Increases in leucocyte numbers secondary to systemic disease are very common in neonates, particularly in association with infection. Careful examination of the blood film often provides additional clues to the underlying cause and is a rapid and sensitive way of identifying likely bacterial or viral infection (Table 3.1). The principal causes of neutrophilia are shown in Table 3.2. In the hospital setting the two most common causes of neonatal neutrophilia are maternal chorioamnionitis and acute bacterial infection. The haematological features of acute bacterial infection are usually marked and distinctive and are described in detail below. It is worth noting that infection with Bordetella pertussis during the neonatal period may be accompanied by marked neutrophilia as well as lymphocytosis.1 Neutrophilia is also seen after a number of specific types of viral infection, particularly chikungunya virus.2,3 Most studies suggest that the neutrophil count is reduced in neonatal coronavirus disease 19 (COVID‐19), although a recent study has shown that the presence of neutrophilia is a predictive factor for severe disease.4 By contrast, the haematological features of chronic bacterial infection in neonates are usually much more subtle. Typically, there is a moderate neutrophilia, often persisting for several weeks, and the majority of neutrophils are mature and lack toxic granulation. This pattern of chronic neutrophilia is seen in association with osteomyelitis or localised skin or wound infections. Neutrophilia also occurs in neonates with constitutional trisomies, including Down syndrome, trisomy 13 and trisomy 18; this suggests that aneuploidy itself might perturb neutrophil development during fetal life, but if so the mechanism of this is completely unknown.5,6 The clinical significance of neutrophilia in neonates with constitutional aneuploidies lies with recognising that neutrophilia is not a useful sign on its own, either of acute infection or of transient abnormal myelopoiesis (TAM) seen in neonates with Down syndrome (discussed in detail below). Neutrophilia secondary to constitutional trisomies is confined to the neonatal period, although follow‐up data for trisomy 13 and trisomy 18 are extremely limited because fewer than 20% of affected neonates survive for more than 12 months.7 Table 3.1 Haematological features of infection in neonatal blood films CRP, C‐reactive protein; HIE, hypoxic ischaemic encephalopathy; NEC, necrotising enterocolitis; WBC, white cell count. Although rare, certain inherited immunodeficiency syndromes classically present with persistent neutrophilia, which is often first evident in the neonatal period. These disorders are important to recognise promptly because of the high frequency of severe infection, which may be fatal.8,9 These include the leucocyte adhesion deficiency (LAD) syndromes caused by bi‐allelic mutations in the ITGB2, SLC35C1 and FERMT3 genes in LAD‐I, LAD‐II and LAD‐III, respectively;10,11 and interferon regulatory factor 8 (IRF8) deficiency caused by bi‐allelic mutations in IRF8.9,12 The co‐occurrence of persistent neutrophilia with delayed separation of the umbilical cord is highly suggestive of a LAD syndrome.11 Table 3.2 Causes of neutrophilia in neonates * Common causes of neonatal neutrophilia. TAM, transient abnormal myelopoiesis; WBC, white cell count; G‐CSF, granulocyte colony‐stimulating factor. Finally, treatment of neonates with corticosteroids or recombinant granulocyte colony‐stimulating factor (G‐CSF) causes neutrophilia that begins a few days after therapy is started and lasts for several days after discontinuation. Another benign cause of neutrophilia, with no neutrophil left shift and no other clinical or laboratory signs of infection, has been reported in neonates born at altitude (≥1550 m).13 The duration of the neutrophilia is not clear as all the neonates in this study were discharged before day 4 of life and the mechanism was not investigated. However, these data suggest that, in relevant countries, care should be taken not to rely on the neutrophil count alone as a marker of infection. Table 3.3 Causes of lymphocytosis in neonates COVID‐19, coronavirus disease 19; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2. The main causes of lymphocytosis during the neonatal period are shown in Table 3.3. Lymphocytosis is not very common in neonates and is usually associated with congenital or nosocomially acquired viral infection, particularly with cytomegalovirus (CMV), respiratory syncytial virus (RSV) or coxsackievirus.14–16 However, it is important to note that the majority of neonates with congenital CMV infection have no clinical findings at birth and that thrombocytopenia is the most common haematological feature of infection (see Chapter 4).15 Neonatal COVID‐19 is also associated with lymphocytosis, in contrast to adults, who tend to have lymphopenia.17 Neonatal infection with Bordetella pertussis, although uncommon, can be associated with very severe infection and a dramatic lymphocytosis.18 In addition, both transplacentally acquired and neonatally acquired malaria cause atypical lymphocytosis similar to that seen in neonatal CMV infection.19 Finally, lymphocytosis may be seen in association with certain inherited immunodeficiency syndromes, in particular Omenn syndrome20 and autoimmune lymphoproliferative syndrome (ALPS).21,22 Eosinophilia is common during the neonatal period, occurring in about 10% of all babies admitted to neonatal intensive care units23 and in up to 75% of the most preterm neonates (<27 weeks’ gestation).24,25 The principal causes are shown in Table 3.4 and a typical example in a preterm neonate is shown in Fig. 3.1. The most common causes of eosinophilia in neonates are bacterial infection and necrotising enterocolitis (NEC) with the eosinophilia developing 1 or 2 days after the onset of these complications.23,24 The eosinophilia is usually mild and transient, but early, persistent eosinophilia in NEC has been found to predict for late complications.28 Eosinophilia is also found secondary to allergic conditions in neonates, such as food protein‐induced enterocolitis, a condition presenting with bloody stools and eosinophilia in breastfed or formula‐fed neonates.29 Finally, eosinophilia is seen in up to 23% of cases of TAM where the eosinophils are part of mutant GATA1 clone30 and may contribute to the liver fibrosis seen in severe cases.31 Table 3.4 Causes of eosinophilia in neonates NEC, necrotising enterocolitis; RSV respiratory syncytial virus. Fig. 3.1 Blood film of a preterm neonate with eosinophilia showing two eosinophils and two neutrophils. May–Grünwald–Giemsa (MGG), ×100 objective. Monocytosis is fairly common in neonates, occurring most often in association with infection (Table 3.5). In general, monocytosis is a non‐specific early feature of acute bacterial infection, the number of monocytes rising with the neutrophil count but usually falling more quickly as the infection resolves. Monocytosis is particularly associated with congenital syphilis, protozoal infections and Candida infections.32–34 More recently, monocytosis together with neutropenia has been noted as a presenting finding in neonates with COVID‐1935 and in neonatal varicella infection.36 There is often a compensatory monocytosis in neonates with either inherited or acquired neutropenia, including neonatal alloimmune neutropenia,37 as discussed later. The other major group of conditions associated with monocytosis are neonatal preleukaemic and leukaemic conditions, including TAM in neonates with Down syndrome, juvenile myelomonocytic leukaemia (JMML) due to inherited rasopathies, and congenital leukaemia. Differentiation of these conditions from infection may be difficult, especially in JMML where the monocytic component of the disease is very prominent.38–42 Neonates with Wiskott–Aldrich syndrome may also present with monocytosis and diagnostic confusion with JMML is increasing recognised.43–45 Table 3.5 Causes of monocytosis in neonates WAS, Wiskott–Aldrich syndrome. Neonatal monocytopenia is rare but may be a useful clue to a diagnosis of certain inherited immunodeficiency disorders, particularly IRF8 deficiency, which is associated with severe and potentially fatal infection in the neonatal period.9 In contrast to infants and older children, neonates have increased numbers of circulating immature ‘blast cells’, most commonly resembling the myeloblasts seen in acute leukaemia (Table 3.6). The numbers of circulating blasts are generally inversely proportional to gestational age and tend to increase in the early stages of acute infection (Fig. 3.2). Most reported cases of increased blast cells are in preterm neonates, are transient and occur as part of a general hyperleucocytosis, often without an underlying cause being identified.46–48 Even in extremely preterm neonates with severe sepsis the percentage of blasts almost always remains below 8%.6 The term ‘neonatal leukaemoid reaction’ (NLR) has been used to describe a white blood cell count ≥50 × 109/l in a neonate. NLR was shown to be associated with sepsis, bronchopulmonary dysplasia, intraventricular haemorrhage (IVH) and a high mortality rate when gestation‐matched neonates with and without NLR were compared.49 Neonates with Down syndrome almost always have circulating blast cells, even in the absence of TAM (discussed later in this chapter).6 Blast cells are also a feature of neonatal JMML seen in some patients with inherited rasopathies.38,39 The main significance of the presence of circulating blast cells is to be able to distinguish the very rare neonates with congenital leukaemia from the vast majority with self‐limiting benign or preleukaemic or spontaneously remitting leukaemic disorders. Table 3.6 Causes of increased blast cells in neonatal blood films ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia. Fig. 3.2 Blood film of a preterm neonate born at 24 weeks’ gestation showing blast cells and promyelocytes. Note the large number of echinocytes typical of a neonate born at this gestation. MGG, ×40. The typical haematological changes associated with bacterial infection at any age are neutrophilia, an increased proportion of immature forms (usually referred to as neutrophil ‘left shift’ or increased band forms) and toxic granulation of the neutrophils. In neonates, one of the most important diagnostic considerations is the postnatal age of the baby at the time that the blood sample is collected as this is crucial to the correct interpretation of the findings. The detection of neutrophil left shift in neonatal blood films is straightforward when the vast majority of circulating neutrophils are non‐segmented ‘band’ forms (Fig. 3.3). However, in many cases the changes are less clear cut; many studies have described attempts to measure the extent of neutrophil left shift in a reproducible and reliable manner. The most sensitive approach is to calculate the ratio of immature band cells to total neutrophils, the (I/T) neutrophil ratio, on the basis of a manual differential count.50,51 In many hospitals this has been superseded by automated counts of immature neutrophils/granulocytes reported as the immature granulocyte percentage (IG%) although both manual and automated approaches appear to give broadly similar results and neither is 100% reliable.52 All of these approaches are greatly affected both by gestational age and by postnatal age and close liaison between the haematology laboratory and the clinical team is crucial to interpreting the significance of the