Anaemia is generally defined as a haemoglobin concentration or haematocrit at least two standard deviations below the mean or below the 5th percentile compared with reference values for age and, if appropriate, gender. For neonates these normal values vary with gestational age at birth (see Table 1.2). For term babies, a haemoglobin concentration (Hb) below 140 g/l is abnormal and could be considered as anaemia, while in preterm babies born before 30 weeks’ gestation the lower limit of normal is 115 g/l. As a first step in assessing the significance and cause of neonatal anaemia, the laboratory result should be considered in light of the clinical findings. Detailed investigations will not be necessary for every neonate with an Hb below the normal range. It is important to consider the possibility of artefactual results, for example due to in vitro blood clotting in the sample tube. In addition, haemoglobin measurements on heel‐prick samples may be misleading as they can be up to 40 g/l lower than venous samples in the first few hours of life, particularly in term babies.1 Another major factor determining the Hb at birth is the timing and position of the baby at the time of clamping of the cord (babies held below the placenta receive more blood). In a term baby, the placental vessels typically contain around 100 ml of blood at birth. Since up to 25% of the placental blood is ‘transfused’ into the baby within the first 15 seconds and 50% by the end of the first minute, even a short delay has the potential to increase the volume of placental blood transfused to the neonate by 50 ml.2 Indeed, as discussed later in this chapter, a policy of deliberate delay to cord clamping has been investigated in several studies as a strategy for reducing neonatal red cell transfusion requirements. The clinical significance of neonatal anaemia depends both upon the haemoglobin concentration and on the ability of the haemoglobin to deliver oxygen to the tissues. This, in turn, depends on the characteristics of the haemoglobin–oxygen dissociation curve. In neonates, the two most important parameters that determine the position of the haemoglobin–oxygen dissociation curve are the concentration of haemoglobin F and the level of 2,3‐diphosphoglycerate (2,3‐DPG) within the red blood cells. At birth, when the predominant haemoglobin is haemoglobin F and the levels of 2,3‐DPG are low, the curve shifts to the left, i.e. the affinity of haemoglobin for oxygen is high and so less oxygen is released to the tissues. This can cause particular problems for the most preterm neonates (<28 weeks’ gestation) in which haemoglobin F forms more than 90% of the total haemoglobin. As the proportion of haemoglobin F falls, either physiologically or following red cell transfusion, and 2,3‐DPG levels rise, the haemoglobin–oxygen dissociation curve gradually shifts to the right and oxygen delivery to the tissues improves. Table 2.1 Mechanisms of fetal and neonatal anaemia Anaemia may be caused by reduced red cell production, increased red cell destruction (haemolysis), blood loss or a combination of these factors (Table 2.1). While simple investigations, in particular the full blood count and blood film, remain the cornerstone for identifying the causes of neonatal anaemias, accurate diagnosis of the rarer inherited anaemias has been greatly facilitated by the increasing speed and availability of next generation sequencing.3,4 Reduced red cell production in the neonate may result either from a virtually complete absence of red cells and erythroid progenitors (red cell aplasia) or from qualitatively abnormal red cell production where erythroid progenitors fail to differentiate normally into mature red cells (ineffective erythropoiesis). Red cell aplasia and anaemias due to ineffective erythropoiesis (often also dyserythropoietic) are uncommon in the neonatal period but are important to recognise promptly because they are often severe and, for inherited disorders, are a cause of life‐long ill health. The most common causes of red cell aplasia are congenital infections (largely due to parvovirus B19) and the genetic disorder Diamond–Blackfan anaemia (DBA). As both parvovirus B19 and DBA cause highly selective effects on the erythroid lineage, the white cell count and platelet count are usually normal in affected neonates. However, reduced red cell production may also occur as part of a general failure of haemopoiesis. In this situation the neutrophil count, platelet count and Hb are all reduced, for example in congenital leukaemias (see Chapter 3). Apart from parvovirus B19, other infections that can cause anaemia due to reduced red cell production include cytomegalovirus (CMV), toxoplasmosis, congenital syphilis, rubella and herpes simplex, although the anaemia and reticulocytopenia are usually relatively mild.5,6 The main diagnostic clues to reduced red cell production in a neonate with anaemia are the combination of a low reticulocyte count (<20 × 109/l) with a negative direct antiglobulin test (DAT or Coombs test) (Table 2.2). The principal causes of anaemia due to reduced red cell production in neonates are shown in Table 2.3. Table 2.2 Diagnostic clues to red cell aplasia in neonates with anaemia Table 2.3 Causes of failure of red cell production in neonates with anaemia * If molecular analysis is readily available and clinical suspicion is high, it may be preferable to perform the bone marrow aspirate after molecular analysis as this may allow additional tests (e.g. electron microscopy for suspected CDA) to be performed at the same time. CDA, congenital dyserythropoietic