin tagged with 125I have been evaluated.237,238 These were small studies and demonstrated feasibility; however, randomized controlled studies have not been performed.
Chemotherapy
In a landmark international trial, patients were randomized to RT with or without concurrent and adjuvant temozolomide. Median and 2-year survival were increased by 2.5 months and 16.1%, respectively, in patients receiving temozolomide, and long-term follow-up showed a persistent survival benefit.190 A companion correlative study demonstrated that methylation of the promoter region of the MGMT gene in the tumor was associated with superior survival, regardless of treatment received, but the benefit was maximal for methylated patients.157 MGMT removes the methyl group from the O6 position of guanine, reversing the cytotoxic effects of methylating agents (such as temozolomide), making the tumor resistant to treatment, while methylation of the promoter region of MGMT results in inactivation of MGMT. MGMT status was strongly associated with survival (Fig. 97.6). Recognizing that a different schedule of temozolomide may overcome chemotherapy resistance, there have been several studies of alternative dosing of temozolomide both at the time of recurrence and in the newly diagnosed setting.191,192 A large phase III randomized international study led by the RTOG compared the standard treatment versus a 21- or 28-day adjuvant temozolomide schedule.181
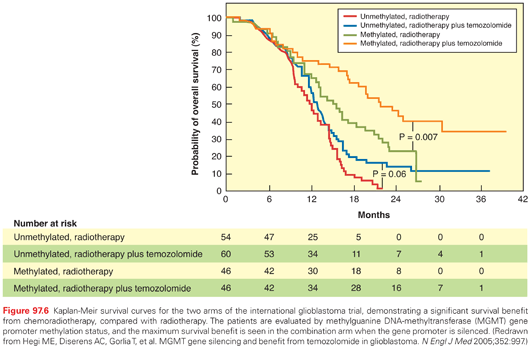
Dose-dense temozolomide failed to result in improved efficacy regardless of tumor methylation status but was associated with more profound lymphopenia and fatigue. Strategies to increase the therapeutic ratio of existing chemotherapies, such as the inhibition of DNA repair enzymes (i.e., poly[ADP-ribose] polymerase [PARP]) are being evaluated. These agents are being combined with radiation and chemotherapy to increase the cytotoxicity of the combination approach.193–195
Although nitrosourea-based chemotherapy is modestly effective for patients with GBM, its use has been supplanted by temozolomide. There is evidence that carmustine-impregnated wafers implanted into the brain at the time of resection provide modest improvement in outcomes in selected patients compared with patients who received placebo wafers.196
Chemotherapy for Recurrent Glioblastoma
Treatment options for recurrent GBM must be tailored to the individual. Few agents have proven activity. A randomized phase II trial of temozolomide versus procarbazine in 225 patients with GBM at first relapse demonstrated that treatment with temozolomide improved median PFS (12.4 weeks versus 8.3 weeks; p <0.006). Radiographic responses were disappointing (5.4% versus 5.3%). Several agents such as the platinoids, taxanes, 5-fluorouracil (5-FU), and irinotecan have been tested, most demonstrating very little activity. In a review of eight clinical trials with 225 recurrent malignant gliomas, the 6-month survival was 15% versus 31% for GBM versus anaplastic astrocytoma.197
Targeted Therapies
As the genetic and molecular pathogenesis of gliomas is better understood, new targets are being identified and inhibitors of associated signaling pathways are being developed. One example is EGFR as a frequently deregulated signaling molecule in GBM, prompting phase I and II trials of erlotinib and gefitinib for recurrent high-grade gliomas. Both have shown limited activity97,198–200 Patients whose tumors demonstrate the variant 3 mutant (EGFRvIII), with resulting constitutive activation of EGFR tyrosine kinase activity, along with intact phosphatase and tensin analog (PTEN), appear to be more responsive to EGFR inhibitors.201 There are two reports of the combination of erlotinib with radiation and chemotherapy for newly diagnosed glioblastoma showing modest additional benefit to standard radiochemotherapy, none of which show convincing survival improvement.202,203 Similarly, RTOG 0211, which evaluated the benefit of RT with concurrent gefitinib, showed median survival similar to that in a historical control cohort treated with radiation alone.204 Irreversible EGFR inhibitors such as afatinib did not show efficacy when used alone or in combination with temozolomide in recurrent GBM.205 There is an ongoing phase II study with the second-generation EGFR inhibitor, dacomitinib.
Preliminary reports using other targeted agents, including the mTOR inhibitor, temsirolimus, and the farnesyl transferase inhibitor, tipifarnib, have shown objective responses in a few high-grade gliomas.206–210
The most promising results have been seen for angiogenic inhibitors. The most important mediator of angiogenesis in GBM is vascular endothelial growth factor (VEGF). Antiangiogenic therapies such as the anti-VEGF monoclonal antibody bevacizumab have produced dramatic radiologic responses and prolonged PFS relative to historical controls.211,212 Based on the results of a randomized phase II study of 167 patients who received bevacizumab with or without irinotecan, the FDA granted accelerated approval to bevacizumab for recurrent glioblastoma in 2009.162 The PFS at 6 months was 43% for single-agent bevacizumab and 50% for the combination arm. The objective response rates were 28% and 38% for the two arms, and median survival times were 9.2 months and 8.7 months, respectively. The most common side effects associated with bevacizumab include fatigue, headache, and hypertension; proteinuria and poor wound healing are also seen. The addition of chemotherapy or targeted therapy to bevacizumab has failed to show any added benefit in recurrent GBM trials, with the exception of the BELOB study, a three-arm multicenter randomized phase II study, in which 148 patients with recurrent glioblastoma were treated with bevacizumab alone, lomustine alone, or the combination of the two. Survival at 9 months was 38%, 43%, and 59%, respectively, and the PFS at 6 months was 16%, 13%, and 41%, respectively, in the three arms.213 The value of the combination of bevacizumab and lomustine in recurrent GBM is currently being evaluated in EORTC 26101, which was initially a randomized phase II study modified into a two-arm phase III trial to address this question.