results. Fig. 3.3 Blood film of a preterm neonate with infection by group B Streptococcus showing two band cells with toxic granulation. MGG, ×100. Neonatal infection remains a major cause of morbidity and mortality throughout the world, accounting for approximately 22% of global annual neonatal deaths.53 The haematological findings can be very useful in making a prompt diagnosis both in well‐resourced counties and in low and middle‐income countries.53–55 Indeed, in our experience, it is often possible to identify the most likely pathogen by integrating the clinical and haematological findings. Neonatal sepsis is often categorised as either early‐onset sepsis (EOS) or late‐onset sepsis (LOS), with EOS variably defined but generally indicating sepsis within the first 3–7 days after birth. This reflects the mode of infection (during labour and delivery for most cases of EOS and nosocomial for LOS). Historically, the most frequent cause of acute bacterial infection presenting at or within 24 hours of birth is group B Streptococcus. This remains a very important cause of EOS but has been overtaken in some countries by nosocomially acquired Gram‐negative infections.56 The blood film appearances of group B Streptococcus infection on day 1 of life are highly characteristic (see Fig. 3.3); the proportion of band cells to mature neutrophils is markedly increased, toxic granulation is minimal and the total neutrophil and leucocyte counts are frequently reduced (for normal ranges, see Table 1.2). By day 2–3 of life, there is typically a neutrophilia with toxic granulation and less marked neutrophil left shift as the neonate responds appropriately to the infection. The presence of increased band cells, in the absence of a more generalised left shift of myeloid cells including myelocytes, promyelocytes and blast cells, is not a normal feature in a neonate at any gestation and is a very sensitive indicator of acute bacterial infection. The only other causes of increased band cells typically seen in neonates are congenital viral infection, such as CMV or coxsackievirus infection, and inherited causes such as the autosomal dominant Pelger–Huët anomaly due to inherited mutations in LBR, which encodes the lamin B receptor.57,58 In heterozygous cases, the neutrophils have unsegmented, dumbbell‐shaped or bilobed nuclei (Fig. 3.4) and the eosinophils have round nuclei (Fig. 3.5), whereas in homozygotes the neutrophil nuclei are also round.59 Fig. 3.4 Blood film of a term neonate with Pelger–Huët anomaly showing three neutrophils with typical dumbbell‐shaped nuclei. MGG, ×100. After group B Streptococcus, the most common cause of acute bacterial infection at birth is a Gram‐negative organism, most often Haemophilus influenzae. In such cases, in our experience, the neutrophils often show cytoplasmic vacuolation as well as toxic granulation and atypical mononuclear cells (i.e. atypical lymphocytes) may also be seen (Fig. 3.6). The presence of atypical mononuclear cells, with or without monocytosis, is also a feature of congenital infection with Listeria monocytogenes (Fig. 3.7), Toxoplasma gondii (Fig. 3.8) and Mycoplasma pneumoniae (Fig. 3.9). Fig. 3.5 Blood film of a term neonate with Pelger–Huët anomaly showing a Pelger eosinophil with a rounded nucleus as well as a neutrophil with a typical dumbbell‐shaped nucleus. MGG, ×100. Fig. 3.6 Blood film of a preterm neonate showing typical features of a Gram‐negative bacterial infection: neutrophil toxic granulation, marked left shift and vacuolation. Note the thrombocytopenia and some red cell fragments consistent with concomitant disseminated intravascular coagulation (DIC). MGG, ×100. Fig. 3.7 Blood film of a neonate with congenital Listeria monocytogenes infection showing peripheral blood monocytosis. Note the erythroblastosis secondary to intrauterine growth restriction (IUGR) which is also a feature of neonatal listeriosis: (a) ×40; (b) ×100, MGG. Although with experience, examination of neonatal blood films is a rapid and useful way of helping to establish the presence of infection in a neonate, there are two important limitations. First, the assessment is fairly subjective and, in our experience, is most accurate when serial daily blood films are available for review as the change in the blood film appearance are often more important than the magnitude of those changes. Secondly, it is important to be aware of a number of pitfalls. In particular, two of the cardinal signs of acute bacterial infection, neutrophilia and toxic granulation, can also be seen in a number of other conditions that are unique to neonates, including meconium aspiration, hypoxic ischaemic encephalopathy (Fig. 3.10) and maternal chorioamnionitis (see Table 3.1). All of these complications cause transient haematological changes, which gradually resolve over the first few days of life. Fig. 3.8 Blood film of a neonate with congenital toxoplasmosis showing atypical mononuclear cells. Note the presence of target cells, which may reflect the liver involvement also present in this neonate. MGG, ×100. Fig. 3.9 Blood film of a preterm neonate born at 26 weeks’ gestation with congenital pneumonia caused by Mycoplasma pneumoniae infection showing neutrophils and atypical mononuclear cells. Note the large number of echinocytes typical of a neonate born at this gestation. MGG, ×40. There have been many attempts to develop more objective ways of identifying acute neonatal infection based on the haematological findings available from the full blood count and blood film review. While most of these rely on various combinations of the white cell count (WBC) and the proportion of immature neutrophils, one of the best developed is the Hematologic Scoring System, which includes neutrophil morphology.60 The developers, Rodwell and colleagues, assigned a score of 1 for each of seven findings: abnormal WBC, abnormal total neutrophil count, elevated immature polymorphonuclear (PMN) count, elevated immature to total PMN ratio, immature to mature PMN ratio greater than or equal to 0.3, platelet count less than or equal to 150 × 109/l and ‘degenerative’ changes in the neutrophils (defined as toxic granulation, vacuolation and/or Döhle bodies). Using a score of ≥3, they could identify almost 100% of neonates with definite or probable infection with a false positive rate of 14%, while with a score of ≤2 the likelihood that sepsis was absent was 99%. They subsequently reported that the scoring system could also be used to identify sepsis in neutropenic neonates when a score of ≥3 was combined with a neutrophil count of <0.5 × 109/l.61 This approach was found to be still valuable in a more recent study54 and, although it is highly reliant on the availability of skilled laboratory staff, expensive equipment is not required. It seems likely that the application of more sophisticated automated methods, including artificial intelligence (AI)/machine learning, will soon provide an alternative approach.55 Fig. 3.10 Blood film of a preterm neonate born at 27 weeks’ gestation and found to have hypoxic ischaemic encephalopathy showing toxic granulation of the neutrophils, a promyelocyte and nucleated red blood cells (NRBC). Note also the thrombocytopenia and occasional schistocyte consistent with DIC. MGG, ×60. Many babies, particularly those who are born before term, have marked neutrophilia and toxic granulation at birth with very few band forms. In the vast majority of cases, these changes are not a sign of acute bacterial infection in the baby but instead are due to maternal chorioamnionitis (Fig. 3.11), which is a very common trigger of preterm delivery. These changes likely reflect the passage of maternal cytokines, such as interleukin‐6 (IL‐6) and IL‐8, across the placenta during labour and delivery.62 In such cases, neonatal blood cultures are negative and C‐reactive protein levels are normal or mildly increased.63 The leucocyte count is usually high and may even exceed 100 × 109/l despite the absence of acute infection in the baby.49 These changes in the blood film are maximal in the first 24 hours after birth and gradually resolve over the first 3–4 days of life. It is important to note that identical findings appearing in a blood film after this time are almost always indicative of acute bacterial infection in the baby. Fig. 3.11 Impact of maternal chorioamnionitis on the blood film at birth showing that toxic granulation of the neutrophils is already present. MGG, ×100. Prominent vacuolation of the cytoplasm of neutrophils, monocytes and sometimes eosinophils can also be a useful diagnostic finding in neonatal blood films (see Table 3.1). As well as a fairly common finding in Gram‐negative infections, this can also be a feature of neonatal Staphylococcus epidermidis infections in association with indwelling vascular catheters (Fig. 3.12). In some cases the organism is visible within these vacuoles (Fig. 3.13); however, systematic evaluation of neonatal blood films or buffy coats suggests that this is not a very sensitive way of detecting bacteraemia and is seen only in very severe sepsis.64 Systemic fungal infection with Candida or Aspergillus species, which are major causes of morbidity and mortality in preterm neonates less than 28 weeks’ gestation at birth,65 also often causes neutrophil and monocyte vacuolation which typically produces a ‘motheaten’ appearance of the cells (Fig. 3.14). Many cases also have thrombocytopenia.65 Blood film evaluation can be a useful diagnostic tool in neonates at high risk of fungal infection as the leucocyte changes may be an early diagnostic sign. One of the most serious complications seen in neonates, particularly preterm neonates, is necrotising enterocolitis, an inflammatory intestinal disease of uncertain aetiology. In the early stages of the disease, the clinical signs are often non‐specific (abdominal distension and feeding intolerance) and the haematological features at this stage include neutropenia and often a fall in the platelet count, initially not to thrombocytopenic levels.66 This is rapidly followed by neutrophil and monocyte vacuolation and thrombocytopenia (Fig. 3.15). In our experience, neutrophil and monocyte vacuolation are almost always present in neonates with NEC and are a very useful early indicator of the diagnosis. Machine learning approaches based on the full blood count are now being developed in order to generate predictive models for diagnosis of the cases of NEC that are best treated by early surgery.66 Fig. 3.12 Blood film of a 3‐week‐old preterm neonate born at 29 weeks’ gestation who developed infection of an indwelling intravascular catheter with Staphylococcus epidermidis. The film shows three vacuolated neutrophils as well as a giant monocyte. MGG, ×100. Fig. 3.13 Blood film of a preterm neonate with Staphylococcus epidermidis infection showing the presence of the organisms within two monocytes. Note also the cytoplasmic vacuolation. MGG, ×100. Neonates with congenital viral infection, most commonly CMV but also enteroviruses and rubella, usually have circulating atypical lymphocytes that can be readily seen in blood films for the first few weeks of life, providing a useful clue to the underlying diagnosis (Figs 3.16 and 3.17) (see Cases 4.1 and 4.2, pages 227 and 230). Similar cells are also seen in neonates who develop nosocomial viral infection, for example due to CMV or RSV. Virus‐associated lymphocytosis in neonates is usually transient and mild. As the viraemia resolves, the numbers of circulating atypical lymphocytes fall. In occasional cases of congenital