anaemia; DNA, deoxyribonucleic acid; Hb haemoglobin concentration; Ig, immunoglobulin; MCV, mean cell volume; NRBC, nucleated red blood cells. Population studies indicate that 25–50% of women of reproductive age remain susceptible to parvovirus B19.7 When the infection occurs in pregnant women who lack immunity, particularly during the first 20 weeks of pregnancy, severe fetal and/or neonatal anaemia due to red cell aplasia develops in up to one‐third of affected pregnancies.7 Although the majority of fetuses are unaffected, it is important to recognise that parvovirus B19 is the cause of 6–7% of cases of non‐immune hydrops fetalis and is the commonest infective cause of this condition.8,9 Typically, the affected fetus or neonate has marked reticulocytopenia (often <10 × 109/l) and in severe cases thrombocytopenia can also occur.5 The red blood cells are morphologically normal and an important diagnostic clue is the absence of polychromasia and circulating nucleated red blood cells (NRBC) despite the presence of severe anaemia (Fig. 2.1). The diagnosis is made by maternal serology and demonstration of B19 DNA in the fetus or neonate by dot‐blot hybridisation or polymerase chain reaction (PCR) of peripheral blood (bone marrow aspiration for morphology and parvovirus B19 PCR may be necessary in difficult cases).9 Fig. 2.1 Blood film of a term neonate with anaemia due to parvovirus B19 infection showing normochromic normocytic red cells with occasional schistocytes and an absence of polychromasia. A normal neutrophil is also shown. May–Grünwald–Giemsa (MGG), ×100. Management of fetal parvovirus B19 infection depends on the severity of the anaemia and whether or not the fetus is hydropic. A recent systematic review and meta‐analysis showed that the presence of hydrops was the main determinant of mortality and adverse perinatal outcome.9 The majority of hydropic fetuses were treated by intrauterine transfusion (IUT) whereas this was only used as treatment for approximately 26% of non‐hydropic fetuses. The outcome was very good for non‐hydropic fetuses, with an in utero mortality rate of about 2% and no neonatal deaths. However, almost 25% of fetuses who presented with hydrops due to parvovirus B19 infection died despite IUT, the majority of them prior to delivery, and a further 10% had a poor neurodevelopmental outcome. In addition, a small number of neonates affected by intrauterine parvovirus B19 infection develop chronic red cell aplasia, with or without evidence of persistent B19 DNA by PCR.5 In such cases, intravenous immunoglobulin (IVIg) has been reported to be effective in treating the anaemia and eradicating persistent viral infection in some,10 but not all, cases.11,12 These two groups of conditions differ in that in red cell aplasia the marrow is hypocellular with reduced progenitor cell numbers, whereas in dysplasia erythropoiesis is active but ineffective and dysplastic. Apart from DBA, the genetic causes of congenital red cell aplasia and dysplasia are extremely rare. They include congenital dyserythropoietic anaemia (CDA) and Pearson syndrome. The other inherited bone marrow failure syndromes, such as Fanconi anaemia, are rarely manifest at birth. Similarly, congenital atransferrinaemia nearly always presents later in childhood or in adulthood, although rare cases presenting as early as 2 months have been described.13 These genetic causes of congenital red cell aplasia/dysplasia can normally be distinguished from each other through a combination of their distinctive bone marrow morphology, family history and the presence, or not, of additional congenital anomalies. However, precise diagnosis increasingly relies on targeted or whole genome sequencing gene panels on neonatal and parental blood samples.14,15 The incidence of DBA is estimated at 5–7 cases per million live births.16 DBA has a clear family history in 20% of cases (autosomal dominant or recessive) and appears to be sporadic in the remaining 80%.16 It usually presents as increasing anaemia over the first few weeks or months of life but approximately 25% of cases present with anaemia at birth16,17 and the most severe cases manifest as second‐trimester anaemia or hydrops fetalis.18,19 Around 40% of neonates with DBA have associated congenital anomalies, particularly craniofacial dysmorphism, neck anomalies, congenital heart disease, urogenital anomalies and thumb malformations, such as a triphalangeal thumb, while around 25% have intrauterine growth restriction (IUGR).17 Several of these anomalies are also seen in Fanconi anaemia. However, the diagnosis is usually clear as Fanconi anaemia very rarely presents in the fetus or the newborn and the blood count usually shows pancytopenia rather than anaemia. Recent studies indicate that about 75% of cases of DBA are due to defects in the synthesis and function of structural ribosomal proteins (RP). To date, heterozygous mutations in 19 different genes with good evidence of pathogenicity have been reported in DBA patients.14 The majority of cases are associated with pathogenic variants in RP genes that encode either the small RP subunit (RPS7, RPS10, RPS17, RPS19, RPS24, RPS26, RPS27, RPS28 and RPS29) or the large RP subunit (RPL5, RPL11, RPL15, RPL18, RPL26, RPL27, RPL35 and RPL35a). RPS19 mutations are the commonest (25% of all DBA). Recently, mutations in two X‐linked genes (GATA1 and TSR2) have also been shown to cause DBA in a small proportion of patients.20,21 In up to 50% of patients, the molecular basis for the disease has proved elusive, even after using a targeted panel to screen all 80 RP genes for mutations11 