There are several reports of small single-arm phase II studies of the combination of bevacizumab with radiation and temozolomide in the newly diagnosed setting.214 Two large randomized trials evaluated the addition of bevacizumab to the initial combined modality therapy of RT and temozolomide. In RTOG 0825, a randomized, double-blinded, placebo-controlled trial, the addition of bevacizumab to temozolomide and radiation resulted in a longer PFS that did not reach the preset level of significance (10.7 months versus7.3 months; HR, 0.79). There was no difference in OS between two arms (16.1 versus15.7 months; HR, 1.13).215 Another phase III, placebo-controlled randomized trial (AVAglio study) in newly diagnosed glioblastoma showed that the addition of bevacizumab to radiation and temozolomide (experimental arm) significantly prolonged PFS (HR, 0.64; p <0.0001; median, 10.6 versus 6.2 months) as compared to radiation and temozolomide (control arm).216 However, the median OS was not significantly different in both arms. One-year OS was 72% and 66%, respectively (p = 0.049); 2-year OS was 34% and 30%, respectively (p = 0.24) in the experimental and control arm. Safety was consistent with known bevacizumab side effects, serious adverse events (AEs) (grade ≥3) were 28.7% versus 15.2% grade in the two arms.
As previously discussed, patients with MGMT-nonmethylated GBM have inferior outcomes as compared to those with MGMT-methylated GBM, and temozolomide is less effective in these patients. The open-label GLARIUS trial was a randomized, multicenter study of 170 patients in which MGMT-nonmethylated GBM patients were randomized (2:1) to radiation with bevacizumab during RT followed by maintenance bevacizumab and irinotecan (experimental arm) compared to standard therapy with daily temozolomide during radiation followed by 6 cycles of temozolomide (control arm).217 The PFS at 6 months rate of 71.1% was significantly higher in the experimental arm compared to 26.2% in the control arm (p <0.0001 log-rank test).
Recognizing that tumors ultimately evade the effect of antiangiogenic agents through various mechanisms, other strategies include the evaluation of the combination of bevacizumab with chemotherapeutic and targeted agents, and the investigation of other VEGF-targeted agents. Batchelor et al.218 reported a reduction in contrast enhancement and edema in 12 of 16 GBM patients who received cediranib (AZD2171), an orally administered pan-VEGF receptor inhibitor, with a median PFS of 3.7 months. However, a phase III randomized trial comparing the efficacy of cediranib failed to show any improvement in PFS with cediranib either as monotherapy or in combination with lomustine compared to lomustine alone in recurrent GBM.219
VEGF Trap (aflibercept), a recombinantly produced fusion protein that captures circulating VEGF and CT-322 (Angiocept, Adnexus Therapeutics, Waltham, MA), a pegylated recombinant peptide with a high affinity for VEGF was tested in the recurrent and in the newly diagnosed setting. The phase II study showed aflibercept had minimal evidence of single-agent activity in unselected patients with recurrent malignant glioma.220
Cilengitide (EMD121974), an integrin inhibitor that showed promise in the recurrent GBM,221,222 was evaluated in two large, newly diagnosed studies. The first study, CENTRIC, a phase III trial investigated the role of cilengitide combined with the standard treatment for patients with newly diagnosed glioblastoma with MGMT promoter methylation.223 The study failed to show any additional benefit with cilengitide in this patient population.223 The other study, CORE, investigated the benefit of cilengitide in the unmethylated MGMT gene promoter in a multicenter, open-randomized phase II trial. There was suggestion of benefit of cilengitide with a median OS of 16.3 months in one of the cilengitide arms compared to the median OS of 13.4 months in the control group (HR: 0.69; p = 0.033). However, this drug is not being further developed.
The other antiangiogenic agents that have undergone investigation in recurrent glioblastoma include XL184, a multitargeted tyrosine kinase inhibitor that acts on the VEGFR, hepatocyte growth factor receptor (MET), and c-KIT; and enzastaurin, an inhibitor of protein kinase C-beta that targets VEGF as well as the mTOR pathway.224 The initial results of these studies have shown similar or inferior outcomes to those reported with other agents.224–226
Other mechanisms of cell growth that are being targeted include epigenetic modulation through histone deacetylase inhibitors, the proteasome inhibitor bortezomib, and the glutamate receptor inhibitor talampanel.227–229
Gene Therapy Strategies
The efficacy and safety of a locally applied adenovirus-mediated gene therapy with a prodrug-converting enzyme (herpes simplex virus thymidine kinase; sitimagene ceradenovec) followed by intravenous ganciclovir was evaluated in 250 patients with newly diagnosed resectable glioblastoma. Temozolomide was not given in all patients. There was no evidence of a survival advantage to this approach.230 Previous strategies have similarly been negative, and the challenges of adequate delivery of the virus and gene transduction into the tumor remain paramount.