CMV infection, the viraemia and thrombocytopenia persist for several months and in such cases the ongoing presence of atypical lymphocytes in the blood film is a useful marker of active infection. Adults with COVID‐19 have also been reported to have atypical lymphocytes67 but their presence in neonates has not yet been reported. Fig. 3.14 Blood film of a neonate with Candida infection showing neutrophil and monocyte vacuolation. Note the large number of target cells, which likely reflect systemic infection involving the liver. MGG, ×100. Fig. 3.15 Blood film of a preterm neonate with necrotising enterocolitis showing vacuolation of the neutrophils and monocytes, which usually appears as one or more rounded punched out ‘holes’ in the cytoplasm of these cells: MGG, (a) ×40, (b) ×100. Fig. 3.16 Blood film of a neonate with congenital cytomegalovirus (CMV) infection showing the presence of typical activated lymphocytes. MGG, ×100. Fig. 3.17 Blood film of a preterm neonate with congenital infection with coxsackievirus showing a large lymphocyte with deeply basophilic cytoplasm and a second cell that may be a binucleated reactive lymphocyte. MGG, ×100. A number of storage disorders cause morphological abnormalities of the white blood cells (reviewed in reference 68). In particular, vacuolation of lymphocytes is seen in two of the most severe of these disorders, Wolman disease (lysosomal acid lipase deficiency) and GM1‐gangliosidosis (β‐galactosidase deficiency). Wolman disease is an extremely rare lysosomal storage disease caused by bi‐allelic mutations in the LIPA gene.69 Deficiency of the lysosomal acid lipase enzyme leads to accumulation of intracellular lipids in the liver, spleen, lymph nodes, intestine and bone marrow, the lipid being visible as vacuoles within the circulating lymphocytes68,70 (Fig. 3.18a). Wolman disease typically presents in the first few months of life with hepatosplenomegaly, jaundice, failure to thrive and malabsorption.71 Although the diagnosis is established using enzymatic analysis and/or deoxyribonucleic acid (DNA) sequencing, evaluation of a neonatal blood film for the typical appearances of lymphocyte vacuolation is a useful screening test in neonates with unexplained hepatosplenomegaly. Prompt diagnosis is important as the disease is fatal by 12 months of age (median age at death 3.7 months)71,72 and effective enzyme replacement therapy with sebelipase alfa is available.70 Type I (infantile) GM1 gangliosidosis is a rare, very severe sphingolipidosis caused by mutations in the GLB1 gene that cause decreased activity of β‐galactosidase and storage of GM1 ganglioside. The disease may present as hydrops fetalis or with severe central nervous system (CNS) dysfunction, developmental delay and hepatosplenomegaly.73 Unfortunately, the disease is uniformly fatal in the first few years of life but the presence of lymphocyte vacuolation on a fetal or neonatal blood film (Fig. 3.18b) may be a useful screening test to guide the specific biochemical and molecular tests for confirmation of the diagnosis.74 Fig. 3.18 Blood films in storage disorders: (a) lymphocyte vacuolation in Wolman disease; (b) lymphocyte vacuolation in β‐galactosidase deficiency; (c) Alder–Reilly bodies in a neutrophil in Sly disease. MGG, ×100. Leucocyte abnormalities can be seen in the blood film of neonates or infants with a number of other inherited storage disorders (Table 3.7). In several of the mucopolysaccharidoses, metachromatic staining of granules known as the Alder–Reilly anomaly is apparent in the neutrophils (Fig. 3.18c). In other storage disorders, such as perinatally lethal Gaucher disease, due to mutations in the GBA gene, the disease manifests with thrombocytopenia or as a ‘blueberry muffin’ skin rash rather than with leucocyte abnormalities.75 Neutropenia is classically defined as a blood neutrophil concentration more than 2 standard deviations below the reference range. However, as discussed in Chapter 1, the neutrophil count varies widely with postnatal age, and different considerations therefore need to be made in interpreting the significance of the neutrophil count in neonates. Although clinicians often worry when a routine blood count reveals a neutrophil count below the normal range (see Table 1.2) because of anxiety about missing cases of severe congenital neutropenia (SCN), in practice, most neutropenia in neonates is mild, transient and of no clinical significance. A pragmatic approach is to consider a neutrophil count at birth of less than 2 × 109/l as abnormal and worth monitoring, and a neutrophil count during the first month of life of <0.7 × 109/l as significant enough to merit further investigation. Table 3.7 Cytological abnormalities in metabolic and storage disorders and other constitutional abnormalities that may present in the neonatal period Causes of neonatal neutropenia are summarised in Table 3.8. The commonest cause of neutropenia at birth in preterm neonates is transiently reduced neutrophil production secondary to intrauterine growth restriction (IUGR) or maternal hypertension.76,77 Most affected neonates also have thrombocytopenia and increased erythropoiesis, manifest as polycythaemia and/or increased circulating nucleated red blood cells (NRBC) (Fig. 3.19). Similar haematological abnormalities (transient neutropenia and thrombocytopenia together with polycythaemia) are seen in the infants born to mothers with diabetes.76 It is important to note that the neutrophils in neonates with IUGR or those with neutropenia secondary to maternal hypertension or diabetes are not left shifted, helping to differentiate this form of neutropenia from sepsis‐associated neutropenia. A retrospective study in 200 neonates with IUGR noted that the NRBC count correlated inversely with the neutrophil count during the first days after birth and also that the most severely weight‐restricted neutropenic neonates had a trend toward higher NRBC counts.77 This suggests that the underlying driver for these haematological abnormalities may be chronic fetal hypoxia, although the precise mechanisms have not been identified. This form of neutropenia resolves spontaneously, usually within a few days of birth and does not persist beyond the first 2 weeks of life.76,77 * Causes of inherited severe congenital neutropenia (SCN) are shown in Table 3.9. Fig. 3.19 Blood film of a preterm neonate with IUGR showing increased numbers of normoblasts and a basophilic erythroblast. MGG, ×60. The commonest cause of neutropenia in term infants is bacterial or viral infection. Neutropenia in the first few hours of life is typically associated with group B Streptococcus infection and can be severe (<0.5 × 109/l) but, in contrast to inherited SCN, the neutropenia is transient and replaced by increasing neutrophilia within 1–2 days. Neutropenia is also an important early sign of NEC and of LOS (i.e. sepsis developing after the first 7 days of life). As infection‐ and NEC‐associated neutropenia is self‐limiting, persistent neutropenia in a term or preterm baby should always be investigated. This includes looking carefully for evidence of other acquired causes, such as congenital or nosocomial viral infection and immune neutropenia (see later). Other important causes of neutropenia are SCN due to a selective failure of neutrophil production (e.g. Kostmann syndrome) (see Table 3.9), marrow replacement due to congenital leukaemia or congenital myelofibrosis, and a number of rare metabolic disorders that are listed in Table 3.8. Finally, severe neutropenia may also be one of the manifestations of rare bone marrow failure syndromes, such as Pearson syndrome and Fanconi anaemia, although in most cases anaemia or thrombocytopenia is more prominent (see Chapter 2). Note that neonates with the Pelger–Huët anomaly are not neutropenic unless they have concomitant infection, although typically automated neutrophil counts are unreliable in these cases (Case 3.1, see page 169). The majority of cases of immune neutropenia in the neonatal period are caused by alloantibodies in the mother directed against neutrophil antigens lacking in the fetus/newborn. Occasional cases arise where the mother herself has autoimmune neutropenia and transplacental maternal autoantibodies cause neonatal neutropenia. True autoimmune neutropenia almost always develops after the age of 6 months but occasional cases presenting in the newborn have been reported.78 This condition is the neutrophil equivalent of haemolytic disease of the fetus and newborn (HDFN) and fetal/neonatal alloimmune thrombocytopenia (FNAIT). In contrast to HDFN and FNAIT, it is not clear whether neutropenia causes any clinically significant disease prior to birth. Alloimmune neutropenia occurs when fetal neutrophils express paternally derived antigens of the human neutrophil antigen (HNA) system that are absent on maternal neutrophils and against which the mother produces immunoglobulin G (IgG) neutrophil alloantibodies. Table 3.9 Inherited causes of severe congenital neutropenia that present in neonates (see also Online Mendelian Inheritance in Man, https://www.omim.org/) * Neutropenia is often intermittent and may be missed in the neonatal period. ** Some cases have isolated severe congenital neutropenia without syndromic features. AD autosomal dominant; AR, autosomal recessive; XLR, X‐linked recessive. Five HNA systems have been recognised as targets for maternal alloantibodies: HNA‐1, HNA‐2, HNA‐3, HNA‐4 and HNA‐5 (reviewed in reference 79). The most common causative antibodies are directed against HNA‐1a and HNA‐1b, followed by HNA‐2. Antibodies directed against HNA‐1c, HNA‐1d, HNA‐3a, HNA‐3b, HNA‐4a, HNA‐4b and HNA‐5a are rare.79 No cases of neonatal alloimmune neutropenia due to anti‐HNA‐5b have yet been reported. Recent data suggest that although anti‐HNA antibodies can be detected in approximately 1% of mothers, only 1 in 10 women with antibodies will deliver babies with documented neonatal alloimmune neutropenia (0.1% of all live births).80 Other studies have reported an even lower frequency of neonatal alloimmune neutropenia, 0.01%81 or even 0.001% (reviewed in reference 80), perhaps because most milder cases are missed unless screening is performed as part of a research study. Like FNAIT, neonatal alloimmune neutropenia can occur in the first pregnancy.82 The full range of clinical presentations of neonatal alloimmune neutropenia is not known as most asymptomatic cases will be missed, given that routine full blood counts are not performed unless neonates are admitted to hospital. Severe cases present in the first few days of life with fever and infections of the respiratory tract, urinary tract and skin, including omphalitis. Retrospective series have identified many cases of neonatal alloimmune neutropenia where there is no evidence of infection and the diagnosis has been made because of an incidental finding of severe neutropenia where a full blood count has been performed for another reason or because of a previously affected sibling.81,82 The majority of cases of neonatal alloimmune neutropenia are diagnosed in term babies but occasional cases in preterm neonates have been reported.83 The main diagnostic clue to neonatal alloimmune neutropenia is a history of unexplained neutropenia that persists for several weeks or months with or without concomitant bacterial infection. The neutropenia is self‐limiting and usually resolves in 1–2 months although occasional cases where the neutropenia has persisted for more than 6 months have been reported.81,84 The reason for prolonged