or using whole exome sequencing.22 The blood film in neonates with DBA typically shows normochromic red cells with an absence of polychromasia and nucleated red cells despite severe anaemia (Case 2.1, see page 88). Reticulocytopenia is usually severe but reticulocyte counts of 20–30 × 109/l are sometimes seen. These findings are not specific for DBA but if investigations exclude parvovirus B19 infection and/or if there are typical DBA‐associated congenital anomalies or a relevant family history, neonates with persistent anaemia and a low reticulocyte count should have specific investigations for DBA. These include molecular testing for RP gene mutations (many laboratories now offer next generation sequencing for DBA diagnosis) and/or a bone marrow examination, which will demonstrate the lack of erythroid precursors characteristic of this disease. In case of doubt, or where molecular testing is not available, measurement of red cell adenosine deaminase (ADA) levels, which are elevated in most affected patients, may be useful.14,23 In the neonatal period, the only treatment of DBA is red cell transfusion, although corticosteroids are used in older infants and children.23 DBA is almost always a life‐long condition although rare spontaneous remissions later in childhood or in adulthood have been reported. Bone marrow transplantation is the only long‐term curative option but carries a significant mortality and this must be balanced against the morbidity of life‐long red cell transfusion or corticosteroids.23 This rare condition nearly always presents within a few days or weeks of birth. It is caused by mutations or deletions in mitochondrial DNA and presents with anaemia, neutropenia, thrombocytopenia and failure to thrive.24 Most affected neonates have low birthweight with metabolic acidosis and exocrine pancreatic deficiency; abnormal liver and renal function are common. The bone marrow is typically hypercellular with erythroid and megakaryocyte dysplasia (Fig. 2.2), including micromegakaryocytes, and basophilic stippling in erythroblasts (Fig. 2.3). The most useful haematological clue to the diagnosis is the highly characteristic vacuolation of early erythroid and granulocytic cells in the marrow aspirate (Figs 2.2 and 2.4). Vacuolated dysplastic normoblasts and neutrophils can also be seen occasionally in peripheral blood films (Figs 2.5 and 2.6). Confirmation of the diagnosis requires mitochondrial genome sequencing, which can be performed very rapidly in specialised centres. It is important to note that whole exome sequencing may miss mitochondrial gene deletions.25 Interestingly, some reports have identified large mitochondrial gene deletions in a small sub‐group of patients considered to have DBA. When these cases were re‐evaluated they were found to be cases of Pearson syndrome which had been misdiagnosed as DBA.26 Unfortunately, the prognosis for children with this condition is very poor, with few surviving beyond the second year of life. Fig. 2.2 Bone marrow film of a neonate with Pearson syndrome showing vacuolation of erythroid and myeloid cells and a dysplastic megakaryocyte. MGG, ×100. Fig. 2.3 Basophilic stippling in a bone marrow erythroblast (centre field) from a neonate with Pearson syndrome. MGG, ×100. Fig. 2.4 Dyserythropoietic erythroblasts in the bone marrow of a neonate with Pearson syndrome showing prominent vacuolation of a proerythroblast. MGG, ×100. Fig. 2.5 Peripheral blood film of a neonate with Pearson syndrome showing vacuolation in a dysplastic neutrophil. MGG, ×100. Fig. 2.6 Peripheral blood film of a neonate with Pearson syndrome showing a dysplastic neutrophil with vacuolation. MGG, ×40 Congenital dyserythropoietic anaemias are now conventionally classified into three main types (I, II and III) with additional variant types of CDA that do not conform to the criteria for types I–III being recognised (reviewed in reference 27). While most cases of CDA present during childhood, type I, II and some variant CDA can all present with neonatal anaemia and/or hydrops fetalis in association with fetal anaemia.28–31 Although in older children the anaemia is typically macrocytic, the blood film in neonates with CDA usually shows normocytic red cells with variable anisopoikilocytosis (Fig. 2.7; Case 2.2, see page 90). The peripheral blood findings are non‐specific and often the most notable feature is the relative absence of polychromasia despite moderate or even severe anaemia. The bone marrow aspirate shows increased erythropoiesis with varying degrees – depending on the type of CDA – of dyserythropoiesis, including megaloblastic changes, binucleate or multinucleate erythroblasts, internuclear chromatin bridges, nuclear irregularity and pale staining chromatin. Diagnosis on the basis of morphology alone is unreliable except in very experienced hands and molecular analysis, with or without electron microscopy, is essential for genetic counselling and planning treatment.3 Next generation sequencing is increasingly used to make a definitive diagnosis.32 Fig. 2.7 Congenital dyserythropoietic anaemia (CDA): (a) blood film of a term neonate with CDA showing normochromic red cells with anisopoikilocytosis (note the absence of polychromasia); (b) bone marrow film of the same child at age 3 months showing cytoplasmic bridging between two basophilic normoblasts and a normoblast with a dumbbell‐shaped nucleus. MGG, ×100. Most cases of CDA type I are autosomal recessive and caused by bi‐allelic mutations of either the CDAN1 gene or the CDIN1 (C15orf41) gene, although the genetic basis for occasional cases remains unknown.32 