Immunotherapies
Immunotherapeutic strategies targeting glioblastomas include recombinant immunotoxins, restoration of local and systemic immunosuppression, one-size-fits-all, and individualized autologous dendritic cell vaccines.89,231–233
Issues in Study Designs for Novel Agents
Several key issues confront the incorporation of new agents in the up-front management of malignant gliomas. First, there is the issue of defining the appropriate end point. In recurrent malignant gliomas, PFS is frequently employed, but because of insufficient evidence linking this to survival in newly diagnosed malignant gliomas, survival remains the gold standard. However, there is considerable heterogeneity in survival outcomes based on clinical and possibly molecular prognostic variables. An adequate staging system has never been developed. The RTOG has analyzed an extensive database of prospectively treated patients (primarily with surgery, radiotherapy, and alkylating chemotherapy), and using a statistical method known as recursive portioning analysis, has developed six prognostic groups, referred to as RTOG recursive portioning analysis classes I to VI. Patients can be segmented into classes using eight variables: age, histology, Karnofsky performance score, mental status, neurologic function, symptom duration, extent of resection, and radiotherapy dose. GBM patients fall in classes III through VI, and their median survival ranges from 4.6 months to 17.9 months (Table 97.5).234
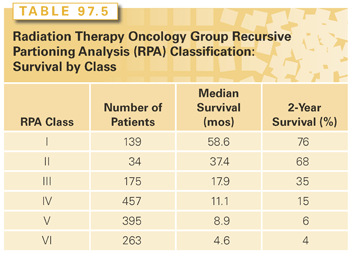
Gliomatosis cerebri is a rare condition with diffuse involvement of multiple parts of the brain (greater than two lobes). On MRI, there is typically diffuse increased signal on T2-weighted and FLAIR images and a low or absent signal in the affected areas on T1-weighted images (Fig. 97.7). Prognostic factors include age and histology as well as Karnofsky performance score. Treatment remains undefined and includes radiation and chemotherapy.291–293
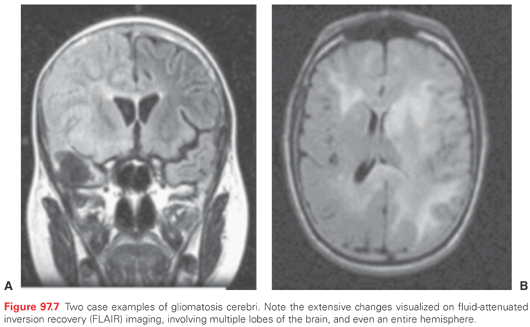
OPTIC, CHIASMAL, AND HYPOTHALAMIC GLIOMAS
Clinical Considerations
Nearly all gliomas of the optic nerve and chiasm are discovered in patients younger than 20 and most occur in those under 10 years of age. Of patients with optic pathway glioma (OPG), 20% to 50% are affected by NF1.235 Patients with NF1 are more likely to have lesions involving one or both optic nerves (anterior), whereas chiasmatic or hypothalamic (posterior) involvement is commonly seen among non-NF1 patients (sporadic). Lewis et al.235 found that gliomas along the anterior visual pathway occurred in 15% of NF1 patients and were occasionally bilateral; 67% of these were neither suspected clinically nor obvious on ophthalmologic examination. In one series, 25% involved the chiasm alone, 33% the chiasm and hypothalamus, and 42% the chiasm and optic nerves or tracts.236 Clinically, they cause loss of visual acuity (70%), strabismus and nystagmus (33%), visual field impairment (bitemporal hemianopsia, 8%), developmental delay, macrocephaly, ataxia, hemiparesis, proptosis, and precocious puberty. Funduscopic evaluation demonstrates a range of findings from normal optic discs, to venous engorgement, to disc pallor because of atrophy. Chiasmal tumors often grow to involve the hypothalamus, causing a diencephalic syndrome characterized by emaciation (especially in children between 3 months and 2 years of age), motor overactivity, and euphoria. In general, optic nerve gliomas have a better prognosis than those involving the chiasm, and tumors confined to the anterior chiasm have a better outcome than posterior chiasmal tumors.
The natural history of these tumors ranges from indolent growth or spontaneous regression (with NF1) to rapid progression or dissemination (with hypothalamic lesions).237–239 Generally, the prognosis of OPG is good with overall 5-year survival rates ranging between 70% and 90%; however, the long-term morbidity is high.239–242 NF1 and age less than 5 years at diagnosis have better PFS.239,243
Pathologic Considerations
Histopathologically, a majority of these tumors are low-grade gliomas, typically pilocytic or fibrillary astrocytomas. They range from primarily piloid and stellate astrocytes (most common), with or without oligodendroglia, through the gamut of malignant astrocytomas to GBM (rarely). Typically, optic gliomas appear as fusiform expansions of any part of the nerve. They may bridge through the optic foramen and expand as dumbbell tumors. The nerve can be infiltrated by tumor originating in the chiasm, the walls of the third ventricle, or the hypothalamus. A subset of optic pathway tumors can show the more aggressive pathologic variant of pilomyxoid astrocytoma as compared to the better known pilocytic astrocytoma.244
Imaging Findings
Diagnosis is best made by MRI, which demonstrates enlargement of the affected optic pathway, often with enhancement. The T2 signal may extend posteriorly along the optic tracts as far as the visual cortex, which may represent tumor infiltration or edema. Cysts and calcification are uncommon, but the hypothalamic component can be cystic.