neutropenia and the mechanism by which this occurs is very unclear. The haemoglobin and platelet count are normal and there are no specific features in the blood film. The diagnosis is made by demonstrating antineutrophil antibodies in the mother and baby, which react against paternal, but not maternal, neutrophil antigens. Investigation typically includes maternal HNA antibody screening, HNA genotyping of the baby and both parents and crossmatching of maternal serum against paternal neutrophils if possible.80 Serological investigation of neonatal alloimmune neutropenia requires particular expertise and is usually performed in specialist laboratories that are often national reference centres. The mainstay of treatment of neonatal alloimmune thrombocytopenia is antibiotics.84 The morbidity and mortality of neonatal alloimmune neutropenia is low but G‐CSF may be useful to boost the neutrophil count in cases with resistant infection and/or prolonged neutropenia,85 although the response is variable86,87 and there are no controlled trials to confirm the benefit of G‐CSF in this setting. This rare condition is caused by the transfer of maternal IgG antibodies across the placenta from mothers with autoimmune neutropenia.88 The clinical manifestations are very similar to those of neonatal alloimmune neutropenia as the neutropenia may be severe. It is therefore important to consider this diagnosis by checking the blood count and blood film in all infants born to mothers with a history of autoimmune neutropenia. The neutropenia is self‐limiting and management is as for neonatal alloimmune neutropenia. Severe congenital neutropenias are a group of inherited diseases characterised by selective failure of neutrophil differentiation. Although the neutrophil count is extremely low (by definition <0.5 × 109/l) and neutrophils are often almost completely absent in the peripheral blood (Fig. 3.20), the bone marrow typically shows maturation arrest of granulopoiesis at the promyelocyte stage of differentiation. Although the disorders are all very rare (estimated incidence 3–8.5 cases per million individuals),89 it is important to identify SCN in the neonatal period because these babies are at high risk of life‐threatening infection from birth. Indeed, the majority of affected infants present with infections in the first few days or weeks of life.89 Typical presentations of neutropenia‐associated infections in the neonatal period include omphalitis, pneumonia, septicaemia, cellulitis and, occasionally, deep tissue abscesses. Infections are often more severe and/or prolonged than in neonates who are able to mount a normal neutrophil response. In most cases, the neutrophil count at diagnosis of SCN is <0.2 × 109/l (see Fig. 3.20), often with a compensatory monocytosis (Fig. 3.21). However, investigations are indicated in all neonates where the neutropenia is severe (<0.5 × 109/l), particularly when this persists beyond the first 2 weeks of life despite resolution of infection and/or if there is a relevant family history.89,90 Fig. 3.20 Blood film of a 3‐week‐old term neonate with severe congenital neutropenia (SCN) showing two monocytes and two lymphocytes (note the absence of neutrophils on this low power view). MGG, ×20. Fig. 3.21 Blood film of a neonate with SCN during an acute infection showing a monocyte and promonocyte, as well as a lymphocyte, but no neutrophils. MGG, ×100. The main causes of SCN that present in the neonatal period are shown in Table 3.9. While most cases present with isolated neutropenia, SCN is also seen in a number of immunodeficiency and multisystem disorders where the presence of the other features may provide clues to the underlying diagnosis. Increasingly, next generation sequencing is used to provide a rapid diagnosis based on gene panels containing known SCN‐associated genes. The most common genetic cause of SCN is mutation of ELANE (previously known as ELA2), the gene that encodes neutrophil elastase, which explains 45–60% of all cases of SCN. The next most frequent causative genes are: the SBDS gene, which causes Shwachman–Diamond syndrome; the G6PT gene, which encodes glucose‐6‐phosphate translocase and causes glycogen storage disease type 1b; and HAX1, which is now known to explain the SCN in the original Kostmann family.89 The number of genes known to cause SCN is steadily increasing with increasing use of whole genome and whole exome sequencing; those that have been reported to present in the neonatal period are shown in Table 3.9 and include mutations in GFI1 (growth factor‐independent protein 1), JAGN1, CSF3R, TCIRG1, SRP54, SRP68, DNAJC21 and several genes affecting the endosomal‐lysosomal system, such as VPS45.89–93 To date the causative genes in SCN have been shown to be important for normal neutrophil differentiation, survival and function, although their role in neutrophil development was sometimes unsuspected prior to the identification of the underlying defects in cases presenting as SCN. In about one‐third of cases no causative gene is identified.90 In that situation, bone marrow examination may be useful in helping to pinpoint the pathophysiology of the disease, for example by demonstrating differentiation arrest, and whole exome sequencing may reveal new candidate causes of SCN, which must always be validated by family and functional studies. Establishing a molecular diagnosis is extremely important, not only for identification of current or future family members, but also because patients with SCN have an increased risk of developing leukaemia of up to 60% in certain subtypes.94,95 The long‐term outlook for patients with SCN has been transformed by the introduction of G‐CSF as the mainstay of treatment. Overall survival is now >80%, although around 10% of patients still die from sepsis or severe bacterial infections.95,96 Up to 60% of cases of SCN are due to heterozygous mutations in ELANE.97 To date, more than 200 different ELANE mutations, either inherited or de novo, have been described.98 The autosomal dominant pattern of inheritance in ELANE‐mutated SCN means that the majority of cases will have a family history. A second clue to the presence of an ELANE mutation as the underlying cause is the absence of congenital anomalies or other syndromic features, which are common in other types of SCN. The treatment of SCN in the neonatal period is a combination of antibiotics for infection and G‐CSF to maintain the absolute neutrophil count above 1 × 109/l, usually starting with a dose of 5 μg/kg/day subcutaneously and titrating according to the neutrophil count.84,89,90 Shwachman–Diamond syndrome is an autosomal recessive disorder caused by bi‐allelic mutations in the SBDS gene.89,90 The syndrome is characterised by the triad of exocrine pancreatic insufficiency, skeletal dysplasia and neutropenia. Although the Shwachman–Diamond syndrome typically presents later in infancy with failure to thrive, severe disease has been reported in term and preterm neonates.99,100 For those cases that do present in the neonatal period, the diagnosis is suggested by the presence of neutropenia in a neonate with IUGR with or without skeletal abnormalities.101 Importantly, Shwachman–Diamond syndrome may present in the neonatal period with severe thoracic dystrophy. Indeed, there are several reports of misdiagnosis of the syndrome as the skeletal disease Jeune syndrome (asphyxiating thoracic dystrophy [ATD]), where the delay in identifying the correct diagnosis as Shwachman–Diamond syndrome, and the associated bone marrow and gastrointestinal failure, may have contributed to the risk of early neonatal death from this condition.101,102 Although neutropenia is almost always present, many cases also have anaemia and/or thrombocytopenia, including in the neonatal period.99,101 The diagnosis is based on the clinical history together with the blood count, evidence of exocrine pancreatic insufficiency, the presence of the distinctive skeletal abnormalities (metaphyseal dysplasia, thoracic dystrophy, short and splayed ribs) and molecular analysis using a targeted next generation sequencing panel or whole exome sequencing to identify the causative mutation in the SBDS gene. Most patients respond well to treatment with pancreatic enzyme replacement, G‐CSF to prevent recurrent infections and antibiotics as necessary. Unfortunately, these patients appear to have a particularly high risk of developing leukaemia later in life.103 It is also worth noting that other very rare bone marrow failure syndromes, such as bi‐allelic mutations in the DNAJC21 or SRP54 genes, may present with a Shwachman–Diamond‐like syndrome.91,92 It is therefore important to perform DNA analysis to establish the correct molecular diagnosis and plan appropriate management. Glycogen storage disease 1b is caused by bi‐allelic mutations in the SLC37A4 gene and deficiency of the glucose‐6‐phosphate transporter (G6PT1).89 Although the clinical presentation may be delayed until later in infancy, the disease has been reported to present in the neonatal period with hepatomegaly, hypoglycaemia, seizures and persistent neutropenia, most likely due to apoptosis of developing and mature neutrophils.104 The diagnosis is based on the clinical history together with the blood count and molecular analysis by targeted next generation sequencing or whole exome sequencing. As with other causes of SCN, treatment with G‐CSF corrects the neutropenia but these patients also have an increased risk of developing leukaemia later in life.105 Although first described more than 60 years ago, the cause of Kostmann syndrome was only identified in 2007 as being due to homozygous mutations of the HAX1 gene.106 In the original series of patients, all from the same large pedigree, the patients presented at birth or shortly thereafter with omphalitis, skin infections, abscesses or sepsis and all of the children died during the first year of life.107 It is now clear that mutations in exon 2 of HAX1 solely affect expression of isoform A of the HAX1 protein and manifest as SCN, while mutations in other exons of HAX1 affect expression of both isoform A and isoform B, resulting in both neurological problems (cognitive dysfunction, developmental delay, seizures) and SCN.108,109 In both types of disease HAX1 deficiency causes ineffective neutrophil production and maturation arrest at the promyelocyte stage. The diagnosis is based on the clinical history together with the blood count and molecular analysis using a targeted next generation sequencing panel or whole exome sequencing. As for other forms of SCN, treatment with G‐CSF is effective in boosting the neutrophil count and preventing infection but these patients are also at risk of developing leukaemia later in life.110 Several retrospective studies have reported an increased frequency of haematological abnormalities in neonates with Down syndrome,111,112 including a markedly increased risk of leukaemia in early childhood and of a neonatal condition known as transient abnormal myelopoiesis (TAM), which may be best regarded as a transient leukaemia rather than as a preleukaemic condition (reviewed in reference 113). The high frequency of other haematological abnormalities in neonates with Down syndrome has only been recognised fairly recently after a prospective study of blood counts and blood films in 200 unselected neonates with Down syndrome documented the haematological findings in detail (see reference 6). This study showed that all neonates with Down syndrome had haematological abnormalities (Table 3.10). Importantly, these