CDA type I is not usually diagnosed in the neonatal period. However, as retrospective series suggest that anaemia and jaundice are common in neonates subsequently diagnosed with the condition, CDA type I should be considered in the differential diagnosis of unexplained, persistent neonatal anaemia.33 CDA type I also occasionally presents with severe anaemia in utero and should be considered in the differential diagnosis of unexplained hydrops fetalis.34–36 Some patients have nail or skeletal abnormalities, particularly syndactyly of the fingers or toes which may provide a useful clue to the diagnosis.37 The characteristic haematological features of CDA type I are macrocytic anaemia together with the presence of binucleate macrocytic erythroblasts and internuclear bridging in the bone marrow.32 Despite the severity of the anaemia in the neonatal period, the majority of infants become transfusion‐independent by the age of 4 months.33 CDA type II is an autosomal recessive anaemia caused by bi‐allelic mutations in the SEC23B gene. The anaemia is typically normocytic with a hypercellular bone marrow containing bi‐ and multinucleated erythroblasts.38 Approximately 60% of patients have anaemia and jaundice in the neonatal period, although the diagnosis is often unclear until later in childhood, and almost half of affected individuals have a history of neonatal red cell transfusion.38 Occasional cases of severe fetal anaemia and hydrops have been reported.28,39 The severity of CDA type II varies from life‐long transfusion dependence (in about 20% of cases) to asymptomatic disease.38 CDA type III, which is very rare, is characterised by moderate to severe macrocytic anaemia and marked erythroblast multinuclearity on bone marrow examination. This type of CDA has not been reported to present in neonates. CDA type III is caused by bi‐allelic mutations in the KIF23 gene.27 Several variant forms of CDA have also been described, some of which present in fetal or neonatal life. These include an X‐linked syndrome of severe anaemia and thrombocytopenia due to somatic mutations in the GATA1 transcription factor gene40 and a severe anaemia presenting in fetal or neonatal life due to bi‐allelic mutations in the KLF1 gene.31,41 Although the majority of affected individuals with bi‐allelic KLF1 mutations present with severe normocytic anaemia and jaundice at or before birth, occasional patients with milder disease do not present until adulthood.31 Despite the severity of the fetal/neonatal anaemia, only occasional neonates with bi‐allelic KLF1 mutations remain transfusion‐dependent.31,42 Variant forms of CDA may also present with neonatal anaemia in conjunction with other syndromic features, including mutations in MVK, the mevalonate kinase gene,43 mutations in COX4I2, encoding a component of the cytochrome c oxidase complex,44 and mutations in the VPS4A gene which predominantly cause severe neurodevelopmental delay.45 Haemolysis is the commonest cause of anaemia in term babies and second only to anaemia of prematurity in preterm babies. The presence of haemolysis is an important sign because, even if mild or transient, this may be the first evidence of a more serious disorder that would otherwise be manifest later in infancy or childhood (e.g. red cell enzymopathies). In addition, there may be implications for future siblings (e.g. haemolytic disease of the fetus and newborn [HDFN]). Initial investigations of suspected haemolysis in a neonate should include a full blood count, a blood film, a reticulocyte count and a Coombs test. The main diagnostic clues suggesting a haemolytic anaemia are increased numbers of reticulocytes and/or circulating nucleated red cells, unconjugated hyperbilirubinaemia and a positive Coombs test (since this is only positive in cases of immune haemolytic anaemia) (Table 2.4). In most cases there are also characteristic changes in the morphology of the red cells in a blood film (e.g. spherocytes in hereditary spherocytosis; see Fig. 2.13). The main cause of immune haemolytic anaemia is HDFN (Table 2.5). Maternal autoimmune haemolytic anaemia (AIHA) may also cause a positive Coombs test in the neonate but clinical evidence of haemolysis or anaemia in the baby is unusual in this situation. The main causes of non‐immune neonatal haemolysis are red cell membrane disorders and red cell enzymopathies (see Table 2.5). Occasionally, neonatal anaemia occurs in haemoglobinopathies, and haemolysis is one of the causative factors (see below). A number of congenital and nosocomial infections can also cause neonatal haemolytic anaemia including CMV, toxoplasmosis, congenital syphilis, rubella, herpes simplex and malaria. Although simple investigations are often sufficient to identify the cause of haemolysis in a neonate, rarer disorders require specialist investigations, including next generation sequencing. Haemolytic disease of the fetus and newborn results from maternal alloantibodies against paternally derived fetal red cell antigens crossing the placenta and binding to the fetal red cells, causing haemolysis and anaemia. Overall, recent estimates indicate that up to 1 in 600 pregnancies is affected by maternal red cell alloimmunisation.46 The principal alloantibodies causing HDFN are those against ABO blood group antigens (anti‐A, anti‐B), Rh antigens (anti‐D, anti‐c or anti‐E), Kell antigens (mainly anti‐K), Kidd antigens (anti‐Jka or anti‐Jkb), Duffy antigens (anti‐Fya) and antigens of the MNS blood group system (including anti‐U). ABO HDFN due to anti‐A or anti‐B antibodies is the most common type of immune haemolytic disease