Treatment Decision Making
In general, children with asymptomatic lesions of the optic pathways found by MRI are not treated unless clinical or radiographic progression is documented. Tumors in children with NF1 tend to be more indolent than sporadic tumors. Only one-third to one-half of children with NF1 with asymptomatic optic pathway tumors found on screening MRIs require treatment for increasing visual symptoms.245 Most children with sporadic tumors undergo imaging because of symptoms and should be treated. Sporadic tumors often present with advanced findings such as hydrocephalus, decreased visual acuity, and endocrinopathies.246 Rarely, both sporadic and NF-associated optic pathway gliomas can regress spontaneously.238
Surgery
Surgery is only rarely indicated for optic pathway gliomas. In appropriate patients, surgery may decrease the recurrence rate and increase the time to recurrence. Patients treated with surgery, followed by radiation and chemotherapy, appear to have the highest long-term control.247 In patients with progressive symptoms (e.g., severe visual loss and proptosis), unilateral anterior tumors that do not involve the optic chiasm may be resected. Biopsy or subtotal resection can be performed for posterior optic pathway gliomas that involve the hypothalamus and optic tract, particularly if they are symptomatic because of local compression and mass effect. Resection of the chiasm is not indicated due to resultant bilateral blindness.
If the tumor involves the chiasm and the MRI raises suspicion of another tumor type, such as an optic nerve sheath meningioma or another parasellar mass, a confirmatory biopsy can be performed. This is rarely needed in patients with NF1, in whom there is a high index of suspicion for an optic nerve glioma. A subtotal resection is indicated if mass effect produces dysfunction of adjacent structures such as the hypothalamus or the nerve itself. Hydrocephalus can be produced by more posteriorly situated tumors and may be alleviated by debulking. If hydrocephalus persists after debulking, CSF shunting (which may need to be biventricular or require fenestration of the septum) becomes necessary. For unknown reasons, hydrocephalus is often persistent after the CSF pathways have been cleared, requiring the insertion of a CSF diversion device even after tumor removal.
Radiation Therapy
Untreated optic gliomas, especially those involving the chiasm or extending into the hypothalamus or optic tracts, progress locally or are fatal in 75% of patients. Tenny et al.248 found that only 21% of patients who were followed after biopsy or exploration survived compared with 64% of those who received RT.
Routine postoperative irradiation is not indicated for gliomas confined to the optic nerve, which can be completely resected.249 RT can prevent tumor progression, improve disease-free survival, and stabilize or improve vision in patients with chiasmal lesions, for whom postoperative residual is the rule. Wong et al.250 reported that 86% of chiasmal gliomas not treated with RT progressed locally, whereas treatment failure occurred in 45% that underwent RT. Furthermore, control was achieved in 87% of the irradiated patients who received a dose of 50 to 55 Gy compared to 55% of those who received 46 Gy or less.
The prognosis for patients with optic nerve tumors may be better than for those with chiasmal-hypothalamic lesions. In a literature review, local control was found to be achieved for 154 of 189 irradiated anterior chiasmal tumors (81%), whereas 92 of 142 posterior tumors (65%) were controlled. Vision improved in 61 of 210 evaluable patients (29%) and remained stable in 118 of 210 patients (56%).251 For chiasmal-hypothalamic tumors, RT produced radiographic shrinkage in 11 of 24 (46%) with a median PFS of 70 months compared with 30 months for patients who did not receive RT.252 Age and tumor location were important prognostic factors, with younger children (less than 3 years), and children with lesions posterior to the chiasm faring less well after radiotherapy.
Three-dimensional conformal radiotherapy, IMRT, and stereotactic techniques are used to minimize the dose to adjacent structures. A report by Debus et al.253 summarized results in patients treated with fractionated stereotactic radiation therapy (FSRT) (52.2 Gy median dose at 1.8 Gy per day).253 All patients remained disease free, and no significant complications or marginal failures were seen despite highly conformal radiation fields. Because these tumors are often focal, techniques like FSRT can offer both excellent local control and decreased late effects.
Chemotherapy
In recent years, chemotherapy has played a pivotal role in the management of OPG in young children in order to spare the developing brain from the adverse effects of irradiation.241,254–257 This is especially important in patients with NF1 who are at significant risk of developing vasculopathy such as moyamoya syndrome and secondary malignancy after receiving radiotherapy.256,258 Retrospective series suggest that cognitive function is preserved better in children who receive initial chemotherapy compared with RT.241,259 Although the appropriate agents are still evolving, vincristine plus carboplatin remains the most common first-line regimen.128 Gnekow et al.260 reported a 5-year PFS of 73% in 55 patients who were treated with this regimen. The randomized Children’s Oncology Group A9952 study showed a 5-year PFS of 35% using carboplatin with vincristine and 48% using thioguanine, procarbazine, lomustine, and vincristine regimen in children with newly diagnosed progressive low-grade glioma.261 Cisplatin-based regimens have shown responses between 50% and 60% and 5-year PFS of 50%.262–264 Other studies have shown temozolomide to be effective.265–267 Vinblastine has also been active in these tumors and is generally a second-line agent.131,132 Collectively, these data suggest that chemotherapy is helpful in delaying tumor progression in a significant portion of children.