abnormalities were seen in the absence of TAM (discussed later). In particular, neonates with Down syndrome had higher haemoglobin concentrations compared with other neonates of the same gestation, increased circulating erythroblasts and abnormal red cell morphology, including macrocytosis, target cells and basophilic stippling (Fig. 3.22). In addition, in the absence of TAM, neonates with Down syndrome had lower median platelet counts than neonates without Down syndrome and 51% had thrombocytopenia (platelets <150 × 109/l). Neonates with Down syndrome had higher numbers of neutrophils and monocytes than neonates without Down syndrome, while the total lymphocyte count was reduced, consistent with previous studies in older children with Down syndrome.114–116 Importantly, circulating blast cells were seen on the blood film in virtually all neonates with Down syndrome (98%).6 In our experience, blast cells are usually <10% of peripheral blood leucocytes in these neonates but they may be as high as 15%, even when very sensitive methods are used to exclude the mutations in the GATA1 gene that define TAM (reviewed in reference 117). The presence of blast cells in the blood film in such a high proportion of neonates with Down syndrome means that making a definitive diagnosis of TAM may be very difficult and mutational analysis of the GATA1 gene is often required to confirm the diagnosis (see later). Leukaemia is rare in neonates, with the exception of neonates with Down syndrome. While congenital leukaemia affects approximately three non‐Down syndrome neonates per million live births, for those with Down syndrome approximately 200 neonates per million live births will have the congenital syndrome known as TAM.118,119 Transient abnormal myelopoiesis, also known as transient myeloproliferative disorder or transient leukaemia, affects around 10% of neonates with Down syndrome while a further 15–20% have a clinically silent form of the disease (silent TAM). As the estimated incidence of Down syndrome worldwide is between 1 in 500 and 1 in 1100 live births, TAM is the commonest leukaemia in neonates. The rarity of neonatal leukaemia suggests that, with the exception of neonates with Down syndrome, fetal haemopoietic stem and progenitor cells are not inherently susceptible to leukaemic transformation. Finally, children with congenital rasopathies, in particular Noonan syndrome, may develop a neonatal leukaemia resembling JMML which, like TAM, is usually transient and resolves without treatment.120 Table 3.10 Haematological abnormalities in neonates with Down syndrome BM, bone marrow; DS, Down syndrome; Hb, haemoglobin concentration; Hct, haematocrit; TAM, transient abnormal myelopoiesis. Fig. 3.22 Blood film of a neonate with Down syndrome showing increased erythroblasts, target cells and macrocytes as well as some spherocytes and echinocytes. A pseudo‐Pelger neutrophil and giant platelet can also be seen. All of these abnormalities are frequent in neonates with Down syndrome even in the absence of a diagnosis of transient abnormal myelopoiesis (TAM) due to acquired GATA1 mutations (see Fig. 3.23). MGG, ×60. Transient abnormal myelopoiesis is a clonal disorder almost unique to Down syndrome, which presents in fetal life, at birth or within the first few weeks of life. The characteristic features are the presence of circulating blast cells, most often resembling megakaryoblasts, as well as dysplastic changes in other peripheral blood cells.6,121 TAM is now known to be caused by mutations in the haemopoietic transcription factor gene GATA1, and is only seen in conjunction with trisomy 21, including cases of mosaicism where some or all of the haemopoietic cells are trisomic for chromosome 21. Somatic mutations in exon 2 or 3 of GATA1 cause production of shorter GATA1 protein (GATA1s) that is leukaemogenic solely in fetal or neonatal blood cells that harbour trisomy 21 and only causes TAM in Down syndrome newborns, including mosaic Down syndrome.122–125 GATA1s protein disrupts fetal blood cell development, causing blast cell accumulation in blood, liver and other tissues and associated fibrosis in severe cases. Importantly, in a proportion of neonates with Down syndrome who have GATA1 mutations at birth, the GATA1‐mutant cells persist and acquire additional mutations, leading to a specific form of acute myeloid leukaemia known as myeloid leukaemia of Down syndrome (ML‐DS) within the first 4 years of life.122,125 In contrast to leukaemias in children without Down syndrome, acute lymphoblastic leukaemia (ALL) has not yet been reported in neonates with Down syndrome.118,119 The clinical findings in TAM vary from life‐threatening disease to an asymptomatic incidental finding. Most cases present within 7 days of birth. All symptomatic cases have increased blast cells in the peripheral blood.6 Severe disease manifests with one or more of: hepatomegaly (40%), splenomegaly (30%), vesiculopapular skin rash, which may give the appearance of a ‘blueberry muffin’ baby (11%), and pleural and/or pericardial effusion (9%).126 Cholestatic jaundice is common (70%) but is not specific for TAM. Mortality in severe cases approaches 20%, usually from progressive liver failure and coagulopathy. Asymptomatic disease may be clinically silent (increased blasts only) or clinically and haematologically silent (silent TAM; blasts not increased but GATA1 mutation present).6 Although the majority of cases of TAM present at or just after birth, later presentations of TAM sometimes occur when the neonate had no relevant clinical features at birth and a blood count and blood film were not performed at that time. However, a small proportion of cases present several weeks after birth with severe conjugated hyperbilirubinaemia, hepatomegaly, liver failure and coagulopathy. In this situation, there may be very few blast cells in the peripheral blood. The reasons for this are not clear but, at least in some cases, it is associated with diffuse blast cell infiltration of the liver and progressive fibrosis.125–129 Finally, around 4% of neonates with TAM present with hydrops fetalis, intrauterine death or death during labour.130,131 Leucocytosis is common in neonates with TAM and may be extreme (WBC >100 × 109/l). In contrast to congenital leukaemia in neonates without Down syndrome, anaemia is uncommon. The platelet count in TAM is extremely variable and may be normal, reduced or increased.6,125,127 It is now clear that thrombocytopenia is not useful in the diagnosis of TAM as there is no significant difference in platelet counts between Down syndrome neonates with and without TAM.6 In some cases of TAM not only is the platelet count increased but the thrombocytosis is extreme.132 The most important haematological finding supporting a diagnosis of TAM is the presence of increased blasts (>10%) in the peripheral blood of a neonate with Down syndrome (Case 3.2, see page 171). It is important to point out that blast cells usually disappear quickly from the peripheral blood over the first 2 weeks of life and therefore blood films should be requested as soon as possible after birth since their detection will not be reliable during and beyond the second week of life. Blast morphology varies from undifferentiated myeloblasts to typical small megakaryoblasts with cytoplasmic blebbing or large, partially differentiated megakaryoblasts (Fig. 3.23). TAM blasts usually express CD117 and variable proportions express CD34, CD7, CD36 and/or CD41/42b.133 Some may express CD235a (glycophorin A).133,134 Fig. 3.23 Blood films of neonates with Down syndrome with TAM: (a) severe TAM with large numbers of pleomorphic circulating blast cells; (b) megakaryoblast, giant platelet and megakaryocyte cytoplasmic fragment; (c) a megakaryoblast, a megakaryocyte and a giant platelet; (d) typical megakaryoblast with cytoplasmic blebbing, two erythroblasts and giant platelets. MGG, (a) ×40, (b–d) ×100. In contrast to congenital leukaemia in neonates without Down syndrome, the blast cells are often very pleomorphic in TAM (Fig. 3.23a,d). In addition, megakaryocytes and giant platelets are often present (Fig. 3.23a–d). This finding, together with the presence of normal or increased platelet counts, in many cases, suggests that the blast cells are able to differentiate into megakaryocytes and platelets, at least to some extent. There are also sporadic reports suggesting that in other cases of TAM there is differentiation into erythroblasts,134 eosinophils30,135 or basophils136 (Fig. 3.24). The current standard for a specific diagnosis of TAM is detection of an exon 2 or 3 GATA1 mutation by DNA sequencing of peripheral blood leucocytes.126 More than 95% of mutations are in exon 2 of the gene.137 As small mutant GATA1 clones may transform to ML‐DS, sensitive next generation sequencing is necessary to avoid false negative results. To avoid false negatives, GATA1 mutation analysis should be performed in the first week after birth while there are circulating peripheral blood blast cells.126 Even using next generation sequencing, large or complex deletions in exon 2 or 3 of the GATA1 gene may be missed or difficult to assess.138 For this reason GATA1 analysis should always be performed in conjunction with a full blood count and morphological review of a peripheral blood film in order to determine the blast cell percentage.126 As the presence of GATA1 mutations in neonatal blood cells in Down syndrome is specific for TAM, blast cell immunophenotyping and bone marrow examination are unnecessary in the neonatal period although both are useful for diagnosis of ML‐DS. An alternative approach to identifying neonates with TAM is to use a flow cytometric method to identify the short GATA1 protein (GATA1s) that is produced by the leukaemic blast cells.139 This is reported to be just as sensitive as next generation sequencing methods although the specific mutation is not identified and the value of this approach has not yet been tested in large series. Fig. 3.24 Blood film of a term neonate with Down syndrome and TAM showing six blast cells including one with basophilic granules and prominent vacuoles. MGG, ×100. Transient abnormal myelopoiesis is known to develop in fetal life although the majority of cases only present at or just after birth.130 It does not present after the age of 3 months. More than 90% of cases of TAM resolve spontaneously within 2–3 months. For the small proportion of neonates with severe disease (5–20%), progressive multiorgan failure carries a high risk of early death due to widespread infiltration of the tissues by GATA1‐mutant blast cells. Risk factors for early death in severe TAM include: preterm delivery, ascites, leucocyte count >100 × 109/l, hepatomegaly and coagulopathy.126,127,133 For such cases, the mainstay of treatment is supportive care and the judicious use of cytarabine as recommended in recent guidelines.126 Importantly, because 20% of cases of clinical TAM later develop ML‐DS, it is important to keep cases of TAM under regular clinical and haematological review until the age of 4 years, the specific time window these children are at risk of this disease.118,121,125,127 At present, no clinical, haematological or molecular features at birth reliably predict which cases of TAM will develop ML‐DS, so any change in the blood count during follow‐up, especially the development of thrombocytopenia, should prompt careful review of the blood film. Although TAM is known to develop during fetal life, the majority of cases do not present until after birth and there have been relatively