in the perinatal period but is usually a fairly mild disease.47 In contrast, anti‐D is the most frequent alloantibody that causes severe haemolytic anaemia, affecting 1 in 1000 pregnancies even in countries where routine antenatal and postnatal anti‐D prophylaxis has been widely implemented.48 Anti‐K antibodies are less common but can cause severe fetal and neonatal anaemia because they inhibit erythropoiesis as well as causing haemolysis.49 Table 2.4 Diagnostic clues to haemolysis in neonates with anaemia and/or jaundice Table 2.5 Causes of neonatal haemolytic anaemias * Equivocal evidence to support haemolysis as a cause of anaemia. EMA, eosin‐5‐maleimide; FBC, full blood count; G6PD, glucose‐6‐phosphate; HPLC, high performance liquid chromatography; HUS, haemolytic uraemic syndrome; Ig, immunoglobulin: PK, pyruvate kinase; TTP, thrombotic thrombocytopenic purpura. This condition is almost always confined to offspring of women of blood group O or A2 who are group A or group B. In recent years, several cases of unanticipated ABO HDFN have arisen in neonates born to surrogate mothers, for example in a baby of blood group AB who was born to a group O surrogate mother and whose biological mother was group B.50 ABO alloimmunisation of the fetus is confined to the 1% of group O women who have high‐titre immunoglobulin G (IgG) antibodies. In contrast to HDFN due to other red cell antibodies, ABO HDFN frequently occurs during a first pregnancy and the recurrence rate in subsequent pregnancies is very high (almost 90%).51 In infants of European ancestry, haemolysis due to anti‐A is more common (1 in 150 births) than that due to anti‐B, which is more common in infants of African ancestry.52 In contrast to cases due to Rh antibodies, where anaemia usually accompanies jaundice, ABO haemolysis is usually characterised by hyperbilirubinaemia without significant anaemia. Several case reports, as well as personal experience, suggest that haemolysis due to anti‐B tends to be more severe than that due to anti‐A and may even cause hydrops fetalis53,54 (Case 2.3, see page 93). The blood film in ABO haemolytic disease characteristically shows very large numbers of spherocytes with little or no increase in NRBC (Figs 2.8 and 2.9) (Table 2.6). Fig. 2.8 Typical blood film of a term neonate with ABO haemolytic disease of the fetus and newborn (HDFN) showing large numbers of spherocytes. Note that no nucleated red blood cells (NRBC) are visible in this image and very few were seen on other fields. MGG, ×40. Fig. 2.9 Blood film of a preterm neonate with ABO HDFN showing large numbers of spherocytes as well as some macrocytes, echinocytes and polychromasia (note the absence of NRBC). MGG, ×100. Table 2.6 Laboratory features of ABO and Rh haemolytic disease of the fetus and newborn Rh haemolytic disease of the fetus and newborn remains the most common cause of severe disease.55 In neonates, Rh HDFN presents with early‐onset, rapidly worsening jaundice together with progressive anaemia. In severe cases there is hepatosplenomegaly. Occasionally, affected neonates develop widespread skin deposits due to extramedullary haemopoiesis, giving an appearance described as ‘blueberry muffin’ baby,56 although this finding is not specific to Rh HDFN as it is also seen in other conditions with extramedullary haemopoiesis, such as congenital leukaemia (see Figs 3.26 and 3.28). In contrast to ABO HDFN, the blood film in Rh HDFN typically shows marked polychromasia due to reticulocytosis, large numbers of circulating erythroblasts, which are often dyserythropoietic, and variable numbers of spherocytes (Figs 2.10–2.12) (see Table 2.6). In the most severe cases, Rh HDFN, presents in utero with rapidly progressive anaemia and hydrops fetalis.52 Fig. 2.10 Blood film of a term neonate with severe Rh HDFN showing large numbers of NRBC, including two early normoblasts, polychromasia and some spherocytes (compare with Fig. 2.8). MGG, ×40. Fig. 2.11 Blood film of a preterm neonate with Rh HDFN showing large numbers of erythroblasts, including several dyserythropoietic cells, and marked polychromasia (note the lower frequency of spherocytes compared with ABO HDFN). MGG, ×100. Fig. 2.12 Blood film of a term baby with severe Rh HDFN showing dyserythropoietic NRBC. MGG, ×100. Effective management of pregnancies affected by red cell alloimmunisation relies on close cooperation between obstetric, neonatal and haematology teams and many countries have developed national guidelines to ensure best practice for management of the fetus and the neonate.57–59 For fetuses at risk, monitoring to assess the degree of anaemia using cerebral middle cerebral artery (MCA) Doppler velocity measurements is typically performed every 1–2 weeks. Once anaemia is suspected, based on increasing MCA velocity, invasive testing via cordocentesis is performed to obtain an accurate measure of the fetal Hb/haematocrit, and IUT and/or delivery is planned, depending on the gestation, previous history and fetal wellbeing.52,59 All neonates at risk should have cord blood taken for measurement of Hb and bilirubin and a Coombs test, and should remain in hospital or under close outpatient supervision until hyperbilirubinaemia and/or anaemia have been properly managed.57–59 Neonates with haemolysis due to Rh HDFN should receive phototherapy from birth, because the bilirubin can rise steeply; this avoids the need for exchange transfusion in some infants. In haemolytic disease due to anti‐K, anaemia is usually more prominent than jaundice and only minimal phototherapy may be necessary despite severe anaemia.60 ABO HDFN usually