Whether chemotherapy alone can improve vision is controversial. Most studies in the literature lack objective data on visual outcome prior to and after chemotherapy. Moreno et al.268 conducted a systematic review of eight reports and found only 14.4% of the children treated with chemotherapy had improvement in their vision. Due to the risk of second malignancy, alkylator-based chemotherapies are generally avoided in patients with NF1.
Clinical and Pathologic Considerations
Brainstem gliomas account for 15% of all pediatric brain tumors but are rare in adults. They can be divided into several distinct types. The diffuse intrinsic pontine gliomas (DIPG) tumors are generally high-grade astrocytomas, either anaplastic astrocytomas or GBM. Completely separate, and clinically distinct are the focal, dorsally exophytic or cervicomedullary lesions that are usually low grade with a better prognosis. Although rare, ependymomas, PNETs, and atypical teratoid-rhabdoid tumors also occur in the brainstem. Nonneoplastic processes that may be confused with a brainstem tumor include neurofibromatosis, demyelinating diseases, arteriovenous malformations, abscess, and encephalitis.
The diagnosis of a DIPG is usually based on a short history of rapidly developing neurologic findings of multiple cranial nerve palsies (most commonly VI and VII), hemiparesis, and ataxia. The initial manifestations of a brainstem glioma are unilateral palsies of cranial nerves VI and VII in approximately 90% of patients. The classic MRI finding is diffuse enlargement of the pons with poorly marginated T2 signal involving 50% or greater of the pons (Fig. 97.8).269 Most are nonenhancing; in children, enhancing lesions could have either a pilocytic or malignant component; in adults, enhancement is worrisome for a malignant glioma.270 Cervicomedullary tumors are nonenhancing, well-circumscribed lesions with an exophytic component. Tectal gliomas are nonenhancing and enlarge the tectal plate, often expanding it into the supracerebellar cistern with associated hydrocephalus. Overall, the prognosis is poor for patients with DIPG, with few patients surviving longer than 1 year.271
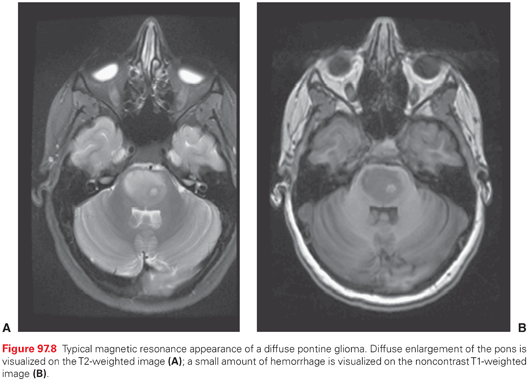
Surgery
Complete resection is almost never possible for the majority if brainstem tumors, and even a biopsy is restricted because of substantial morbidity and mortality.272,273 In most centers, biopsy of DIPG is not undertaken for very typical cases. Stereotactic needle biopsy can be performed if atypical imaging findings or clinical characteristics suggest another diffuse brainstem disorder. Resection has no place in the treatment of diffuse pontine gliomas in children or adults. For the rare focal astrocytic lesions of the adult or pediatric brainstem, surgery may play a larger role. Tectal gliomas have a typical imaging appearance, and biopsy is neither necessary nor safe. However, the accompanying noncommunicating hydrocephalus (from compression of the aqueduct of Sylvius) can be treated with CSF diversion, either by third ventriculocisternostomy or by ventriculoperitoneal shunting.274 Dorsally exophytic astrocytomas within the fourth ventricle or at the cervicomedullary junction are often partially resectable with low morbidity and excellent long-term results.275 These dorsally exophytic brainstem tumors arise from the substance of the pons and are very dangerous to resect in totality. Because many of them will remain indolent after partial resection, complete removal at the first surgery is likely not warranted and associated with severe neurologic deficits. Intrinsic astrocytomas or ependymomas at the cervicomedullary junction can often be completely removed through a posterior midline approach.276 In a retrospective review of 28 patients with juvenile pilocytic astrocytoma of the brainstem treated with resection in 25 cases and biopsy in 3,277 the 5- and 10-year PFS rates were 74% and 62%, respectively, after gross total resection or resection with linear enhancement and 19% and 19%, respectively, when solid residual tumor was present, suggesting that long-term survival after resection of these tumors may relate to the extent of initial excision.
Radiation Therapy
RT, the primary treatment for brainstem tumors, improves survival and can stabilize or reverse neurologic dysfunction in 75% to 90% of patients. The GTV is usually best defined using T2-weighted or FLAIR MRI. A margin of 1.0 to 1.5 cm is added to create a CTV. These lesions should be treated to 54 to 60 Gy using daily fractions of 1.8 to 2.0 Gy. In a multi-institutional survey by Freeman and Suissa,278 the 1-, 2-, and 5-year survival rates of children treated with conventional RT techniques were 50%, 29%, and 23%, respectively. Hyperfractionation, designed to deliver higher tumor doses, has been evaluated, without a significant survival advantage (median survival, 8.5 months versus 8.0 months for conventional versus hyperfractionated regimens).279 Several drugs, such as topotecan, and motexafin-gadolinium have been investigated as radiosensitizers, without clear evidence of benefit, and therefore, the role of sensitizers remains investigational.