few detailed case reports describing the findings of fetal disease. A recent systematic review identified 38 cases of which just over half survived beyond the neonatal period: 12 (32%) resulted in stillbirth and 4 (11%) in neonatal death.130 Diagnosis in utero was usually prompted by abnormal ultrasound findings, particularly hepatomegaly, with or without splenomegaly (n = 31; 79.5%), hydrops fetalis (n = 12; 30.8%) and pericardial effusion (n = 9; 23.1%); a smaller number of cases had cardiac abnormalities (n = 5; 12.8%), fetal ascites (n = 4; 10.3%), pleural effusion (n = 3; 7.69%) or peripheral oedema (n = 1; 2.56%). Where fetal blood sampling was performed, this showed leucocytosis and large numbers of circulating blast cells. There is currently insufficient evidence to determine the most effective way of managing TAM in utero. The majority of cases have been managed using supportive therapy (for example red cell transfusion, if indicated for anaemia) or early delivery and no cases have been treated in utero with cytarabine. In the index case described by Tamblyn and colleagues130 the clinical and haematological features of TAM were already largely resolved before birth, despite the absence of treatment, suggesting that even severe fetal TAM should be very carefully monitored rather than treated with chemotherapy before birth. In neonates without Down syndrome, acute leukaemia may present with a range of immunophenotypes, including acute myeloid leukaemia (AML), ALL, mixed phenotype acute leukaemia (MPAL) or, rarely, blastic plasmacytoid dendritic cell neoplasm. The majority of cases of neonatal leukaemia are AML rather than ALL (Fig. 3.25a). However, there appears to be no family history of leukaemia or childhood cancer in most cases. Neonatal AML differs from AML later in childhood in several ways. First, in a recent review of published cases, we found a striking predominance of monocytic, megakaryocytic and erythroid leukaemias in the neonatal setting compared with any other time of life119 (Fig. 3.25b). Secondly, there have been many well‐documented examples of spontaneous remission of neonatal AML, although in some cases this is transient (discussed in more detail later). Finally, the spectrum of genetic abnormalities associated with neonatal AML differs from that in older children. Of the 110 cases we identified in our recent review, just over half had one of three molecular genetic abnormalities: rearrangement of the KMT2A gene (formerly known as the MLL gene) on the long arm of chromosome 11; a translocation between chromosome 8 and chromosome 16, t(8;16)(p11.21;p13.3), creating a KAT6A‐CREBBP fusion; and t(1;22)(p13.3;q13.1), creating an RBM15‐MRTFA (previously RBM15‐MKL1) fusion gene (see Fig. 3.25). Neonatal ALL also differs from ALL later in childhood. First, cases are nearly all CD10‐negative B‐lineage (Pro‐B) leukaemias. Secondly, virtually all cases are associated with KMT2A gene rearrangement (see Fig 3.25). Thirdly, there appears to be a low frequency of additional mutations, suggesting that expression of a KMT2A fusion gene alone is sufficient to cause ALL in fetal/neonatal cells. The role of KMT2A in neonatal leukaemia is therefore of particular interest. It is only in this age group that a single molecular abnormality is responsible for the majority of leukaemias. Furthermore, it is only in neonates that KMT2A rearrangement causes such a wide spectrum of leukaemias: ALL in about 60% of cases and AML in about 40%, as well as a small number of MPAL or, even more rarely, blastic plasmacytoid dendritic cell neoplasm (Table 3.11).119 Fig. 3.25 Summary of the phenotypic, immunophenotypic and genetic features of neonatal acute leukaemia. (a) The majority of leukaemias in neonates without Down syndrome are acute myeloid leukaemias (AML). Of these, the three most common genetic subtypes are cause by rearrangements of the KMT2A (previously MLL) gene (KMT2A rearranged) or translocations between chromosome 8 and 16 (t(8;16); KAT6A‐CREBBP fusion gene) or chromosomes 1 and 22 (t(1:22); RBM15‐MRTFA fusion gene). In contrast, acute lymphoblastic leukaemia (ALL) in neonates is almost always associated with rearranged KMT2A. (b) The majority of neonatal AML have monoblastic characteristics but acute megakaryoblastic (MK) and erythroblastic leukaemias also occur and are proportionately more common in neonates than in older children or adults. Almost all neonatal ALL has B‐lineage characteristics. These data are summarised from Roberts et al. (2018)119 and are based on a literature review of 144 cases of neonatal acute leukaemia. Table 3.11 Congenital leukaemias excluding those associated with Down syndrome)* * For a more exhaustive list and supporting references, see reference 119. † Spontaneous remission is common. ‡ Spontaneous remission occurred in the one reported case. ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia; BPDCN, blastic plasmacytoid dendritic cell neoplasm; MPAL, mixed phenotype acute leukaemia. Based on reference 119. The most common clinical signs of neonatal leukaemia are hepatosplenomegaly and skin lesions (leukaemia cutis), which are seen in two‐thirds of congenital AML and 50% of congenital ALL cases,140 as well as in the even rarer blastic plasmacytoid dendritic cell neoplasm.141,142 Leukaemia cutis characteristically manifests as a generalised red, brown or purple nodular rash, leading to the term ‘blueberry muffin’ baby (see Fig. 3.27).143 Skin involvement is a particular feature of neonatal leukaemias compared with leukaemia in older children and may be the sole sign of neonatal leukaemia, without any apparent peripheral blood or bone marrow involvement.144,145 However, neonates who present with leukaemia cutis need to be monitored carefully as the disease often evolves to systemic disease over the following months.146 It is therefore important to consider a diagnosis of leukaemia in any neonate who presents with a ‘blueberry muffin’ rash even if the blood count and blood film are normal. Other causes of a ‘blueberry muffin’ rash in the newborn are shown in Table 3.12. The majority of affected neonates have systemic disease and are unwell at presentation.148 As well as hepatomegaly and/or splenomegaly, many will present with or develop jaundice, progressive pallor, pleural effusions and/or ascites and some have lymphadenopathy. In the most severe cases, this is followed by hepatic failure which can lead to rapid death even if the leukaemia regresses.149 Although CNS disease has been reported to occur in around 50% of cases of neonatal leukaemia overall, a recent series of 12 neonates with AML found only two cases with CNS disease.148 The signs of CNS involvement include a bulging fontanelle, reduced level of consciousness, papilloedema and retinal haemorrhages. As the leucocyte count is often very high (see Fig. 3.25), affected neonates may develop signs of leucostasis, including increasing respiratory distress, cardiac failure, renal failure and severe acidosis.150–153 Table 3.12 Causes of a ‘blueberry muffin’ rash in a neonate The haematological features of neonatal leukaemia include hyperleucocytosis due to large numbers of circulating blast cells (Fig. 3.26), with leucocyte counts as high as 830 × 109/l being reported,150 as well as anaemia and thrombocytopenia, which often worsen very quickly over the first few days or hours of life. Some neonates develop coagulation abnormalities secondary to hepatic infiltration or disseminated intravascular coagulation (DIC).148 In the majority of cases of AML, immunophenotyping of peripheral blood or bone marrow blasts or immunohistochemistry of biopsied skin nodules shows monocytic/monoblastic or megakaryoblastic features (Fig. 3.27). Occasional cases of acute basophilic leukaemia154 and erythroleukaemia are also reported in neonates.149,153,155–158 Neonatal ALL (Fig. 3.28) is nearly always of B‐lineage, usually with a pro‐B immunophenotype (CD19+, CD45low, CD22low, CD79a+, CD10−, CD20−) and often with co‐expression of CD33 or CD15.159–161 However, rare cases of T‐lineage ALL have been reported.162 Importantly, a number of cases have been reported where neonates present with B‐lineage ALL but at relapse, often soon afterwards, are found to have ‘switched’ lineage, relapsing as AML, almost always with a monoblastic immunophenotype.163 The dramatic clinical presentation and haematological features, particularly the presence of very large numbers of circulating blast cells, usually makes the distinction of neonatal leukaemia from other conditions straightforward, although expert immunophenotyping and genetic analysis may be required to establish the precise diagnosis, especially in MPAL cases. Fig. 3.26 Blood film of a newborn with congenital AML showing large numbers of myeloblasts and two normoblasts. Note the absence of neutrophils, lymphocytes and monocytes. MGG, ×100. Fig. 3.27 ‘Blueberry muffin’ rash due to leukaemia cutis in a neonate with congenital AML associated with KMT2A rearrangement. From Roberts et al. (2018)119. Fig. 3.28 Blood film of a neonate with congenital ALL showing hyperleukocytosis, a single neutrophil and pleomorphic blast cells. Some blast cells are large and have a diffuse chromatin pattern. Others are smaller with some chromatin condensation, a feature that is far more likely to be seen in ALL than in AML. Note also the cytoplasmic blebs, which could lead to confusion with acute megakaryoblastic leukaemia. The blast cells expressed CD19, CD34, HLA‐DR and CD33 and were negative for CD10, CD15 and CD38. A KMT2A‐MLLT10 fusion was detected. MGG, ×100 (with thanks to Dr Georgina Hall for sharing this film). Blast cells are commonly seen in preterm neonates, particularly those that are less than 26 weeks’ gestation at birth (see Fig. 3.2) and especially those with infections or NEC, but they rarely exceed 8% of the total circulating white blood cells.119 Circulating blast cells are seen in almost all neonates with Down syndrome but rarely exceed 15%. By contrast, in TAM (see earlier) the frequency of circulating blasts in neonates with Down syndrome may exceed 80% and hyperleucocytosis (white cell count >100 × 109/l) is seen in a small proportion of cases (reviewed in reference 113). It is important to note that TAM can occur in neonates with mosaic Down syndrome who have none of the characteristic clinical features of the syndrome. The most common molecular genetic abnormality in neonatal leukaemia is rearrangement of the KMT2A gene and the associated formation of an abnormal fusion gene (see Fig. 3.25). The vast majority of cases of neonatal ALL involve KMT2A fusion genes. Up to 120 KMT2A partner genes have been described in acute leukaemia.164 In neonatal leukaemias the most common is the formation and expression of a KMT2A‐AFF1 fusion gene as a result of a t(4;11)(q21.3;q23.3) chromosomal translocation. Typically, KMT2A‐AFF1 causes early precursor or pro‐B ALL in neonates and infants and AML in adults, although AML and MPAL have both been reported in neonates.140,159 Lineage switch from a B‐lineage ALL at presentation to AML at relapse is particularly associated with neonatal leukaemia due to the KMT2A‐AFF1 fusion gene and is well recognised, though uncommon.163 The natural history of lineage switching is consistent with a leukaemia‐initiating mutation in a fetal stem or progenitor cell with lymphoid and myeloid lineage potential, although it is also possible that KMT2A‐AFF1 fusion gene is able to reactivate myeloid genes in fetal/neonatal progenitors that are already committed to the B‐lineage.165 