just requires phototherapy, as significant anaemia is uncommon. The indications for exchange transfusion in HDFN are: Irradiated blood must be used for infants who have previously received IUT, to prevent the risk of transfusion‐associated graft‐versus‐host disease (TA‐GvHD). IVIg has been used to reduce the need for exchange transfusion but the value of this approach remains to be proven in clinical trials.62 Neonates with haemolytic disease due to anti‐Rh antibodies may develop ‘late’ anaemia at a few weeks of age, requiring ‘top‐up’ transfusion. Late anaemia occurs particularly in neonates who have received one or more IUTs.63 This is seen both in HDFN due to anti‐D and that due to anti‐K and is attributable, at least in part, to suppression by transfusion of the anaemia‐induced compensatory increase in erythropoiesis64 and possibly also by red cell alloantibodies present in maternal breast milk.65 Although recombinant EPO has been reported to reduce the need for top‐up transfusion for late anaemia, it is not effective when haemolysis is brisk and there are no data from randomised trials to support the routine use of EPO in this situation.66 Folic acid (500 μg/kg daily) should be given to all babies with haemolysis at least until they reach 3 months of age,60 but iron supplementation is unnecessary as the vast majority of neonates with red cell alloimmunisation have iron overload at birth.67 A number of genetic red cell membrane disorders can present in the neonatal period with haemolytic anaemia (Table 2.7). In neonates, haemolysis due to a red cell membrane disorder usually presents with jaundice, with or without anaemia, and the blood film shows abnormal red cell morphology. The changes in red cell morphology are often very characteristic and allow a provisional diagnosis to be made simply by reviewing the blood film. As for other genetic red cell disorders, the family history is usually extremely useful. In the majority of cases, it is possible to elicit a history of neonatal jaundice or anaemia in several generations of close family members or sometimes of splenectomy or cholecystectomy during childhood or early adulthood. A summary of the main features of red cell membrane disorders presenting with anaemia and/or jaundice is shown in Table 2.7. It is important to note that occasional cases of coinheritance of a red cell membrane disorder with a genetic defect in bilirubin metabolism, such as Gilbert syndrome, may have very severe hyperbilirubinaemia in the neonatal period.68 The main clues to the type of red cell membrane disorder are the family history and the characteristic shape of the red cells in a blood film. Many of the classical biochemical and functional tests used for the diagnosis of red cell membrane disorders, such as red cell membrane electrophoresis and osmotic fragility, have largely been replaced by molecular analysis using next generation sequencing, which is generally much more precise. However, newer functional tests, such as flow cytometric analysis of eosin‐5‐maleimide (EMA) dye binding or osmotic gradient ektacytometry, still play an important role, especially in cases where the pathogenicity of newly identified molecular variants in red cell membrane genes is uncertain.369–71 Hereditary spherocytosis is the most common of the red cell membrane disorders causing symptomatic anaemia, affecting 1 in 2000 to 1 in 5000 live births to parents of northern European extraction.72 The disorder is caused by mutations in one or more of five genes that encode red cell cytoskeleton and transmembrane proteins: α spectrin (SPTA1), β spectrin (SPTB), ankyrin‐1 (ANK1), band 3 anion transport protein (SLC4A1) and erythrocyte membrane protein band 4.2 (EPB42) (reviewed in reference 70). In 75% of families, hereditary spherocytosis is inherited in an autosomal dominant fashion. In the remaining cases, inheritance is non‐dominant and there is usually no family history, either because of autosomal recessive inheritance or because of the presence of a de novo autosomal dominant mutation. Autosomal recessive hereditary spherocytosis can be caused by bi‐allelic mutations in any of the five membrane genes, whereas autosomal dominant disease is seen only for mutations in SPTB, ANK1 and SLC4A1 (Table 2.8).73 The autosomal recessive forms of hereditary spherocytosis are typically very severe, presenting as hydrops fetalis or severe neonatal haemolytic anaemia (see Table 2.7).74–76 Table 2.7 Red cell membrane disorders presenting with anaemia and/or jaundice in the neonatal period Hb, haemoglobin concentration; HE, hereditary elliptocytosis; HS, hereditary spherocytosis; MCH, mean cell haemoglobin; MCHC, mean cell haemoglobin concentration; MCV, mean cell volume. Table 2.8 Genes mutated in autosomal recessive and autosomal dominant or sporadic hereditary spherocytosis (HS) * Proportion of cases in Europe and the USA; these frequencies are very different in the Japanese population, where 45–50% of HS cases are due to mutations in EPB42. Data summarised from reference 73. HS, hereditary spherocytosis. Most neonates with hereditary spherocytosis will have jaundice, although this is not usually severe and is easily controlled with phototherapy. However, neonates with hereditary spherocytosis who coinherit the trait for Gilbert syndrome, a common polymorphism in the promoter region of the uridine diphosphate–glucuronosyltransferase gene (UGT1A1), often develop severe jaundice and may need exchange transfusion to prevent the development of kernicterus.77,78 The Hb in hereditary spherocytosis is usually normal at birth but around