Fewer data exist with respect to brainstem glioma in adults, but there is some evidence that these tumors may be less aggressive in adults, with OS that ranges from 45% to 66% at 2 to 5 years, perhaps because of a greater frequency of more favorable tumor types.280 In the series from ANOCEF, 48 adult patients with brainstem gliomas were grouped on the basis of their clinical, radiologic, and histologic features.270,281 Nearly half had nonenhancing, diffusely infiltrative tumors and had symptoms that were present for longer than 3 months. Eleven of these 22 patients underwent biopsy, and 9 had low-grade histology. Nearly all underwent radiotherapy and had a median survival of 7.3 years. A second group of 15 patients who had presented with rapid progression of symptoms and had contrast enhancement on MRI were described. Fourteen of these patients underwent biopsy, and anaplasia was identified in all 14 specimens. Despite radiotherapy, the median survival in this group was 11.2 months, which approximates the survival in pediatric series.
Chemotherapy
Despite numerous clinical trials, there is no clear evidence to show increased survival for patients with DIPG who receive radiation and chemotherapy as compared to radiation alone. The recent discovery that the majority of DIPGs harbor mutations of lysine 27 (K27) in the histone 3.3 gene (so called K27M mutations) may allow for the development of targeted therapies.282,283 Similarly, a subset of DIPGs have amplifications of receptor tyrosine kinase family members (i.e., PDGFR-α) suggesting other avenues to targeted therapy.284 Clinical trials using temozolomide during and after RT have not shown improvement in the outcome.285–287 Thus, no agent used either during or after radiation treatment has been shown to have benefit over radiation alone.
Clinical and Pathologic Considerations
Cerebellar astrocytomas, which occur most often during the first 2 decades of life, arise in the vermis or more laterally in a cerebellar hemisphere. They are usually well circumscribed and can be cystic, solid, or some combination of both. It is not uncommon to have a small tumor (mural nodule) associated with a large cystic cavity.
Histologically, most are low-grade pilocytic astrocytomas that lack anaplastic features. In a series of 451 children, cerebellar astrocytomas accounted for 25% of all posterior fossa tumors, and 89% of the 111 cerebellar astrocytomas were low grade.288 Approximately 75% of these tumors are located only in the cerebellum, with the remainder involving the brainstem as well. Because these tumors usually arise in the vermis or median cerebellar hemisphere, the clinical presentation is similar to that of medulloblastoma, with truncal ataxia, headache, nausea, and vomiting. In infants, head enlargement from hydrocephalus is seen. The majority of cerebellar pilocytic astrocytomas have an oncogenic fusion gene (KIAA1549-BRAF) that results in the activation of the BRAF oncogene, and which might be a rational target for future therapy.289,290
Surgery
Gross total resection is tantamount to a cure for these lesions.291,292 In most cases, incomplete removal should be managed by conservative monitoring because the majority of remnants will not grow, will remain low grade, and are easy to remove if they progress in the future.
Radiation Therapy
Nearly all completely resected cerebellar astrocytomas do not require RT. Even when they progress, repeat resection is reasonable if a majority of the tumor can be removed. Only in the most exceptional circumstances would RT be necessary for the treatment of child with a true cerebellar pilocytic astrocytoma.
Chemotherapy
In general, chemotherapy is not indicated. Based on the experience with optic pathway gliomas, several of which have pilocytic features, carboplatin has been used for recurrent tumors.293,294 There is limited experience with the use of temozolomide in this setting. High-grade gliomas that arise in the cerebellum are treated with regimens identical to their supratentorial counterparts.
Clinical and Pathologic Considerations
Gangliogliomas, along with pilocytic astrocytomas, pleomorphic xanthoastrocytomas, and subependymal giant cell astrocytomas, are considered astroglial variant forms of low-grade gliomas.295 They are more circumscribed than diffuse low-grade gliomas, are classified as grade 1 or 2, and do not typically invade the normal brain. Because they less frequently progress to higher grade lesions, surgery alone is often curative. Gangliogliomas are more common in children than adults. They are the most common neoplasms to cause chronic focal epileptic disorders, and they typically arise in the temporal lobe but may also occur in the brainstem, spinal cord, and diencephalon.296 They may include a cystic component, and the solid portion is free of normal brain parenchyma. Unlike diffuse low-grade gliomas, gangliogliomas enhance on MRI scans. They contain both glial and neuronal elements. The glial elements, which stain for glial fibrillary acidic protein, are almost always astrocytic and often pilocytic, but fibrillary astrocytes are also common. The glial elements dictate whether the lesion is grade 1 or 2. The neurons in the tumor are neoplastic and are characteristically large and relatively mature (i.e., they contain ganglion cells). The presence of neoplastic neurons may be confirmed by immunostaining for neuron-specific enolase and synaptophysin. Grade 2 lesions have rarely been observed to progress to a higher grade.297,298
Surgery
Surgical resection is directed at removal of the contrast-enhancing portion of the tumor. Nevertheless, although lesions located within eloquent brain regions are resectable, they may present significant surgical challenges because the boundary between tumor and functional brain may be difficult to define, even with the aid of modern surgical adjuncts (e.g., operating microscope, computer-assisted navigation, functional brain mapping). Although no phase 3 prospective studies have documented the superiority of surgery over other approaches (e.g., radiotherapy), retrospective studies have indicated that complete resection is associated with a very favorable long-term survival.297–299 Resection of gangliogliomas also can result in seizure control.300 Grade 2 gangliogliomas may recur, and some patients do poorly. The degree of anaplasia determines the prognosis.