Indeed, recent data from single‐cell ribonucleic acid (RNA) sequencing studies in KMT2A‐rearranged infant ALL suggest that ‘lineage plasticity’ is a specific property of fetal/neonatal haemopoietic cells.166 It is now clear that all cases of acute leukaemia occurring within the neonatal period arise in utero and therefore in fetal cells. This is shown by the presence of the causative fusion genes, such as KMT2A‐AFF1, in neonatal blood spots167–169 and through twin studies which describe genetically identical leukaemias in monozygotic twins who present in the first month of life, indicating that leukaemia‐initiating cells must have been transmitted from one twin to the other by intrauterine placental sharing.160,170–172 In neonatal AML, around half of the cases are caused by three specific fusion genes: KMT2A‐AFF1, KAT6A‐CREBBP and RBM1‐MRTFA (previously RBM15‐MKL1). While KMT2A‐AFF1 and KAT6A‐CREBBP both cause predominantly monocytic/monoblastic leukaemias, the RBM15‐MRTFA fusion gene resulting from a t(1;22)(p13.3;q13.1) translocation is always megakaryoblastic. These leukaemias are often accompanied by fibrosis in the bone marrow and/or liver.173–176 The KAT6A‐CREBBP fusion gene results from a translocation between chromosomes 8 and 16, t(8;16)(p11.21;p13.3), and is of particular importance because of the relatively high frequency of cases that undergo spontaneous remission.177–184 Many of these cases subsequently relapse, but there are several well‐documented cases who have remained in long‐term remission.177,179,181,182 It is therefore very important that otherwise well neonates with AML due to the KAT6A‐CREBBP fusion gene seen following t(8;16) translocation are carefully monitored before embarking on chemotherapy. Occasional spontaneous remissions have also been reported in neonates with KMT2A‐associated neonatal AML,185,186 but not in those with KMT2A‐associated ALL. Overall, about 40% of cases of neonatal AML that appear to undergo spontaneous remission ultimately relapse (reviewed in reference 119). The mechanism of spontaneous remission of neonatal leukaemia is not clear. It seems likely that differences between the fetal and postnatal microenvironment, with the latter lacking key factors needed for the maintenance of the clone of leukaemic cells, play a role together with intrinsic properties of the transformed fetal leukaemic stem or progenitor cells. In neonates with symptomatic disease, expert supportive care and chemotherapy are essential as the mortality of neonatal leukaemia remains very high (up to 60% for AML and 80% for ALL).148 Careful management of hyperleucocytosis, including exchange transfusion, may be needed to reduce the risk of CNS haemorrhage and leucostasis in lungs and other organs.149,187–189 It is essential to monitor fluid balance and renal and liver function closely, especially in preterm neonates, who are more at risk of severe complications.149,188–190 Coagulation problems are common and blood product support is often required.140,170,174,189,190 Tumour lysis is common because of the leucocytosis and uric acid depletion with rasburicase should be used,191 provided that glucose‐6‐phosphate dehydrogenase (G6PD) deficiency has been excluded, because rasburicase has been reported to trigger fatal haemolysis in G6PD deficiency;192 allopurinol can be used where rasburicase is contraindicated.140 Neonates have been eligible for several large collaborative treatment studies in infant ALL193,194 and AML,195,196 although reduced doses compared with those used in older children are often employed because of treatment‐related toxicity in neonates.188,195 These studies show that a small proportion of children with neonatal leukaemia can be cured by chemotherapy.188,197,198 For neonates with AML, combination chemotherapy is supplemented with CNS‐directed therapy119 and data from the largest study indicated comparable survival for infants aged 0–3 months compared with infants aged more than 3 months at diagnosis (51% 5‐year overall survival).198 For neonatal ALL, most cases published since 2000 have been treated with the combination chemotherapy regimens used for infant ALL.119 Haemopoietic stem cell transplantation has been shown to cure a small number of children with relapsed neonatal leukaemia148,159 but the number of cases is too low to properly define the role of transplantation in first or second remission. Despite the possibility of spontaneous remission in some cases of neonatal AML, other neonates with the same molecular genetic abnormalities have an aggressive disease course199 and therefore there is currently no clear guidance as to which children can avoid chemotherapy.140 In addition, one series of t(8;16)‐associated neonatal AML highlighted the high rate of relapse in neonates who were not treated initially because of apparent regression of their leukaemia; four of these seven neonates subsequently required chemotherapy.183 Neonates with Noonan syndrome are at risk of developing either JMML or a related transient myeloproliferative disorder. JMML is a rare childhood leukaemia that usually presents in the first 5 years of life and is more common in boys.200 It is caused by somatic or germline mutations in RAS‐pathway genes, most commonly in PTPN11, KRAS, NRAS, CBL or NF1.201,202 Although JMML typically presents in infancy or childhood, in Noonan syndrome it may manifest in the neonatal period when it carries a particularly bad prognosis.203 Neonates with Noonan syndrome due to germline mutations in PTPN11, or occasionally CBL, KRAS or NRAS, can develop either JMML or a JMML‐like myeloproliferative disorder often referred to as Noonan syndrome‐associated myeloproliferative disorder (NS/MPD) in the first few days or weeks of life.38,39,204 Differentiating NS/MPD from JMML is important as the prognosis is very different; this may be difficult as both have the same underlying genetic cause (Case 3.3, see page 174). A diagnosis of JMML is made in all cases that meet the internationally defined consensus criteria, which include a monocyte count more than 1 × 109/l, splenomegaly, less than 20% blasts in the blood or bone marrow and a somatic mutation in either the PTPN11, KRAS or NRAS genes.205 The clinical features of NS/MPD include splenomegaly, hepatomegaly, rash, fever and, occasionally, hydrops fetalis. The haematological features include anaemia, thrombocytopenia and leucocytosis with increased numbers of monocytes, many with dysplastic features, and increased circulating blast cells (Fig. 3.29). The percentage of blasts is usually <20%, helping to distinguish NS/MPD from other forms of neonatal leukaemia, including many cases of TAM, and other causes of monocytosis (see Table 3.5), particularly sepsis. Where a diagnosis of Noonan syndrome is strongly suspected in a neonate, it is important that a blood count and blood film are performed and evaluated by an experienced haematologist. In any neonate where the blood film is suggestive of JMML, it is important to perform molecular analysis to identify the precise mutation and determine whether or not this is germline by analysing both peripheral blood and germline DNA (for example from a skin biopsy). Although the majority of cases of NS/MPD are not unwell and the condition undergoes spontaneous remission within a few weeks or months of birth, some germline PTPN11 mutations and CBL mutations are associated with more rapidly progressive disease in the neonate.39,203 Fig. 3.29 Blood film of a neonate with Noonan syndrome‐associated myeloproliferative disorder, showing large numbers of abnormal monocytes and occasional blast cells. MGG, ×20. The mainstay of treatment for NS/MPD is good supportive care since the majority of cases resolve spontaneously and chemotherapy is rarely necessary.206 The majority of neonates with NS/MPD will survive, in contrast to infants who are diagnosed with JMML after the neonatal period, where survival is 50–60% as the disease is refractory to chemotherapy, and the only curative treatment is haemopoietic stem cell transplantation, which is associated with a high rate of relapse.200 It is therefore important to offer genetic counselling and molecular testing to families of patients with germline mutations. However, in contrast to TAM, neonates with NS/MPD in whom the neonatal MPD resolves do not have an increased risk of subsequent leukaemia.38,203 (For abbreviations used in the text of the illustrative cases, see page 176.)
3
Neonatal infection and leucocyte disorders
Leucocyte abnormalities in neonatal systemic disease
Increases in leucocyte numbers
Causes and significance of neonatal neutrophilia
Features of infection on neonatal blood film
Differential diagnosis
Pitfalls/comments
Band cells
Acute bacterial infection
Congenital viral infection
Rare congenital causes (e.g. Pelger–Huët anomaly)
Band cells in the absence of other leucoerythroblastic features are not a normal feature in preterm babies
Toxic granulation of neutrophils
Acute bacterial infection
Chronic bacterial infection (e.g. osteomyelitis)
HIE (toxic granulation may persist for up to 2 weeks after birth)
Maternal chorioamnionitis without acute infection in the baby (may persist for up to 1 week after birth)
Meconium aspiration (may persist for up to 1 week after birth)
Toxic granulation after the first week of life is usually due to infection (apart from cases of severe HIE)
Neonates born to mothers with severe chorioamnionitis may have very marked leucocytosis (WBC up to 80 × 109/l), even in the absence of acute neonatal infection (normal or minimally raised CRP and lack of band cells being the most helpful distinguishing features)
Vacuolation of neutrophils
Staphylococcus epidermidis infection (typically line infections)
Candida spp. infection
NEC
Other acute bacterial infections, e.g. due to Gram‐negative species
Neonates with NEC nearly always have neutrophil vacuolation
Acute bacterial infection and NEC often co‐exist; neutrophil vacuolation is not specific to NEC but the absence of neutrophil vacuolation makes NEC unlikely
Leucoerythroblastic features
Normal feature in the first week of life in preterm babies (≤28 weeks’ gestation at birth)
HIE (features present at birth and may persist for up to 2 weeks)
Neonates with Down syndrome in the first 2 weeks of life
Neonates with leucoerythroblastic features due to infection nearly always have a peak in band cells as well
Condition
Diagnostic notes
Infection
Acute bacterial infection*
Often preceded by transient neutropenia and with an increased proportion of band cells
Usually accompanied by toxic granulation of the neutrophils
Chronic bacterial infection (e.g. osteomyelitis)
Toxic granulation often mild
Neutrophil left shift usually absent
Some viral infections (e.g. chikungunya virus)
Neutrophilia, usually mature; occasional cases with severe thrombocytopenia
Maternal chorioamnionitis*
Mature neutrophilia with toxic granulation
Constitutional trisomies
Trisomy 21 (Down syndrome)
Trisomy 13 (Patau syndrome)
Trisomy 18 (Edwards syndrome)
Neutrophilia is commonly seen in neonates with Down syndrome even in the absence of infection or TAM
Transient abnormal myelopoiesis associated with Down syndrome
Blast cells are >10%
Leucocytosis (WBC >30 × 109/l) is common
Inherited immunodeficiency disorders
LAD syndromes, IRF8 deficiency
Persistent neutrophilia; delayed separation of the cord (LAD syndromes); hypofunctional/dysplastic neutrophils (IRF8 deficiency)
Other
Benign altitude‐related neutrophilia
Neutrophilia without left shift in neonates born at altitude (≥1550 m above sea level) who have no clinical or laboratory evidence of infection