one‐third of neonates will develop significant anaemia in the neonatal period.79 The anaemia is usually moderate (70–100 g/l). The diagnosis of hereditary spherocytosis may be made from the neonatal blood film alone where the family history is known, the film is good quality and other causes of spherocytosis have been excluded. Typically, the blood film in neonates with hereditary spherocytosis shows spherocytes with little or no polychromasia (Figs 2.13 and 2.14). Only one‐third of neonates with hereditary spherocytosis will have a reticulocyte count of >10% and circulating NRBC are not usually increased unless there is moderate or severe anaemia.80 The diagnosis of hereditary spherocytosis is sometimes difficult in neonates, especially in milder cases, because low numbers of spherocytes may be seen in healthy babies for the first few weeks of life. In addition, the blood film typically has an appearance identical to that of ABO HDFN (see Fig. 2.14b). Although the Coombs test is nearly always positive in ABO HDFN, occasional neonates will have both hereditary spherocytosis and ABO incompatibility. For these reasons, it is advisable to confirm suspected cases of hereditary spherocytosis. The most reliable and widely used method, which has been extensively validated in neonatal samples, is the EMA binding test. This is a rapid, flow cytometry‐based test that measures the uptake of the fluorescent EMA by band 3 in the red cell membrane. Reduced EMA dye binding is a very sensitive and specific screening test for certain red cell membrane defects, which is positive in about 90% of cases of hereditary spherocytosis, including neonates.81,82 In most cases no other diagnostic tests will be required. However, in difficult cases, for example where the clinical features are atypical and/or the family history is either unavailable or does not fit the clinical picture, further investigations should be considered, including next generation sequencing.70 Fig. 2.13 Blood film of a neonate with hereditary spherocytosis showing spherocytes and one button‐mushroom poikilocyte. MGG, ×50. Fig. 2.14 (a) Blood film of a neonate with hereditary spherocytosis showing moderate numbers of spherocytes (note the absence of polychromasia and NRBC in this case where the haemolysis was very mild). MGG, ×100. (b) Blood film of a neonate with ABO HDFN showing almost identical appearance with moderate numbers of spherocytes. MGG, ×100. The management of hereditary spherocytosis depends on the severity of the anaemia and the jaundice. For those neonates that do develop anaemia, up to 75% will require one or two transfusions during the neonatal period before a transfusion‐free plateau Hb of 80–100 g/l is achieved after several months.79 Jaundice is normally readily controlled by phototherapy and exchange transfusion is rarely required. Folic acid supplementation (500 μg/kg daily) is recommended for all affected infants with evidence of chronic haemolysis. The majority of children and adults with hereditary spherocytosis are not transfusion‐dependent. However, chronic haemolysis, even when mild, predisposes these patients to a number of long‐term complications including gallstones, splenomegaly, leg ulcers, parvovirus‐associated red cell aplastic crisis and episodic acute anaemia due to haemolytic crises, usually provoked by infection (reviewed in detail in reference 73). These disorders are caused by autosomal dominant mutations in the genes encoding α or β spectrin (SPTA1, SPTB) or erythrocyte membrane protein band 4.1 (EPB41).70 The prevalence of hereditary elliptocytosis has been estimated at 1 in 2000 to 1 in 4000 of the population but this varies considerably in different parts of the world.73 For example, hereditary elliptocytosis is more common in areas of endemic malaria, particularly in people of African and Mediterranean origin.83 The clinical manifestations of hereditary elliptocytosis and related syndromes are complex in that at least three different types of disease manifestation are recognised; any of these may present in the neonatal period. The most frequent presentation, particularly in neonates of African descent, is common hereditary elliptocytosis, an autosomal dominant condition which rarely has any clinical manifestations apart from the presence of elliptocytes in the blood film. The diagnosis in the neonatal period is therefore made as an incidental finding when a blood film is reviewed for another reason, such as screening for the presence of infection. No further investigations or haematological follow‐up are necessary and folate supplementation is not indicated. The more severe forms of hereditary elliptocytosis are summarised in Table 2.7. Neonates who are homozygous or compound heterozygous for hereditary elliptocytosis mutations have severe, transfusion‐dependent haemolytic anaemia that presents in fetal life or within a few days of birth. Almost all of these neonates have hereditary pyropoikilocytosis (HPP), which is typically caused by bi‐allelic (homozygous or compound heterozygous) variants, especially in SPTA1, that disrupt spectrin self‐association.84,85 Rare cases of HPP caused by bi‐allelic SPTB mutations86 or digenic inheritance of SPTA1 and SPTB mutations have been reported.85,87,88 Hereditary pyropoikilocytosis is the most common form of severe hereditary elliptocytosis. It presents in the neonatal period with marked, persistent transfusion‐dependent haemolytic anaemia. The diagnosis is usually easily made from the low mean cell volume (MCV), which may be as low as 25 fl, and the blood film, which show extreme poikilocytosis with