Radiation Therapy
Because resection has the potential to cure most of these lesions, radiotherapy is generally reserved for subtotally resected cases or for recurrences.301 It is also used for lesions in complex locations where further resection may result in significant morbidity. To determine the optimal strategy for gangliogliomas, Rades et al.302 conducted a literature-based retrospective study of more than 400 patients treated for ganglioglioma. They examined four different treatment strategies (GTR or subtotal resection [STR] with or without radiotherapy) in 402 patients identified from reports published between 1978 and 2007. Surgery was found to be the mainstay of therapy, with 209 patients undergoing GTR and 193 undergoing STR. Adjuvant radiotherapy was used in 101 patients (20 following GTR and 81 following STR). Patients who underwent GTR had higher rates of OS and PFS than individuals who underwent STR. For patients undergoing GTR, the 10-year rates of local control and OS were 89% and 95%, respectively, better than the 52% and 62% observed for patients undergoing STR. This indirectly indicates that GTR is the most effective treatment strategy for gangliogliomas. For patients undergoing STR followed by postoperative radiotherapy, the 10-year rate of local control was 62%, better than the 52% for patients undergoing STR without postoperative radiotherapy; although the 10-year survival also improved from 65% to 74% with the use of postoperative radiotherapy in patients with subtotally resected tumors, this did not reach statistical significance. For the 40 patients undergoing STR for whom radiotherapy details were known, local recurrence was observed in 6 of 22 (27%) receiving 54 Gy, compared to 7 of 18 (39%) receiving greater than 54 Gy, implying no specific dose–response relationship.
Chemotherapy
Chemotherapy for gangliogliomas is generally reserved for young children who have undergone subtotal resection and who demonstrate disease progression. In older patients, it is typically used as salvage therapy to treat recurrent tumors after the failure of surgery and RT. In general, for astroglial variants such as gangliogliomas, no optimal chemotherapeutic regimens have been defined, and most researchers consider disease stabilization (rather than a complete tumor response) to be a successful outcome.
Clinical and Pathologic Features
Ependymomas were originally thought to arise from the ependymal cells lining the cerebral ventricles and the vestigial central canal of the spinal cord as they resemble this tissue under the microscope, although more recently it has been accepted that they arise from radial glial cells, a type of CNS stem cell.303,304 Ependymomas can arise throughout the nervous system, and are usually divided into those from the supratentorial, infratentorial (posterior fossa), and spinal regions. Those in the spinal region are broken down into the intramedullary lesions and the myxopapillary ependymomas of the conus medullaris and cauda equine. Although these tumors look very similar under the microscope (histology), they are demographically, clinically, transcriptionally, and genetically distinct and should not be regarded as the same entity.304,305 More recently, it has been shown that there are two clear groups of posterior fossa ependymoma, the posterior fossa type A (PFA) tumors and the posterior fossa type B (PFB) tumors.306,307 PFA tumors occur in young children (infants), are more likely to be lateral (CP angle), and have a terrible prognosis. PFB tumors are diagnosed in older children, found in the midline, and have a much better prognosis than PFA tumors. Figure 97.9 shows the typical magnetic resonance appearance of a midline, the posterior fossa ependymoma. In the past, much was made of the pattern of anaplasia in ependymoma histology, with the diagnosis of anaplasia taking an ependymoma from WHO grade II to WHO grade III. More recently, a number of luminaries in the field of neuro-oncology have shown that the intra- and interobserver reliability in the diagnosis of anaplasia in ependymoma is very high and its clinical utility is, therefore, very limited.308
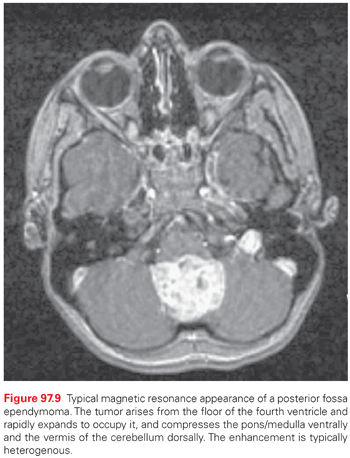
Clinical presentation depends on location. Tumors with ventricular involvement often cause increased ICP and hydrocephalus by obstruction of CSF pathways. Headaches, nausea and vomiting, papilledema, ataxia, and vertigo are frequent. Focal neurologic signs and symptoms are seen with supratentorial ependymomas that involve the parenchyma. The presence of calcification in a fourth ventricular tumor on CT is very suggestive of an ependymoma. Supratentorial parenchymal tumors cannot be readily distinguished from other gliomas by imaging. Posterior fossa tumors in infants that protrude below the foramen magnum are more likely to be ependymomas, as are posterior fossa tumors that cause a head tilt (due to compression of the XIth cranial nerve as it crosses the foramen magnum).
Metastatic dissemination of ependymomas occurs in the leptomeningeal space in a similar pattern to that seen in medulloblastomas, albeit at a much lower rate (<5% of patients at presentation). This low rate of observable dissemination at diagnosis has lead to the almost universal use of local, rather than craniospinal radiotherapy at diagnosis for patients with ependymoma.
Subependymomas are benign tumors with an admixture of fibrillary subependymal astrocytes. They are distinct from subependymal giant cell astrocytomas, which occur in the lateral ventricles in tuberous sclerosis. Subependymomas occur most often in the floor or walls of the fourth ventricle in older men. Most are asymptomatic and slow growing, and treatment is rarely needed except for hydrocephalus or demonstrated growth. They are often incidentally found at autopsy.