Drugs (e.g. corticosteroids, G‐CSF)
Congenital infection
Cytomegalovirus
Coxsackievirus
Acquired neonatal infection
Cytomegalovirus
Respiratory syncytial virus
SARS‐CoV‐2 (COVID‐19)
Bordetella pertussis
Malaria
Immunodeficiency disorders
Omenn syndrome
Autoimmune lymphoproliferative syndrome
Causes and significance of neonatal lymphocytosis
Causes and significance of neonatal eosinophilia
Condition
Diagnostic notes
Infection
Usually mild and transient
Develops 1–2 days after the onset of infection
Average duration 6 days
Necrotising enterocolitis
Seen in ~50% of cases
Develops within 24 hours of the diagnosis of NEC
Allergic/inflammatory conditions
Eosinophilia is persistent and may be marked
Clinical features of the underlying condition will be present
Transient abnormal myelopoiesis in neonates with Down syndrome
Seen in up to 23% of cases but not usually prominent
May be a later feature (median age at presentation 41 days)
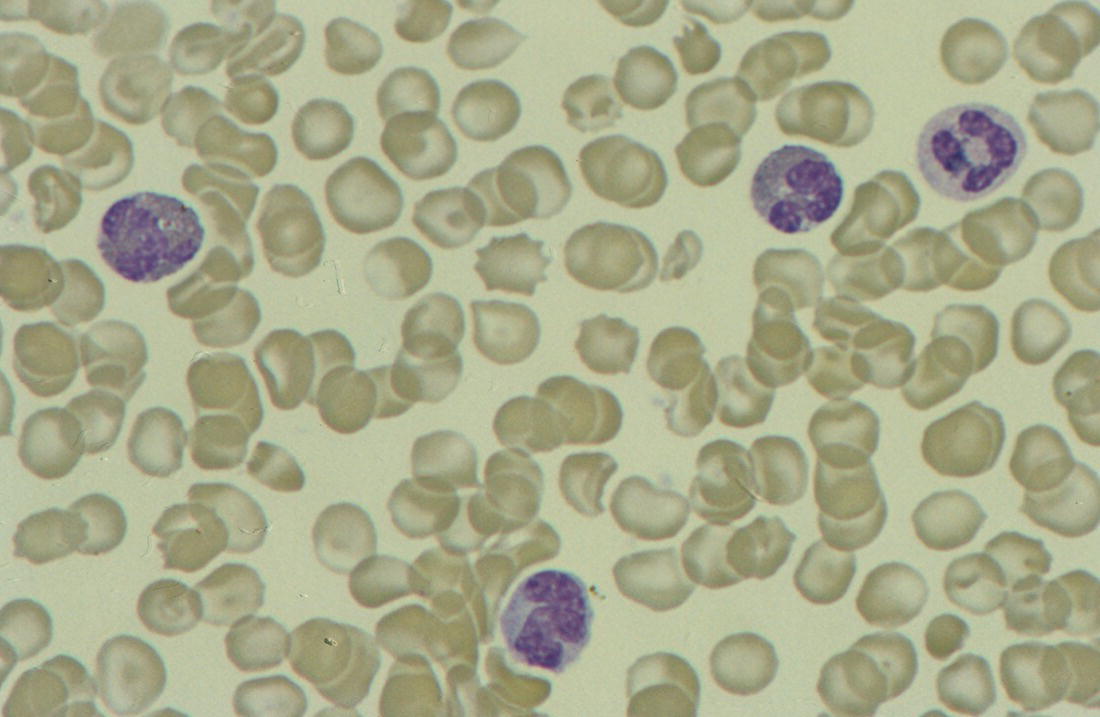
Causes and significance of abnormalities in monocyte number in neonates
Condition
Diagnostic notes
Infection
Particularly seen in congenital protozoal infections, congenital syphilis and Candida infections
Common early feature of bacterial infection which resolves within a few days
Blasts nearly always <8%
Transient abnormal myelopoiesis/transient myeloproliferative disorder associated with Down syndrome
Acquired mutations in the GATA1 gene always present
Peripheral blood blasts variable (1–90%)
Also occurs in neonates with mosaic Down syndrome and no other clinical features of Down syndrome
Other features usually present (see Table 3.10)
Transient juvenile myelomonocytic leukaemia associated with congenital rasopathies
Most commonly seen in association with Noonan syndrome (PTPN11 mutations)
Peripheral blood blasts variable
Increased numbers of abnormal monocytes always present
Family history may be helpful (e.g. of neurofibromatosis)
Congenital neutropenia
Isolated severe neutropenia from birth,
recurrent bacterial infection, family history
monocytosis increases during infections
Isolated severe neutropenia at birth,
No family history; recovers in 1–2 months
Other features of WAS (e.g. eczema, microthrombocytopenia, recurrent infections) in a male infant
Circulating blast cells in neonates
Condition
Diagnostic notes
Non‐specific normal feature in preterm neonates, particularly if unwell due to infection or other causes
Transient feature which resolves within a few days
Blasts nearly always <8%
Transient abnormal myelopoiesis/transient myeloproliferative disorder associated with Down syndrome
Acquired mutations in the GATA1 gene always present
Peripheral blood blasts variable (1–90%)
Also occurs in neonates with mosaic Down syndrome and no other clinical features of Down syndrome
Other features usually present (see Table 3.10)
Transient juvenile myelomonocytic leukaemia associated with congenital rasopathies
Most commonly seen in association with Noonan syndrome
Peripheral blood blasts variable
Increased numbers of abnormal monocytes always present
Family history may be helpful (e.g. of neurofibromatosis)
Congenital leukaemia (in neonates without Down syndrome)
40% acute myeloid leukaemia
60% acute lymphoblastic leukaemia
Peripheral blood blasts usually >20%
Characteristic molecular and cytogenetic abnormalities (e.g. KMT2A rearrangements in ALL and t(1;22) in AML)
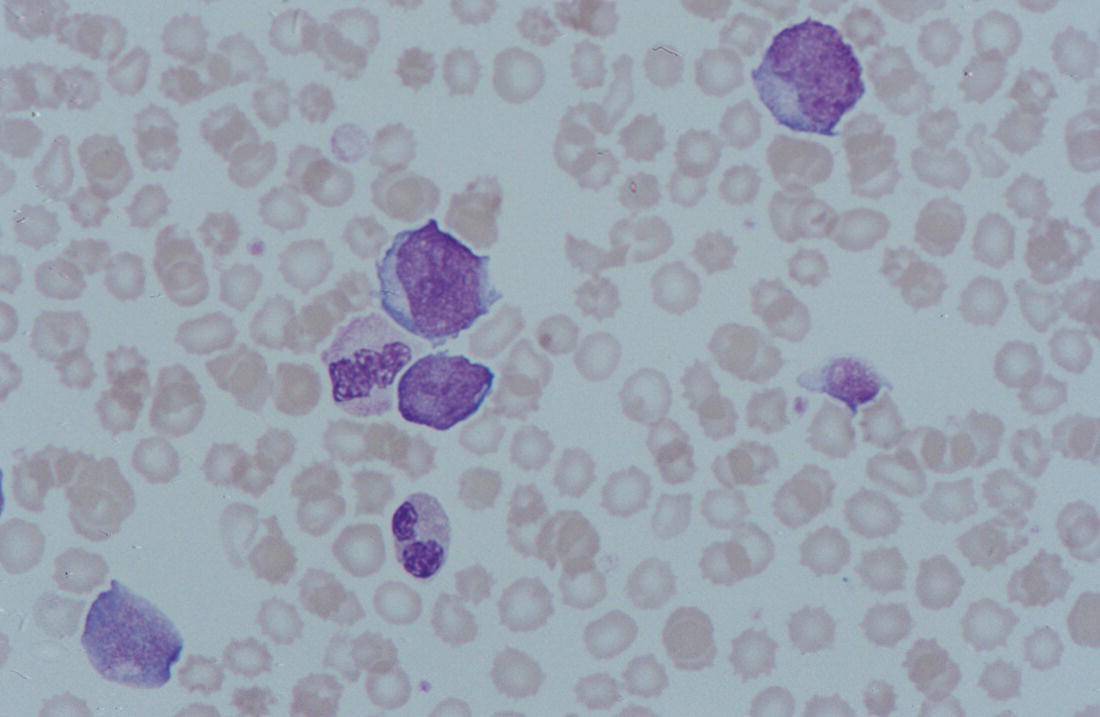
Neonatal infection and its differential diagnosis: clues from the blood count and blood film
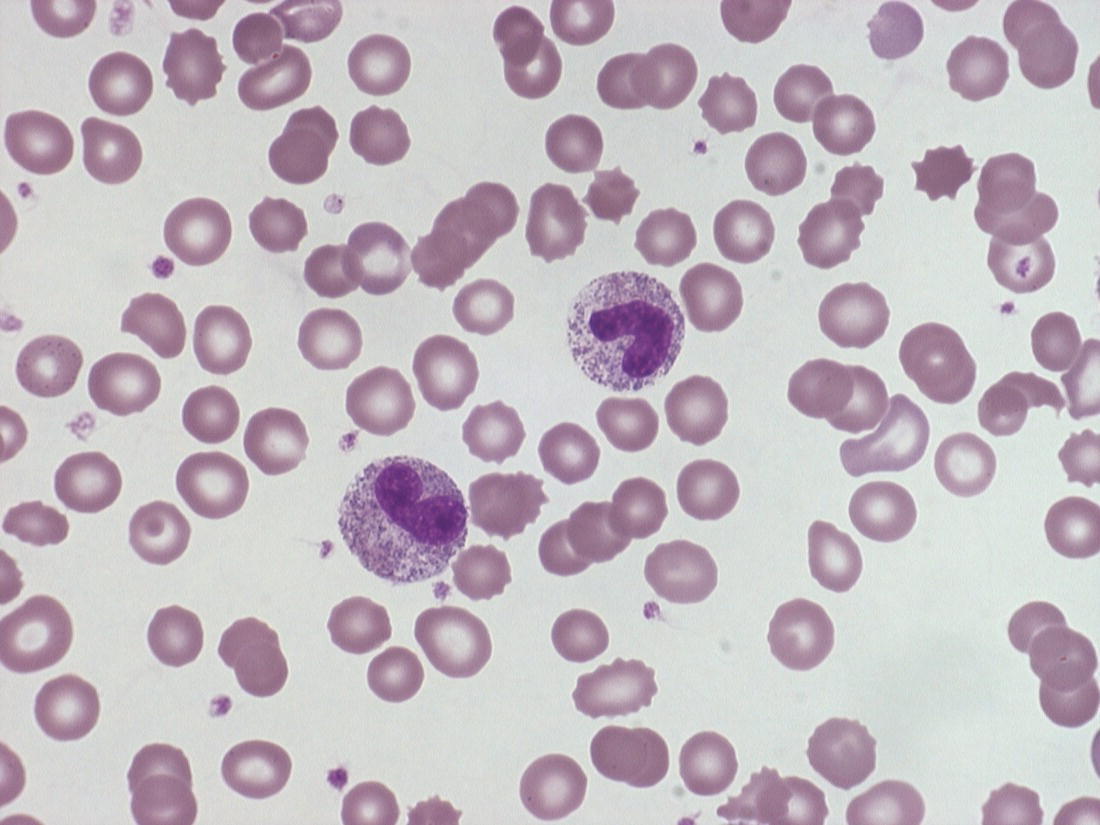
Acute bacterial infection
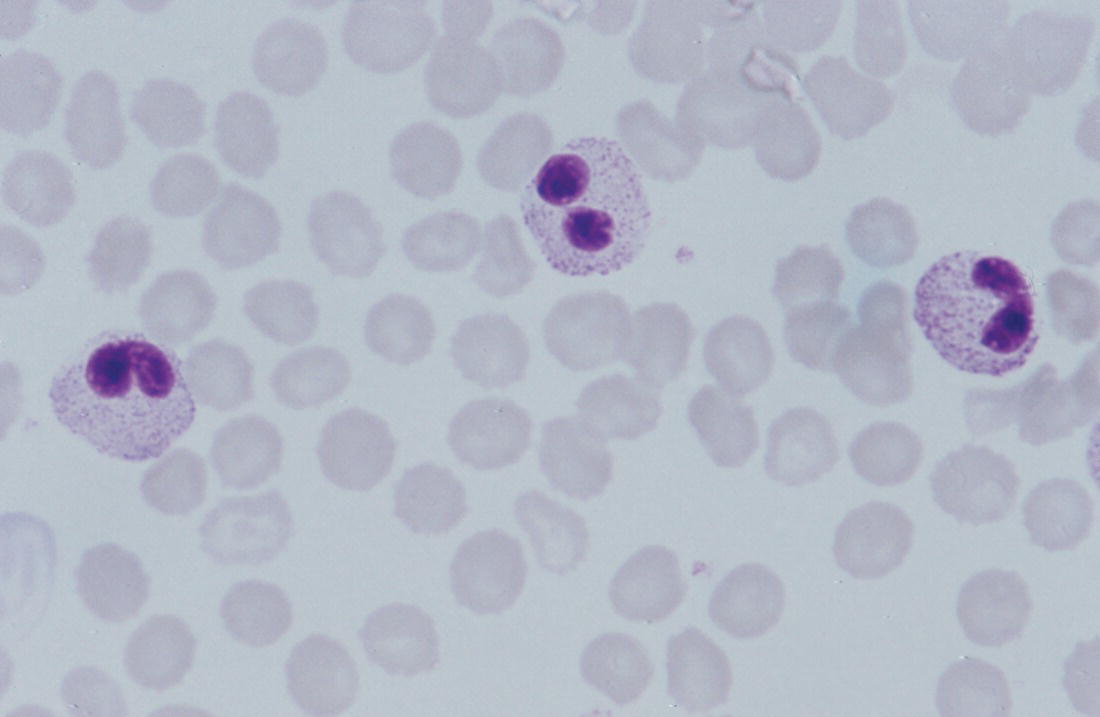
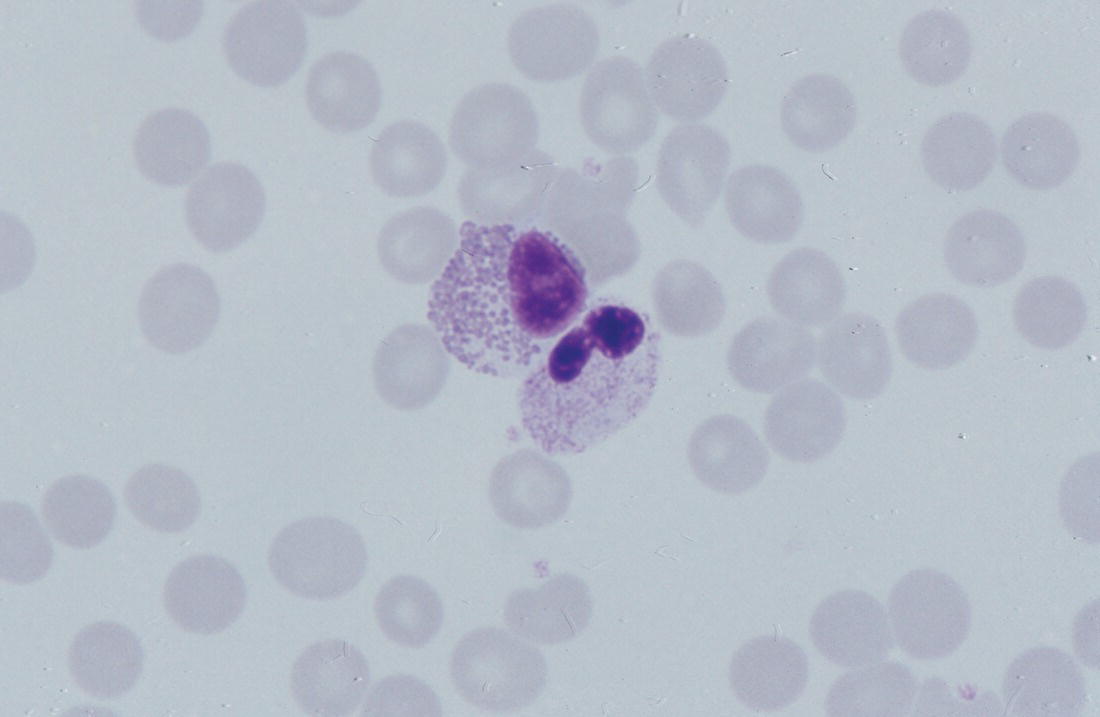
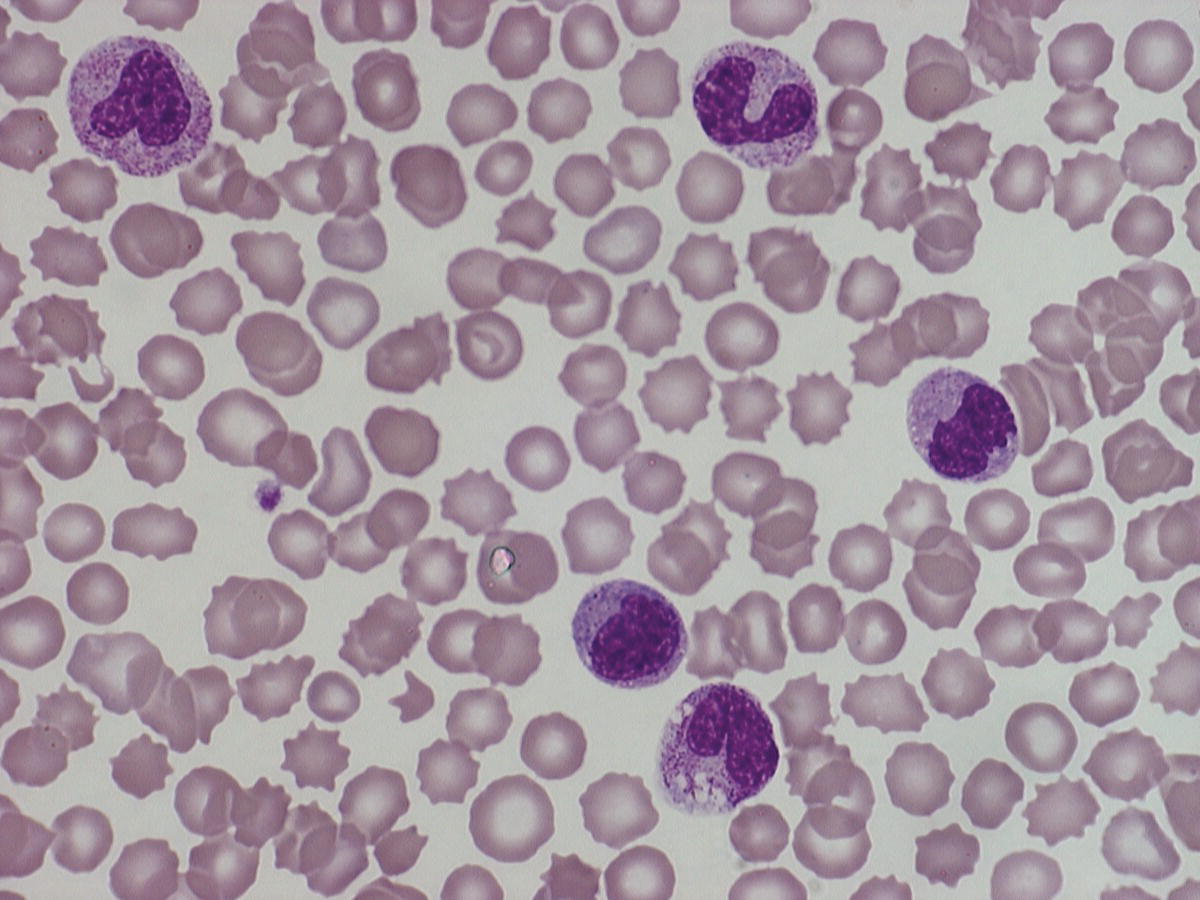
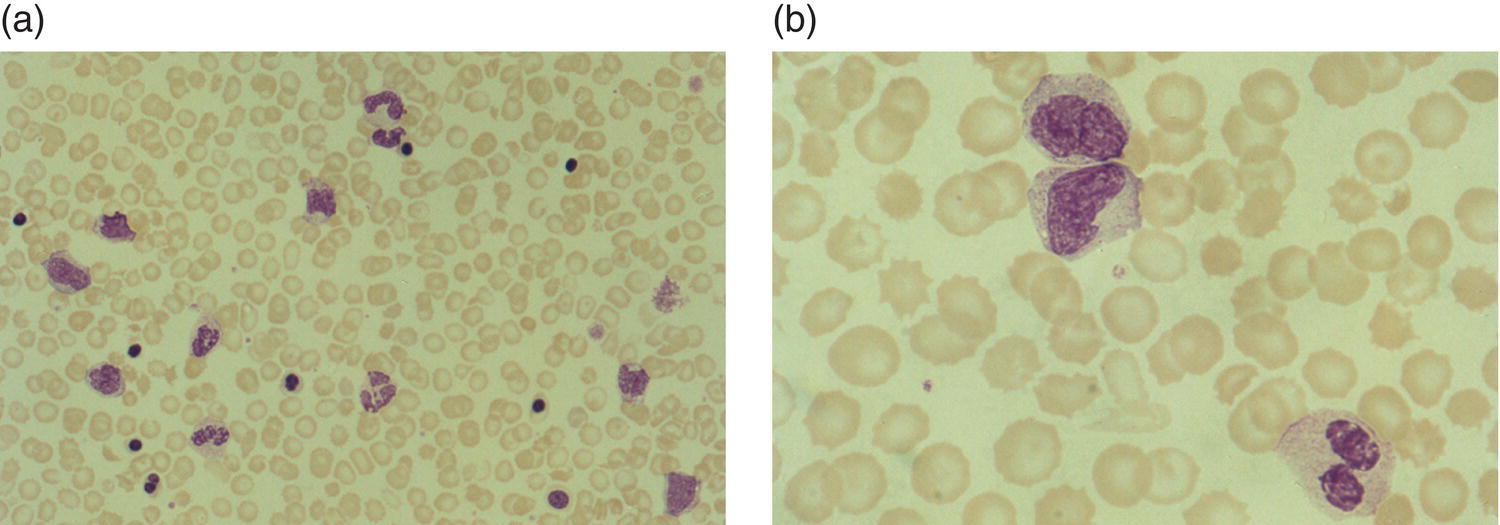
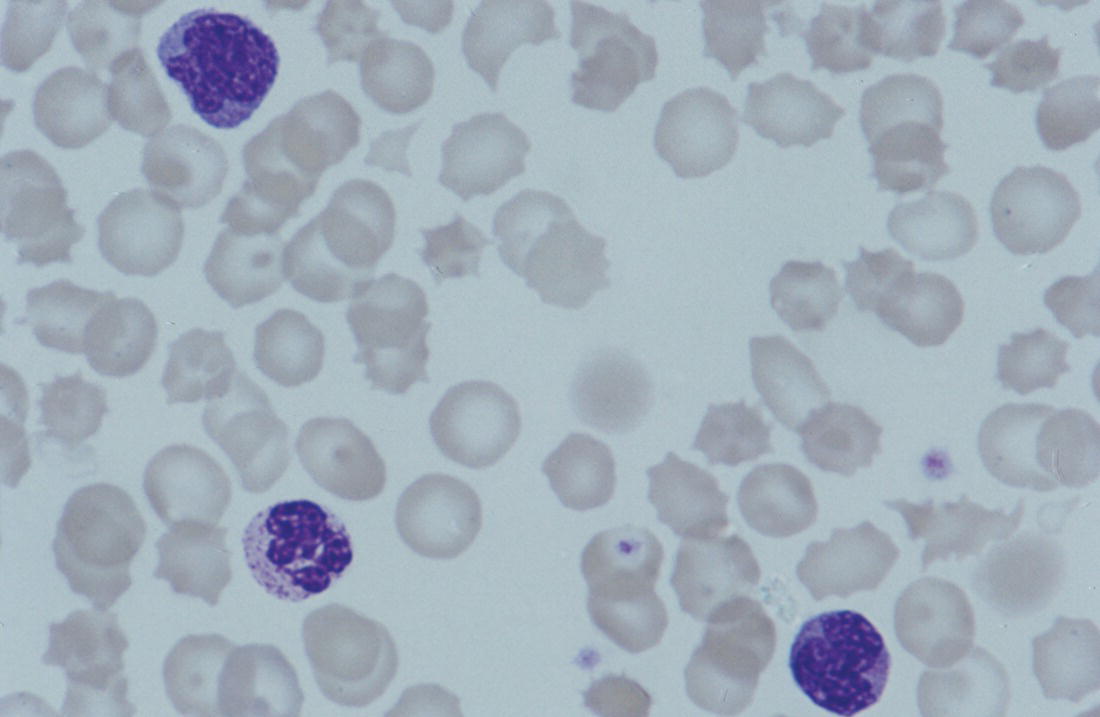
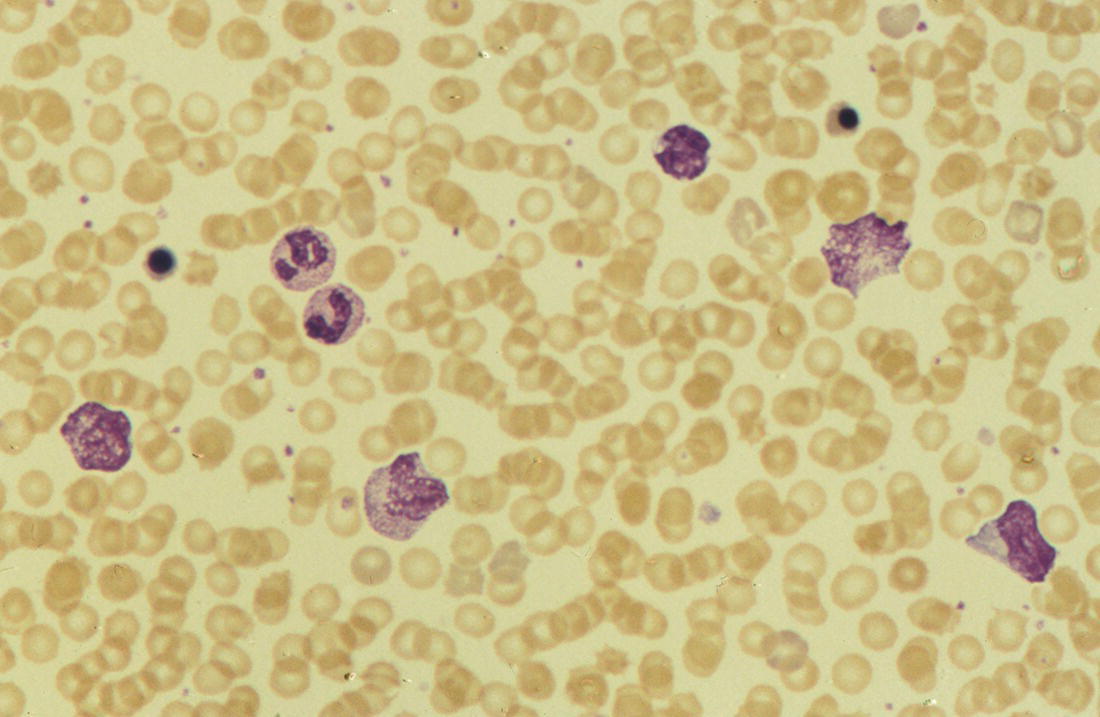
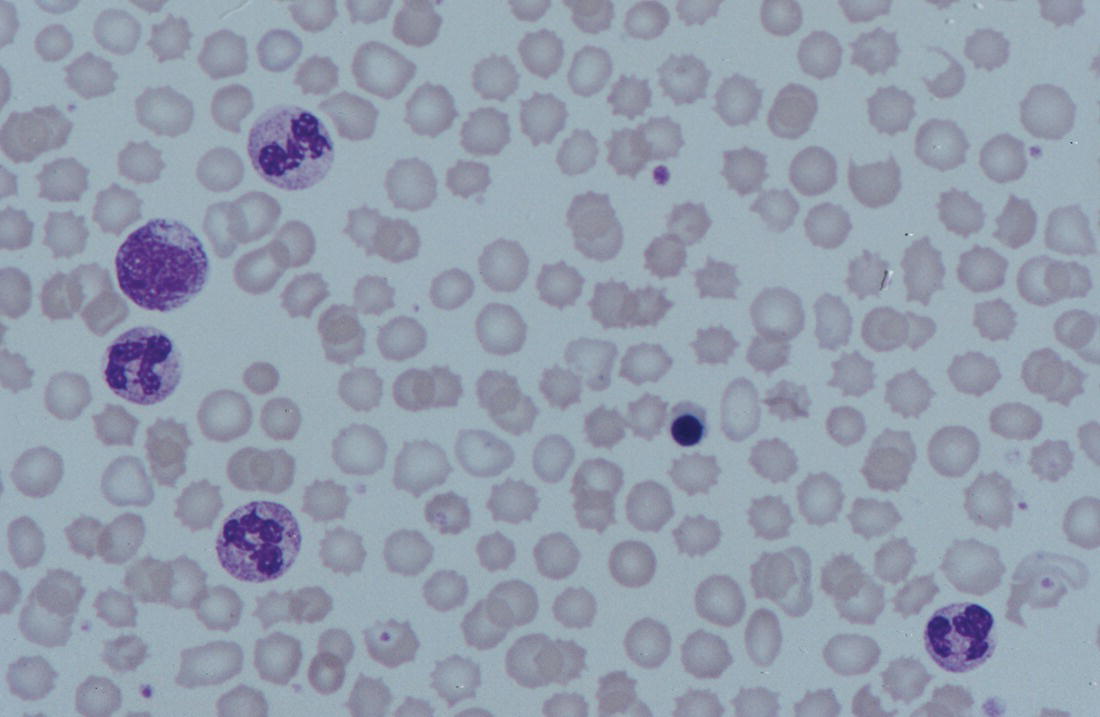
Maternal chorioamnionitis
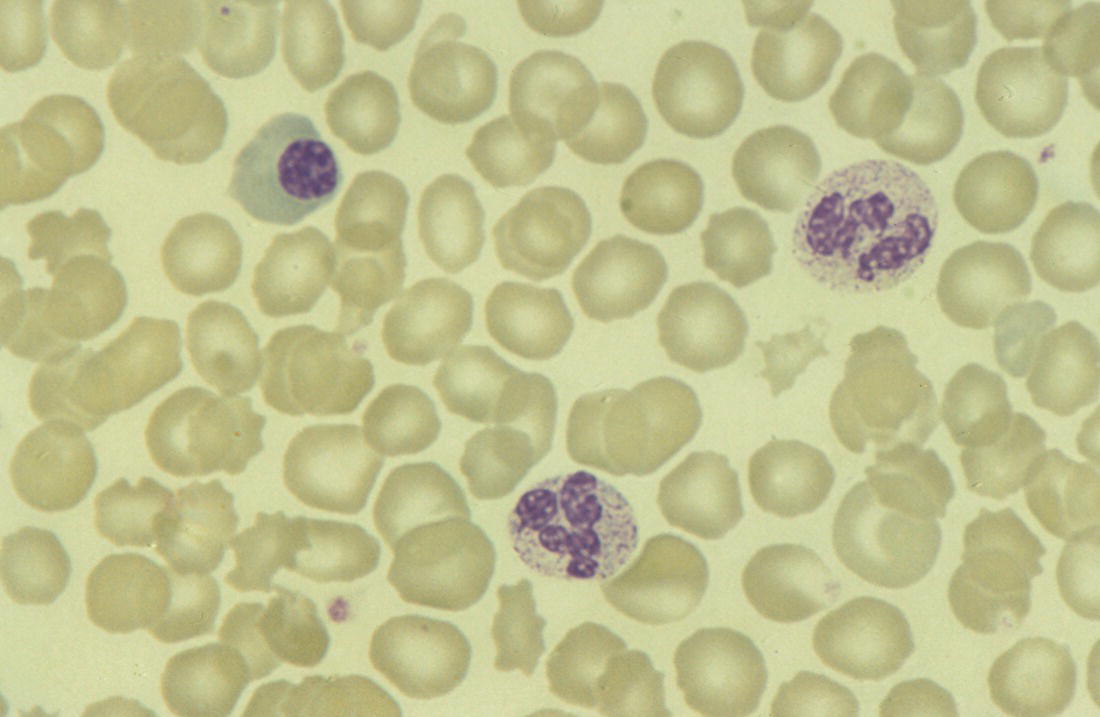
Leucocyte vacuolation
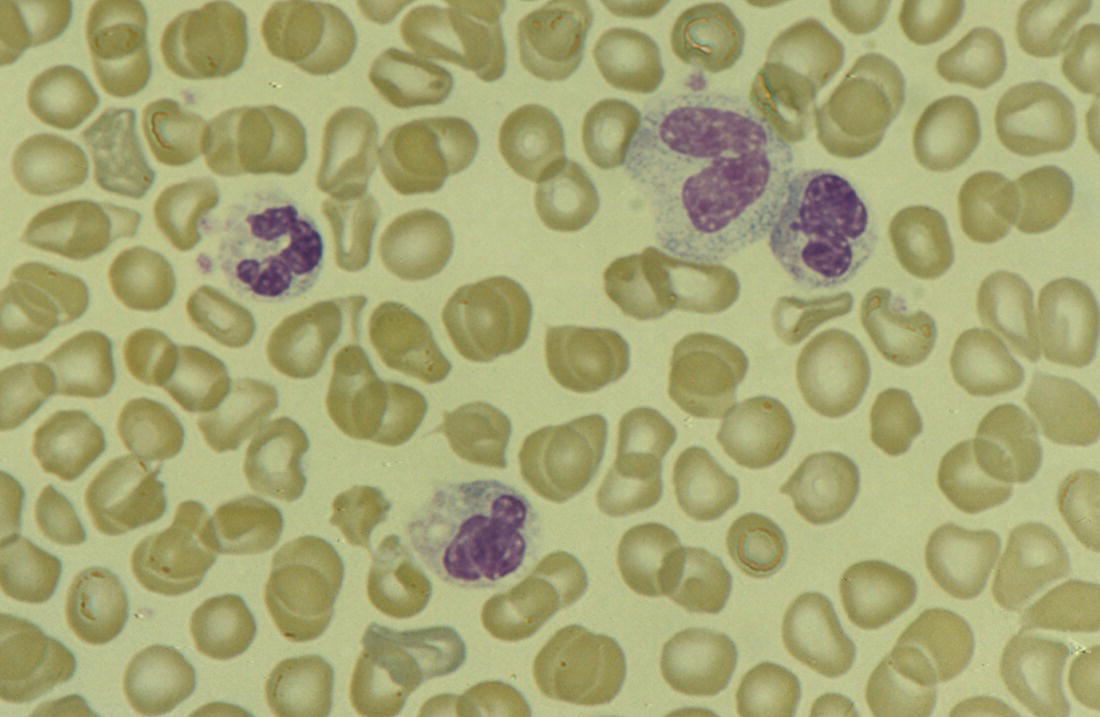
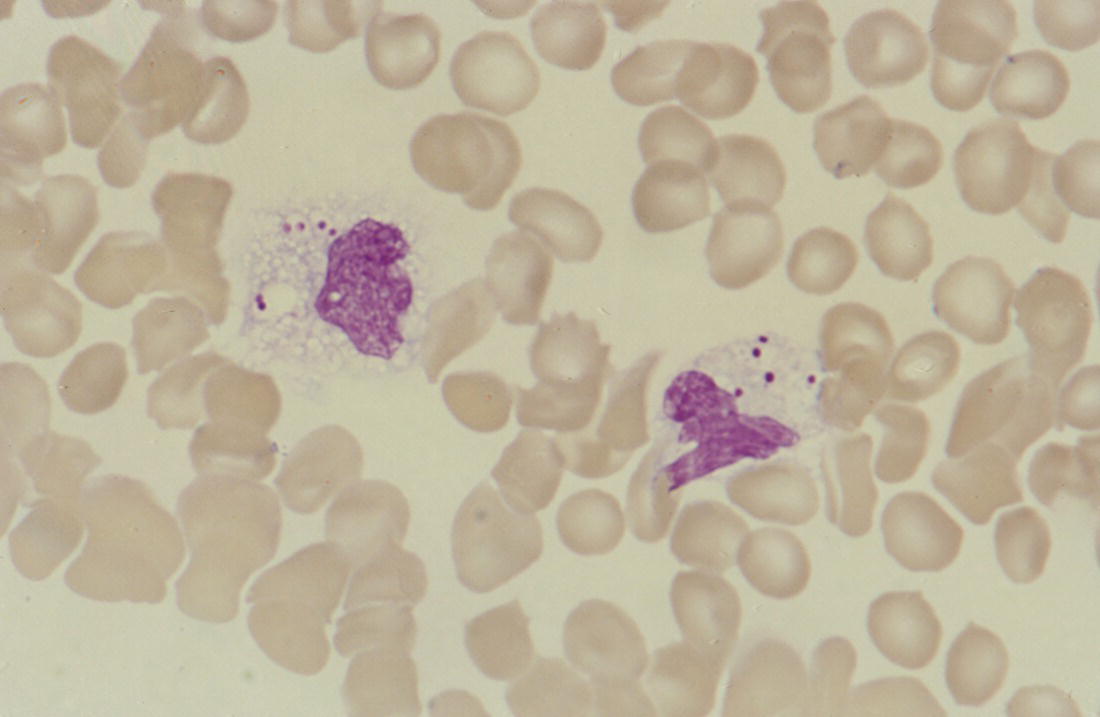
Viral infection
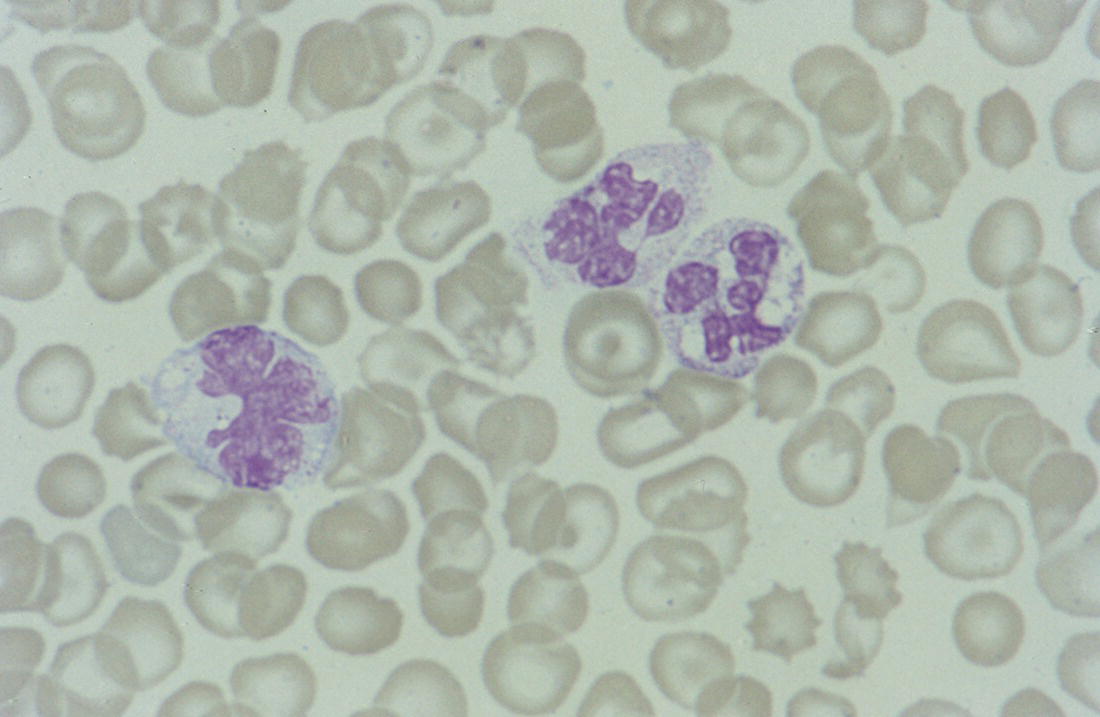
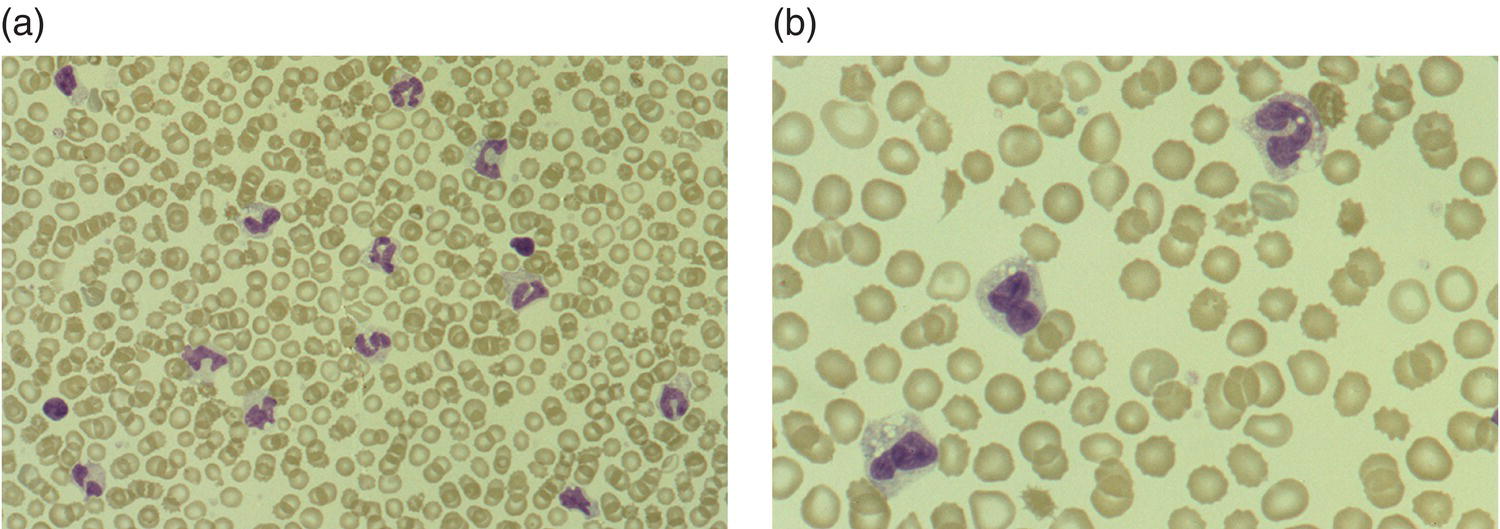
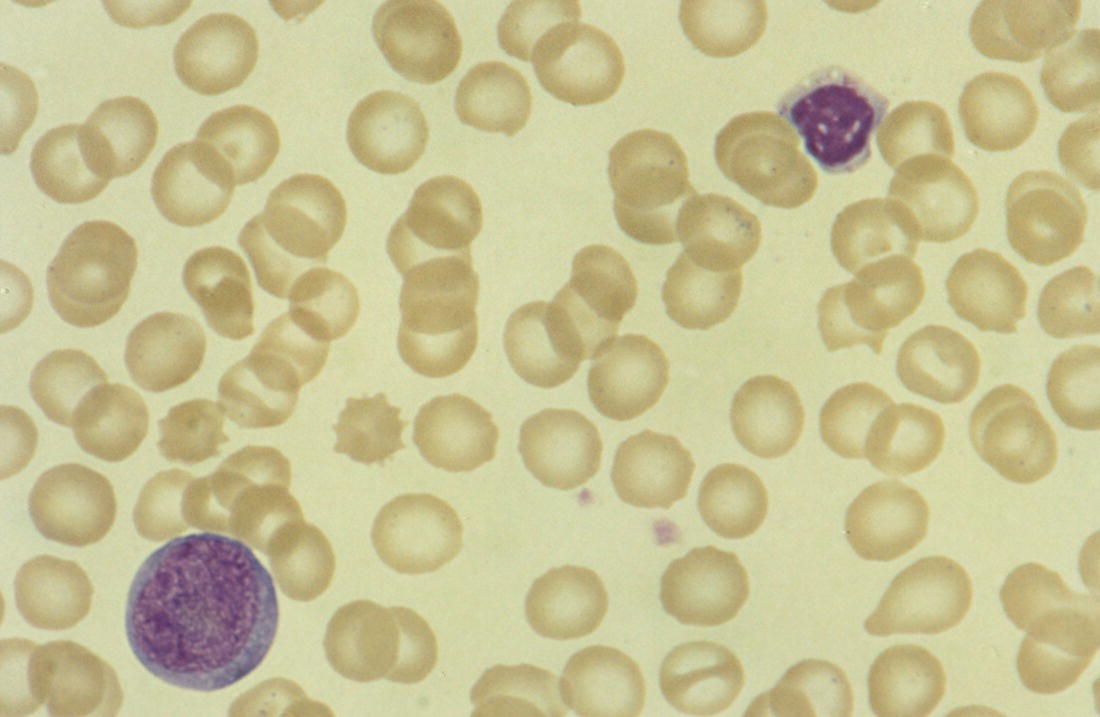
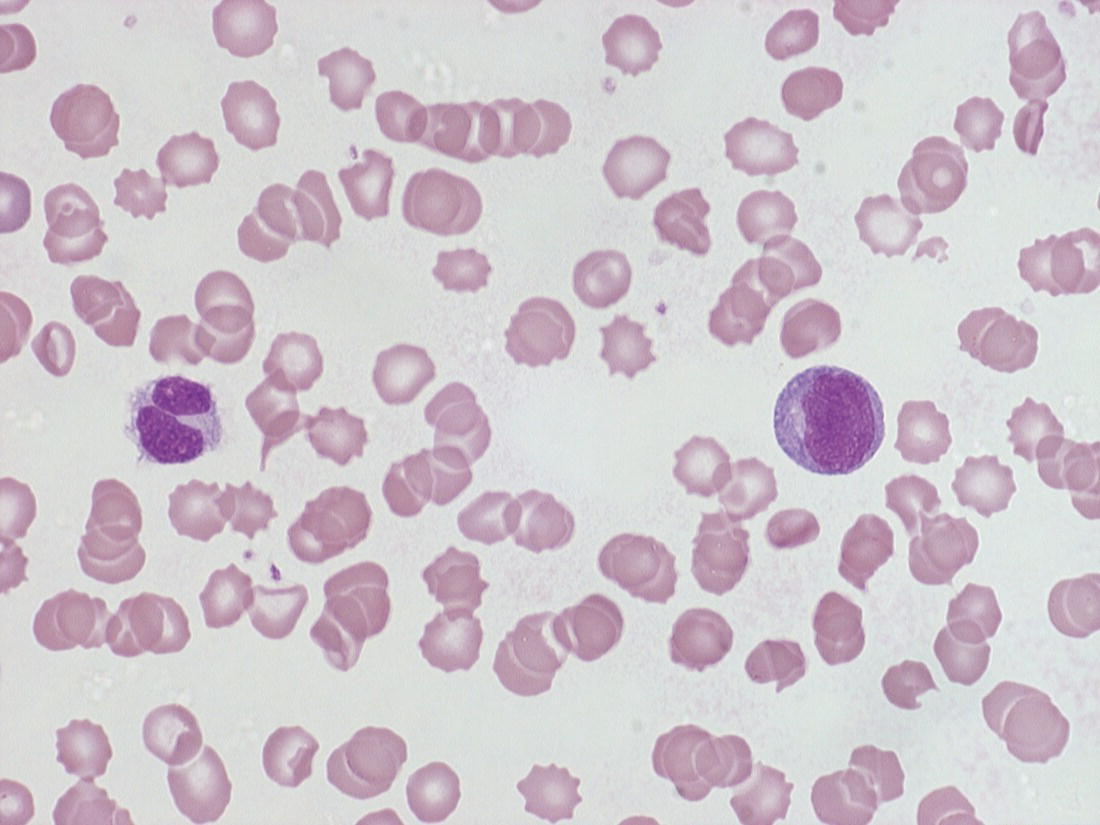
Storage disorders: diagnostic clues from the blood film
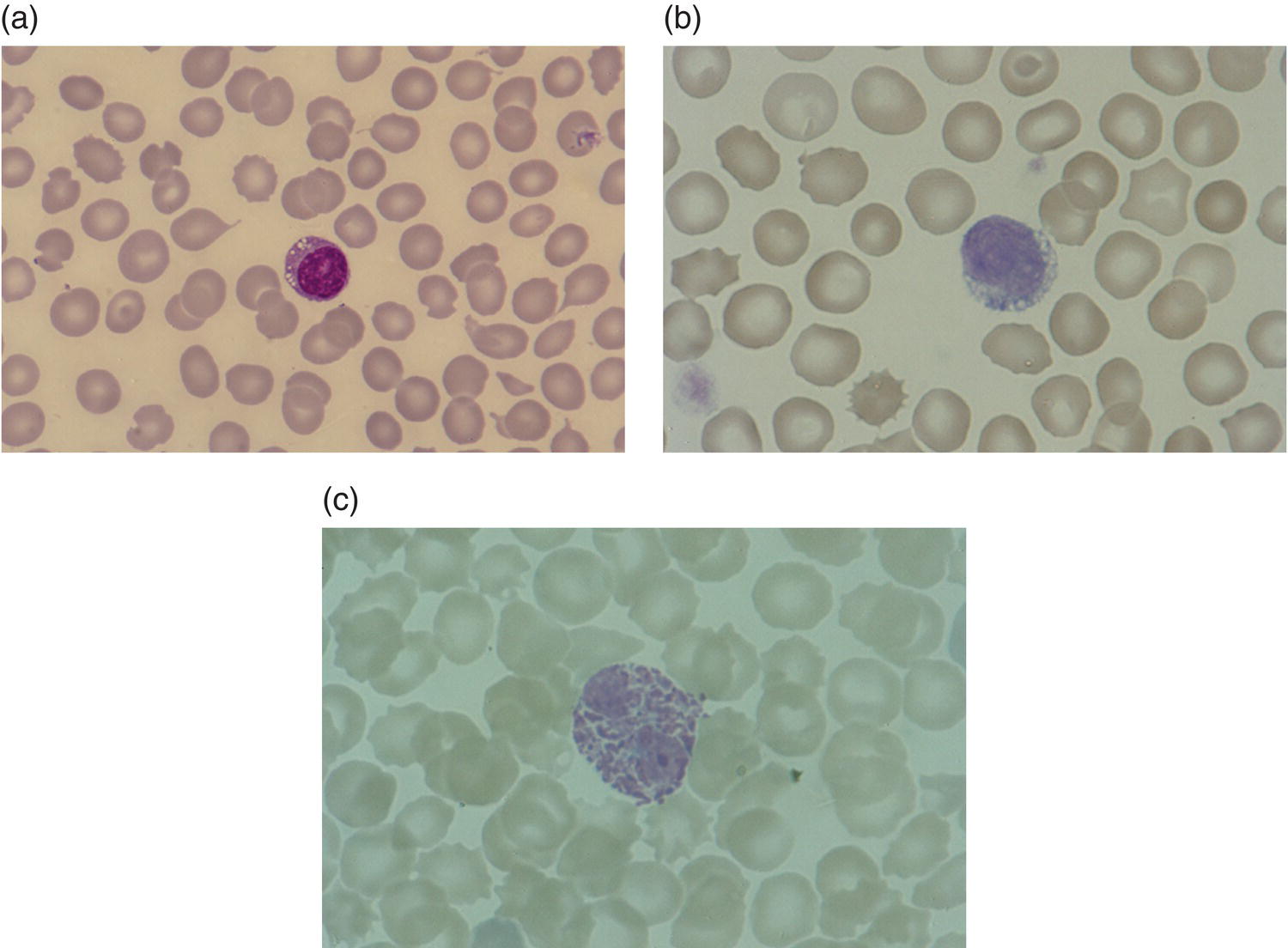
Neonatal neutropenia
Definition and causes of neutropenia
Cytological features
Possible diagnoses
Diagnostic clues
Large, brightly staining granules, which can also be larger than normal, in neutrophils and other granulocytes (Alder–Reilly anomaly); abnormal lymphocyte and monocyte granules are sometimes also present
As an isolated anomaly or associated with one of the mucopolysaccharidoses (e.g. Sly syndrome, Sanfilippo syndrome, Hurler syndrome, Hunter disease), mucosulphatidosis, infantile type neuronal ceroid lipofuscinosis
Family history, features of a storage disorder – dysmorphism, hepatomegaly, slow development
Giant and aberrantly staining granules in all granulocytes, monocytes and lymphocytes
Chédiak–Higashi anomaly
Family history, pancytopenia, infection, jaundice, albinism and neurological abnormalities
Cytoplasmic inclusions in granulocytes (Döhle body‐like)
MYH9‐related disorders including May–Hegglin anomaly
Family history, macrothrombocytopenia; other disease features (e.g. deafness and nephropathy) are absent in the neonatal period
Cytoplasmic vacuolation
Carnitine deficiency, neutral lipid storage disease, Pearson syndrome
Pancytopenia in Pearson syndrome
Lymphocyte vacuolation
Mucopolysaccharidoses (e.g. Hunter disease), mucosulphatidosis, mucolipidosis type II (I‐cell disease) and type IV, Niemann–Pick disease type A, Wolman disease, GM1‐gangliosidosis, mannosidosis, fucosidosis, Pompe disease, sialic acid storage disease
Family history, features of a storage disorder – dysmorphism, hepatosplenomegaly,
CNS dysfunction, developmental delay
Chronic in utero hypoxia
Maternal hypertension
Intrauterine growth restriction
Maternal diabetes
Infection
Acute, perinatal bacterial infection, e.g. group B Streptococcus
Congenital viral infections, e.g. cytomegalovirus
Neonatal bacterial infections
Neonatal viral infections, e.g. cytomegalovirus
Necrotising enterocolitis
Immune
Autoimmune (maternal or neonatal)
Alloimmune
Chromosomal anomalies
Trisomy 21
Trisomy 18
Trisomy 13
Metabolic disorders
Hyperglycinaemia
Isovaleric acidaemia
Propionic acidaemia
Methylmalonic acidaemia
3‐Methylglutaconic aciduria type II (Barth syndrome)
Pearson syndrome
Marrow replacement
Congenital leukaemia (see Table 3.11)
Congenital primary myelofibrosis
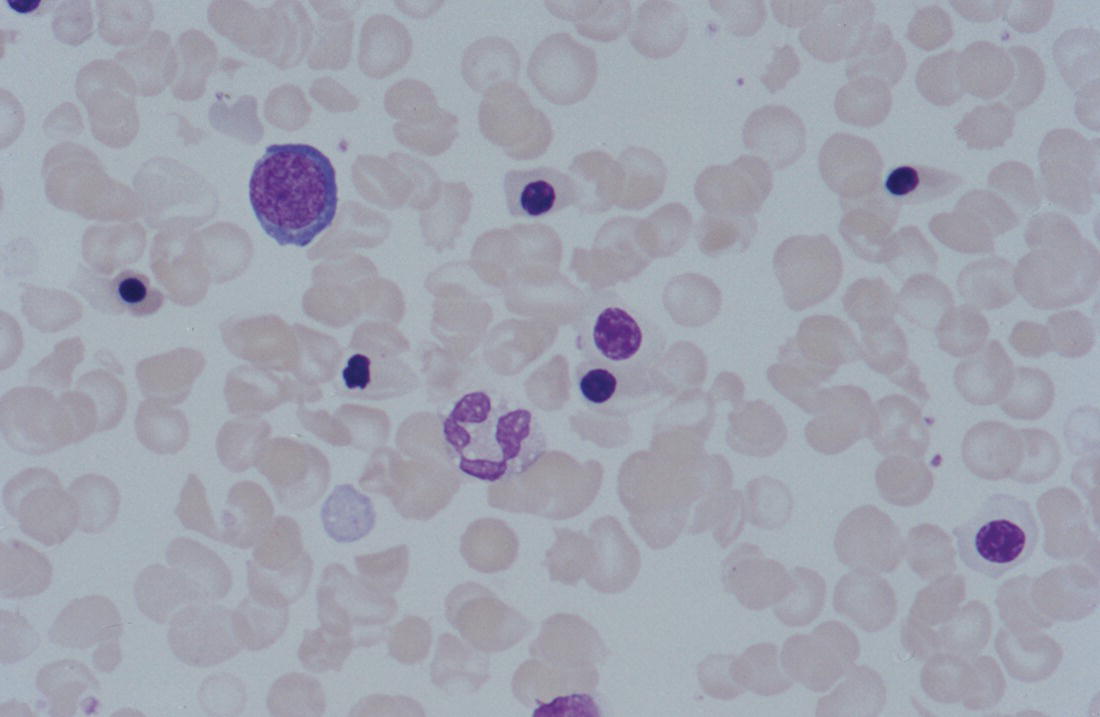
Immune neutropenia
Alloimmune neonatal neutropenia
Nature of condition
Cytogenetic or genetic abnormality implicated (inheritance)
Isolated severe congenital neutropenia
ELANE (AD)
HAX1 (AR)
CSF3R (AR)
GFI1 (AD)
VPS45 (AR)
JAGN1 (AR)
TCIRG1 (AD)
SRPS4 (AD)
CSF3R (AR or AD)
Congenital neutropenia associated with immunodeficiency or syndromic features
Shwachman syndrome*
SBDS (AR) or DNAJC21 (AR)
Reticular dysgenesis
AK2 (AR)
Dursun syndrome
G6PC3 (AR)
Barth syndrome
TAFAZZIN (XLR)
CLPB syndrome**
CLPB (AD)
Wiskott–Aldrich syndrome
WAS (XLR)
Neonatal neutropenia due to maternal autoantibodies
Inherited congenital or neonatal neutropenia
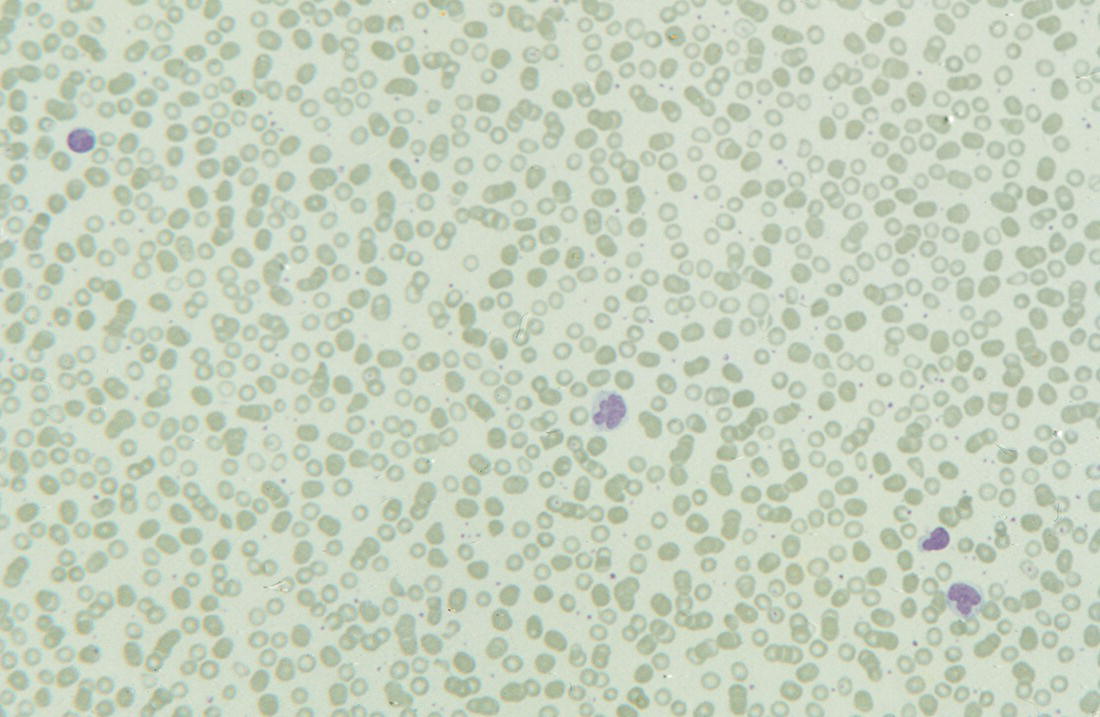
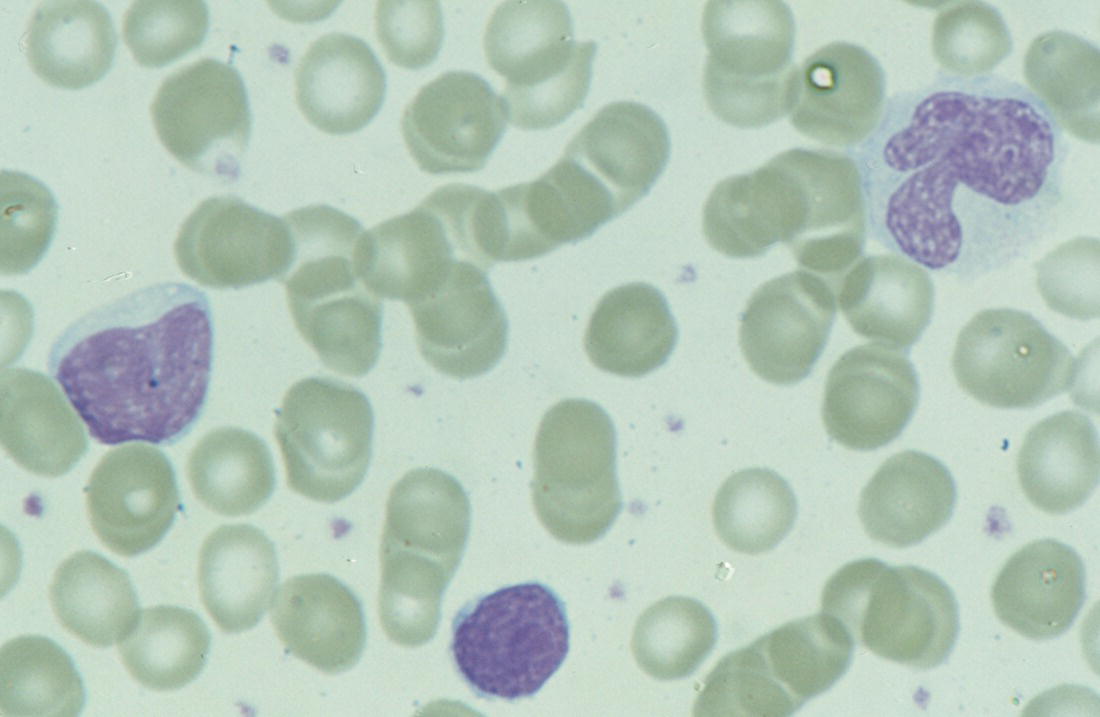
Severe congenital neutropenia due to ELANE mutation
Shwachman–Diamond syndrome
Severe congenital neutropenia due to glucose‐6‐phosphate transporter (SLC37A4) mutation
Severe congenital neutropenia due to HAX1 mutation
Haematological features of neonates with Down syndrome
Congenital leukaemia
Condition
Acquired GATA1 mutation
Haematological features
Clinical features
Transient abnormal myelopoiesis
Yes
100% of cases
Hb usually normal (anaemia rare)
Platelet count may be increased, in the normal range or reduced
Leucocytosis usual but WBC range overlaps with DS neonates without TAM
Blasts >10%
Megakaryocyte fragments usually present in blood film
BM blasts usually not increased (BM examination unhelpful)
Hepatosplenomegaly 40%
Skin rash 20%
Pleural/pericardial effusion ± ascites 10%
Jaundice 70–80%
Silent transient abnormal myelopoiesis
Yes
100% of cases
Hb increased or normal
Platelets normal or reduced
WBC normal
Blasts <10%
No increase in frequency of hepatosplenomegaly, jaundice, skin rash, pleural/pericardial effusions or ascites compared to neonates with DS who lack GATA1 mutations
Trisomy 21 (no evidence of TAM or silent TAM)
No
Compared with neonates without DS:
Hb increased (24% polycythaemic, Hct >0.65)
Platelets lower (40% thrombocytopenic)
WBC counts higher due to increased neutrophils, basophils and monocytes
98% have peripheral blood blasts (<15%)
No clinical features are specific for TAM as ~60% of neonates with trisomy 21 have jaundice and up to 10% have hepatosplenomegaly, rash, pleural/pericardial effusion or ascites secondary to medical complications in the absence of GATA1 mutations
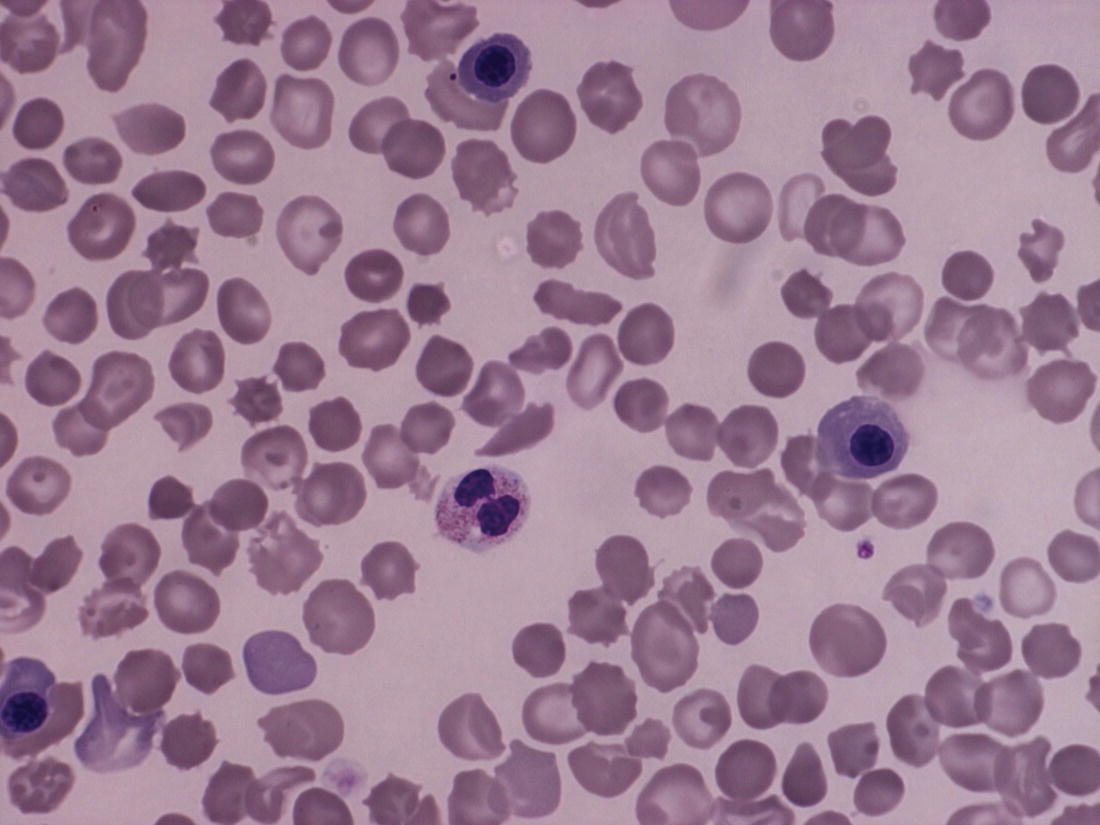
Leukaemia and preleukaemia in neonates with Down syndrome
Overview and definition
Clinical features
Haematological features
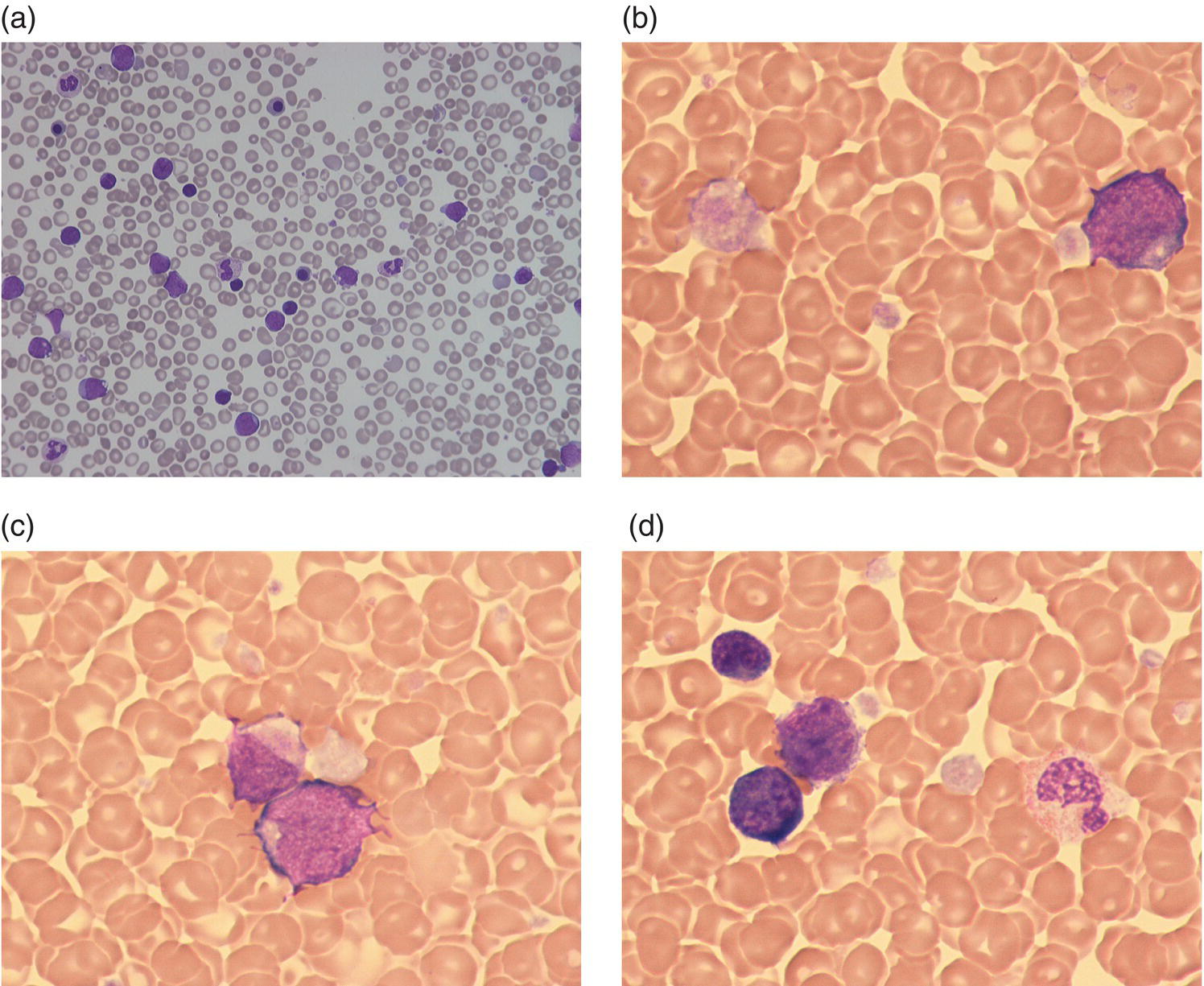
Molecular genetics
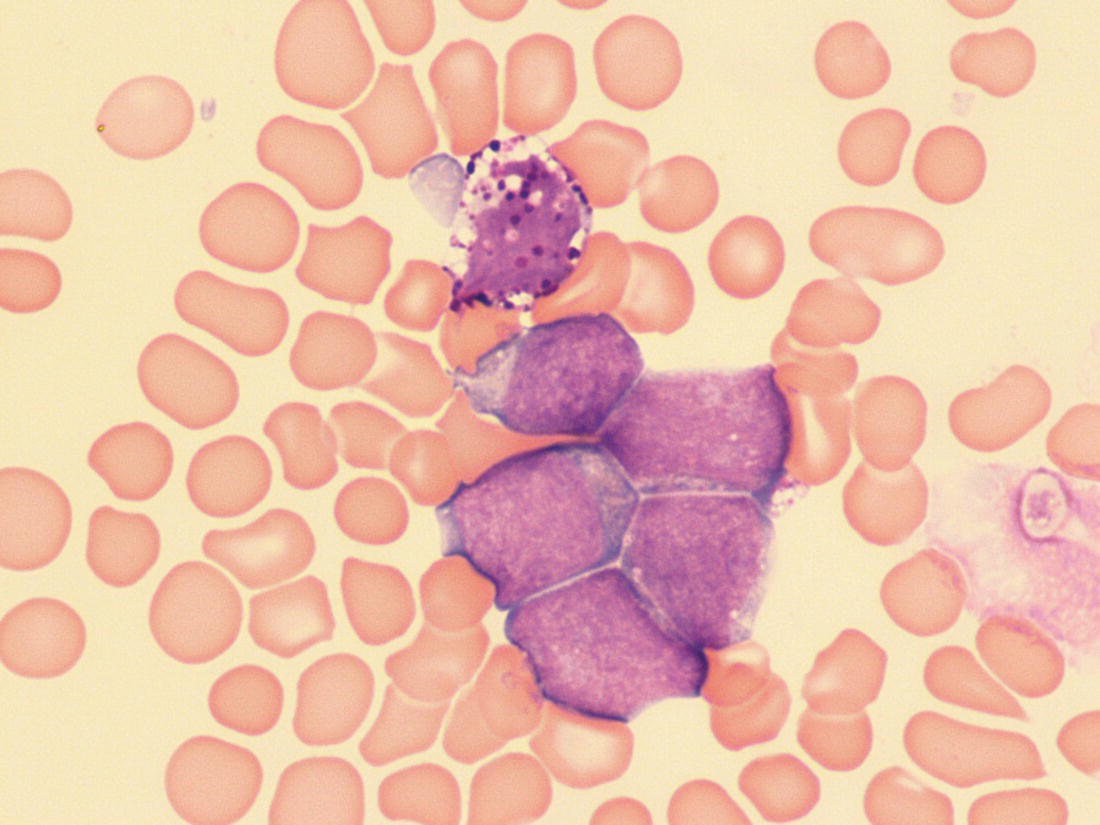
Natural history and prognosis
Fetal presentation of transient abnormal myelopoiesis
Acute leukaemia in neonates without Down syndrome
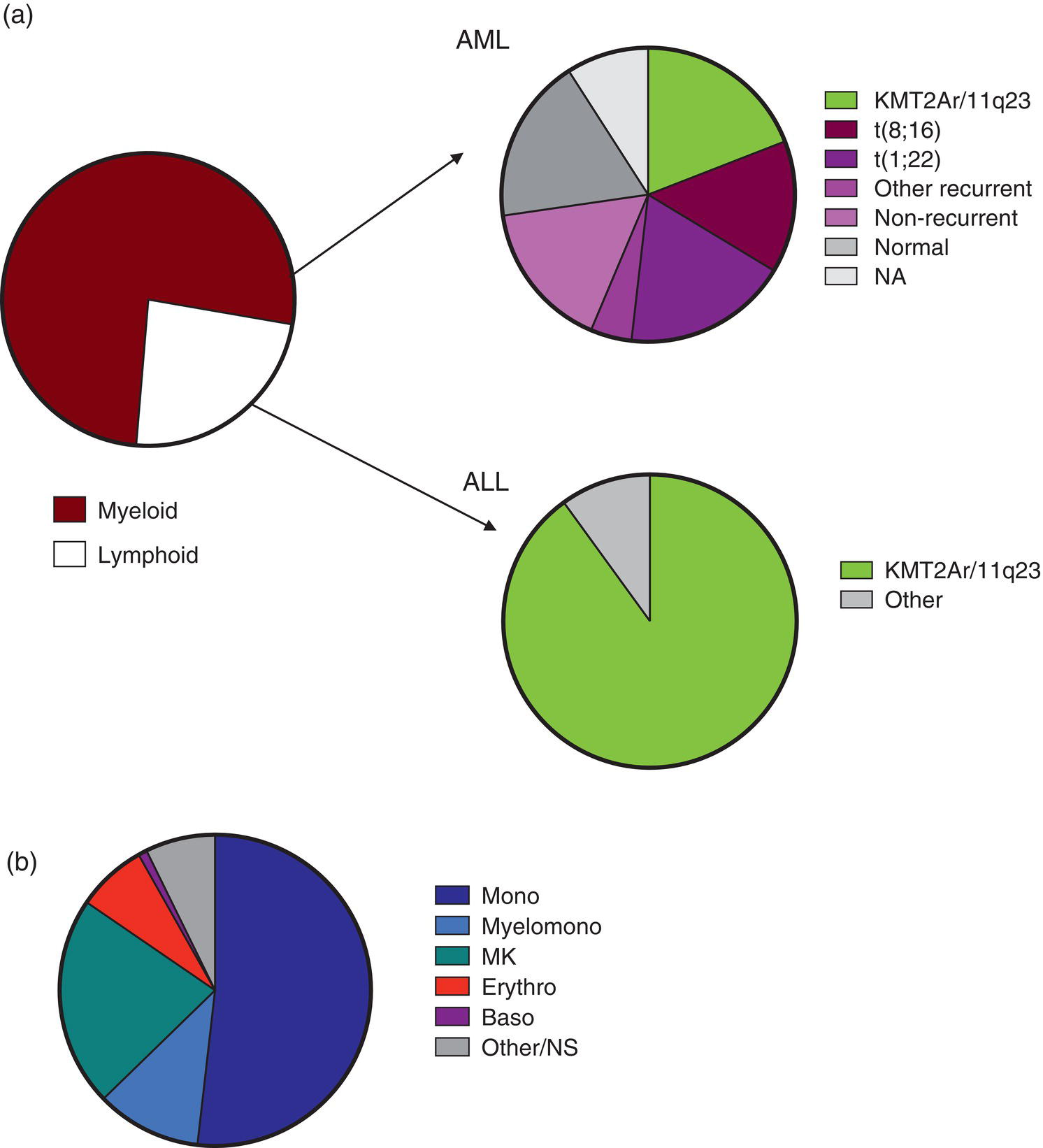
Condition
Molecular abnormality
Characteristic features
AML
With rearrangement of KMT2A at 11q23.3
t(4;11)(q21;q23.3); KMT2A‐AFF1
Acute myelomonocytic or monoblastic leukaemia
t(9;11)(p21.3;q23.3); KMT2A‐MLLT3
Mainly acute monoblastic leukaemia
t(10;11)(p12.31;q23.3); KMT2A‐MLLT10
Acute myelomonocytic, monocytic or monoblastic leukaemia
t(11;19)(q23.3;p13.11); KMT2A‐ELL
Acute myelomonocytic or monoblastic leukaemia
t(11;19)(q23.3;q13.3); KMT2A‐MLLT1
Acute monocytic leukaemia
t(1;11)(p36;q23); KMT2A rearranged
Acute monoblastic leukaemia
t(1;11)(q21;q23); KMT2A rearranged
AML not further specified
Other
t(1;22)(p13.3;q13.1); RBM15‐MRTFA (previously RBM15‐MKL1)
Acute megakaryoblastic leukaemia
t(8;16)(p11.2;p13.3); KAT6A‐CREBBP and variants
Mainly acute monoblastic leukaemia†
t(8;21)(q22;q22.1); RUNX1‐RUNX1T1
Miscellaneous including:
t(5;6)(q31;q21)‡
RUNX1 duplication
Acute monocytic/monoblastic leukaemia
ALL
t(4;11)(q21;q23.3); KMT2A‐AFF1 and variants
Pro‐B (early precursor) ALL
t(11;19)(q23.3;p13)
ALL (pro‐B or common/pre‐B)
MPAL
t(4;11)(q21;q23.3); KMT2A‐AFF1
Pro‐B/acute monoblastic leukaemia
BPDCN
t(2;17;8)(p23;q23;p23); CLTC‐ALK1
Blastic plasmacytoid dendritic cell neoplasm
Clinical features
Malignant and premalignant disorders
Leukaemia cutis
Transient abnormal myelopoiesis
Haemophagocytic lymphohistiocytosis
Langerhans cell histiocytosis
Metastatic neuroblastoma
Disseminated juvenile xanthogranuloma147
Congenital infections
Syphilis
Toxoplasmosis
Rubella
Cytomegalovirus
Coxsackievirus
Severe fetal anaemia
Haemolytic disease of the fetus and newborn
Hereditary spherocytosis
εγδβ thalassaemia
Twin‐to‐twin transfusion
Fetomaternal haemorrhage
Haematological features
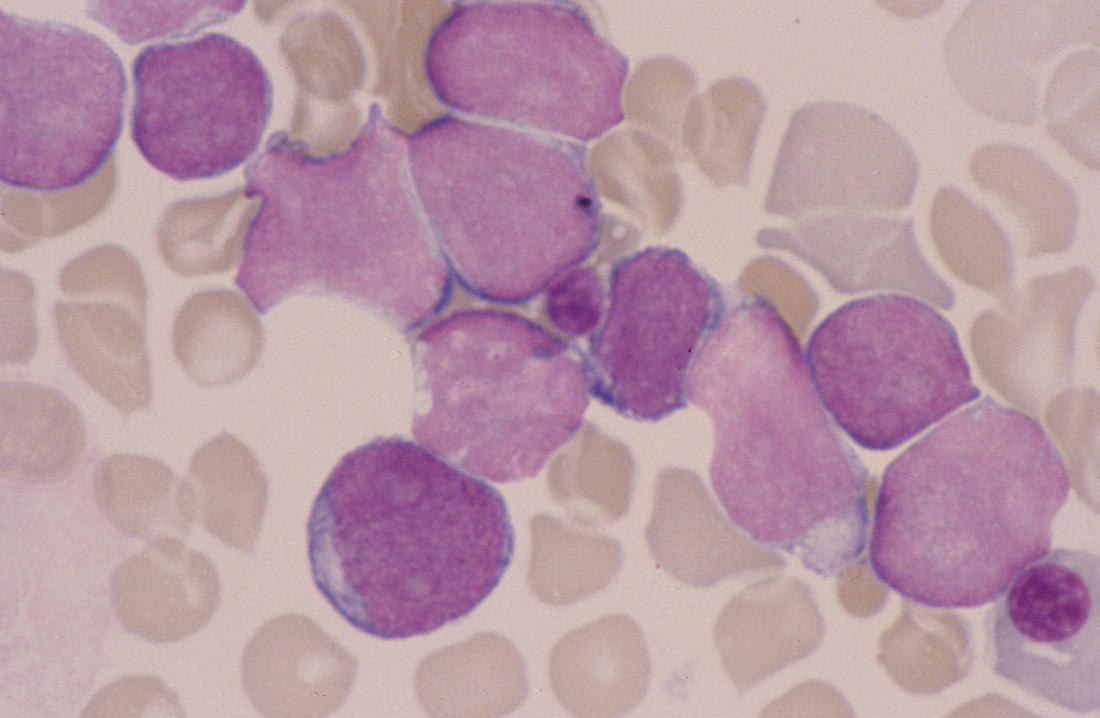
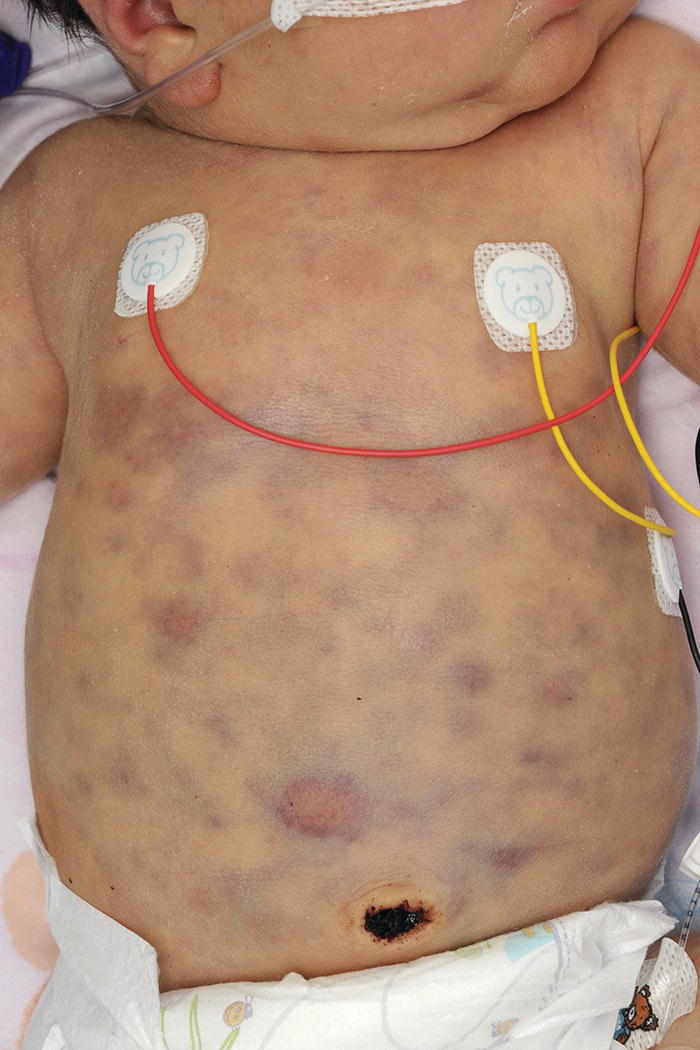
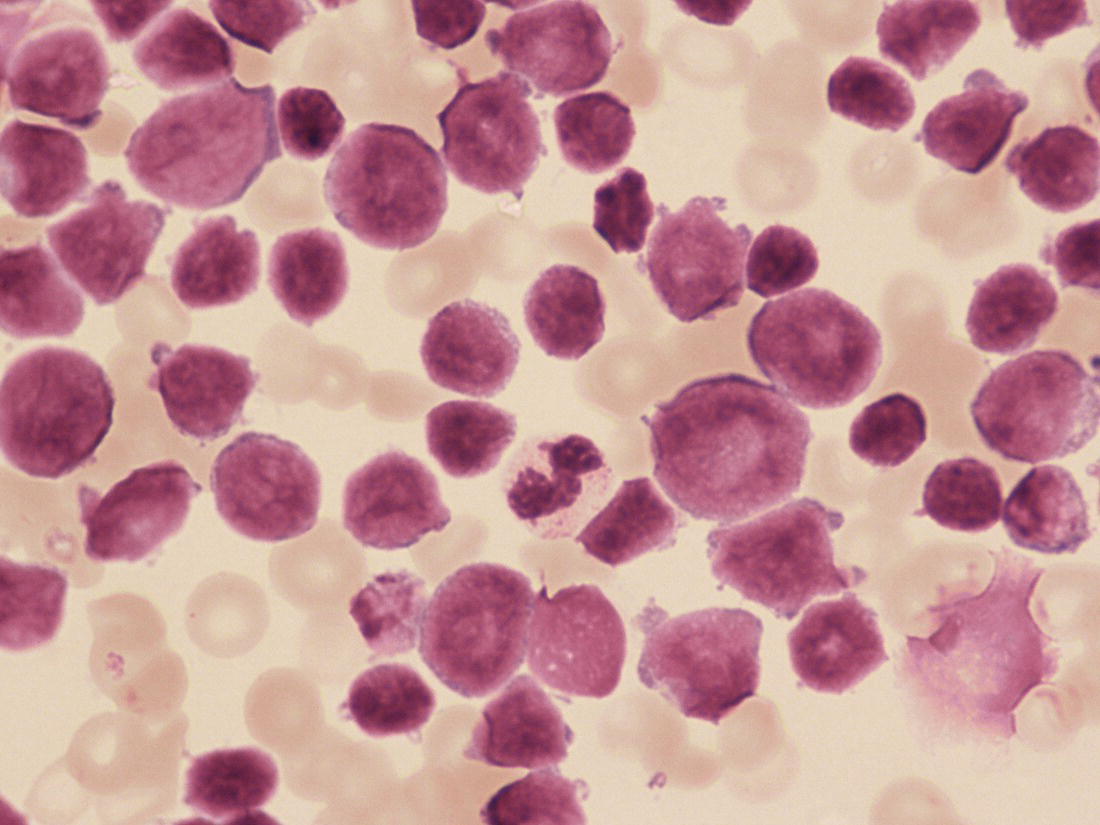
Molecular genetics
Treatment
Supportive care
Chemotherapy and haemopoietic stem cell transplantation
Juvenile myelomonocytic leukaemia and Noonan syndrome myeloproliferative disorder
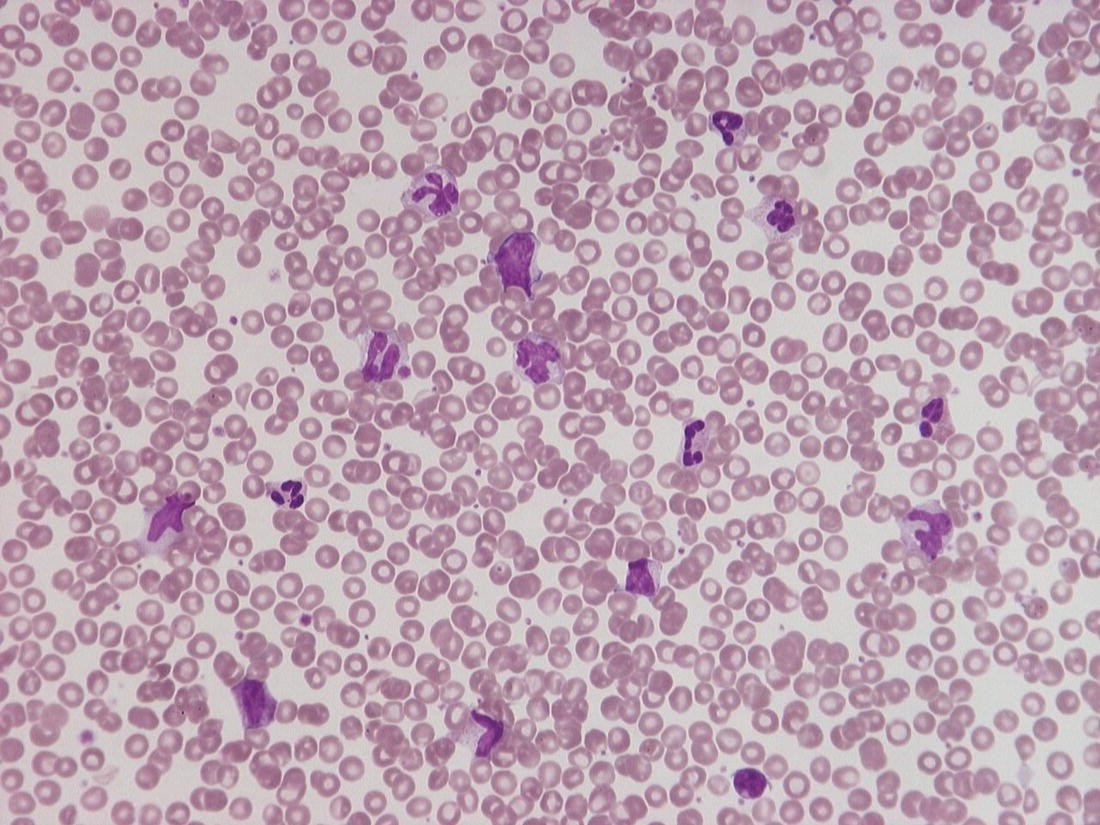
Illustrative cases
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