Surgery
Several retrospective studies support the relationship between postsurgical residual ependymoma and a poorer outcome, and therefore, maximal safe resection is the goal.309–311 These tumors may also extend through the foramen of Luschka, entangling the cranial nerves in the basal cisterns, which also precludes a complete resection. The less common supratentorial tumors are removed as with any glioma. Avoidance of bleeding into the ventricular system is important to prevent postoperative hydrocephalus.
Radiation Therapy
Postoperative irradiation improves the recurrence-free survival of patients with intracranial ependymomas, and 5-year survival rates with doses of 45 Gy or more range from 40% to 87%.152 Therapeutic utility of local radiation is established for ependymoma patients, even in infants.312 Because local failure usually dominates the recurrence patterns, low-grade supratentorial ependymomas are typically treated using partial brain fields with a dose of approximately 54 Gy. Low-grade infratentorial ependymomas are also treated using limited fields. The best survival results in retrospective series have been shown for patients who undergo gross total resection followed by radiotherapy.374,375 For most patients, a more usual volume consists of the tumor bed and any residual disease plus an anatomically defined margin of 1 to 1.5 cm to create a CTV. Larger margins may be required in areas of infiltration, and special attention must be paid to areas of spread along the cervical spine because 10% to 30% of fourth ventricular tumors extend down through the foramen magnum to the upper cervical spine.313,314 Patients with neuraxis spread (positive MRI or positive CSF cytology) should receive craniospinal irradiation (40 to 45 Gy) with boosts to the areas of gross disease and to the primary tumor to total doses of 50 to 54 Gy.
Chemotherapy
There is no evidence that any type of chemotherapy improves survival in children with ependymomas.315 Single-agent carboplatin, cisplatin, and etoposide, as well as multiagent chemotherapy, have been evaluated in small series and, to date, few if any drugs have shown even modest consistent activity in ependymomas.377–380
Although a complete removal of the ependymoma has a positive impact on the outcome, a complete resection is achieved in only 40% to 60% of cases.316,317 Therefore, responsiveness to preirradiation chemotherapy was investigated in a Children’s Oncology Group (COG) study where an objective response rate of 58% to preirradiation chemotherapy, consisting of cisplatin, etoposide, cyclophosphamide, and vincristine, in children with incompletely resected ependymoma was seen.383 The 3-year event-free survival in patients assigned to preirradiation chemotherapy because of incomplete resection was 58% and was comparable to those who had a complete resection and were assigned to irradiation alone. However, 15% of the children who received preirradiation chemotherapy experienced progression prior to RT. Therefore, a subsequent COG study was carried out that aimed to decrease the progression rate prior to radiotherapy by employing a strategy of second-look surgery following the preirradiation chemotherapy in children with residual disease. In this study, patients who had a complete resection of a differentiated supratentorial ependymoma were observed without any further therapy. The results of this study are pending. The recently opened randomized COG study is exploring whether maintenance chemotherapy following radiation will improve event-free survival and OS.
The primary application of chemotherapy, therefore, is investigational, and it is within the realm of neoadjuvant therapy to improve resectability, as primary adjuvant therapy in young children to delay radiotherapy and as possible salvage. In the Baby Pediatric Oncology Group study, a 48% response rate was reported to two cycles of vincristine and cyclophosphamide in 25 children younger than 3 years of age with ependymoma, allowing a delay in radiotherapy by 1 year without impacting the outcome.318 However, the use of chemotherapy to delay radiotherapy has to be approached cautiously. In a trial of 34 patients with anaplastic ependymoma, 25 patients relapsed relatively rapidly and only 3 patients who did not receive radiotherapy survived.319
Despite multimodal therapy, 50% of the patients with ependymoma will experience a relapse. The majority of the recurrences are local, and prognosis is poor after relapse.320 Resection, reirradiation, and chemotherapy are the common treatment modalities for relapsed ependymoma. Various antineoplastic agents such as etoposide, cyclophosphamide, temozolomide, cisplatin, and irinotecan have failed to improve survival in these patients.287,321,322 Novel therapies to target molecular pathways are currently under investigation.
Clinical and Pathologic Considerations
Meningiomas are believed to arise from epithelioid cells on the outer surface of arachnoid villi in the meninges, also known as arachnoidal cap cells. The most frequent locations are along the sagittal sinus and over the cerebral convexity (Fig. 97.10). Meningiomas are extra-axial, intracranial, and sometimes intradural—extramedullary spinal tumors that produce symptoms and signs through the compression of adjacent brain tissue and cranial nerves. They often also produce hyperostosis; bony invasion does not indicate malignancy. They rarely metastasize except after multiple resections when they may spread to the lung, where growth is typically slow.
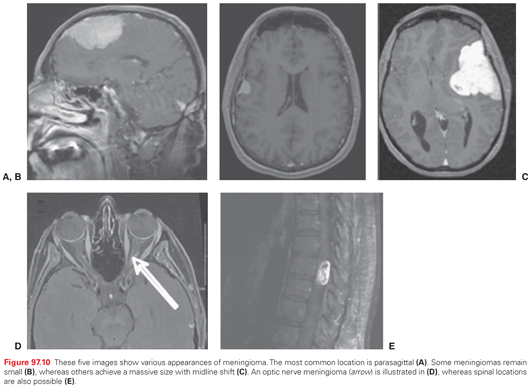
The WHO categorizes this tumor into three grades.4
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