large numbers of bizarre, fragmented red cells, microspherocytes and elliptocytes (Fig. 2.15a). Examination of the parental blood films is very helpful since one or both parents typically has common hereditary elliptocytosis (Figs 2.15b,c).89 It is important to note that HPP can also occur when both parental blood films appear to be normal, for example as a result of a de novo mutation in combination with a silent spectrin mutation, such as the αLELY variant (Case 2.4, see page 95). The treatment of neonates with HPP is red cell transfusion. Transfusions should continue on a regular basis until the child is old enough to undergo splenectomy, to which there is an excellent response with all patients rendered transfusion‐independent.85 Hereditary elliptocytosis with infantile poikilocytosis is the second most severe form of hereditary elliptocytosis in the neonatal period. It occurs in a very small proportion of neonates with common hereditary elliptocytosis due to mono‐allelic α spectrin variants. Typically, affected neonates have moderately severe haemolytic anaemia with marked poikilocytosis as well as elliptocytes. Occasionally the jaundice is severe enough to require exchange transfusion. Most neonates require red cell transfusion for anaemia but become transfusion‐independent as the poikilocytosis and anaemia resolve over the first 3–6 months of life; apart from elliptocytes in the blood film the blood results are completely normal by the age of 12 months. The diagnosis is suggested when one, but not both, parents have typical common hereditary elliptocytosis with elliptocytes in the blood film and no anaemia or jaundice. However, diagnosis is often finalised only when the natural history confirms that this is not a case of HPP and/or molecular diagnosis is performed. Fig. 2.15 Blood films of a neonate with hereditary pyropoikilocytosis and her parents. The baby was noted to be jaundiced at 36 hours of age and on day 5 had a haemoglobin concentration (Hb) of 133 g/l, a low mean cell volume (MCV) of 85 fl and a reticulocyte count of 125–175 × 109/l. (a) Blood film of the neonate showing marked anisopoikilocytosis with ovalocytes, elliptocytes and schistocytes; a single NRBC and occasional spherocytes can also be seen. (b) Blood film of the mother who was haematologically normal but was heterozygous for the low expression allele SPTA1 c.[5572C>G:6531‐12C>T]; p. (Leu1858Val), known as α spectrinLELY (low expression Lyon allele). (c) Blood film of the father who had hereditary elliptocytosis and was heterozygous for SPTA1 c.460_462dup; p. (Leu155dup). The neonate showed compound heterozygosity for these two SPTA1 mutations. MGG, ×100. Fig. 2.16 Blood film of an older patient with Southeast Asian ovalocytosis showing ovalocytes, macro‐ovalocytes and distinctive stomatocytes. MGG, ×100. This condition is characterised by striking morphological abnormalities in the blood of children and adults with ovalocytes, macro‐ovalocytes and distinctive stomatocytes (Fig. 2.16). It results from homozygosity for a mutation in SLC4A1, which causes haematological abnormalities but without clinical consequences beyond the neonatal period. However, about half of heterozygous neonates have haemolytic anaemia and hyperbilirubinaemia.90 Homozygosity causes hydrops fetalis and usually intrauterine death.91 There are many aspects of the clinical and haematological presentation of the inherited red cell enzymopathies that are unique to the neonatal period. These differences are important for tailoring our diagnostic approach in this age group and also for our understanding of their clinical and haematological impact. First, from a diagnostic point of view, the initial presentation of these disorders in neonates is usually with jaundice. Anaemia, if it occurs, is not usually present at birth and many neonates never develop anaemia even when the jaundice is severe. Secondly, in contrast to red cell membrane disorders, there are usually no diagnostic changes on the neonatal blood film. Thirdly, with the exception of glucose‐6‐phosphate dehydrogenase (G6PD) deficiency, inherited red cell enzyme disorders are rare. G6PD deficiency, on the other hand, is common, with millions of affected individuals worldwide. Importantly, the G6PD gene is on the X chromosome and therefore the vast majority of neonates with G6PD deficiency are male. Furthermore, the geographical distribution of G6PD deficiency maps to the prevalence of malaria and therefore most, but not all, affected neonates are of African, Mediterranean, Asian or Arabic descent. Although G6PD deficiency is by far the most common of the inherited red cell enzymopathies that present in the neonatal period, awareness of the rarer disorders is extremely important as they are often difficult to diagnose and yet can be associated with very severe disease (Table 2.9).92 Table 2.9 Red cell enzymopathies presenting with neonatal anaemia and/or jaundice G6PD, glucose‐6‐phosphate dehydrogenase; Hb, haemoglobin concentration; PK, pyruvate kinase. The prevalence of G6PD deficiency is particularly high in individuals from Central Africa (20% of males) and the Mediterranean (10% of males) but is also seen in the Indian subcontinent, the Far East and the Middle East (reviewed in reference 93). In neonates, G6PD deficiency typically presents with jaundice during the first few days of life, with a peak incidence on day 2 or 3.94 Neonatal jaundice due to G6PD deficiency is often severe, despite an Hb that is normal or only slightly reduced and severe anaemia is rare.95,96 Although the majority of affected neonates are boys, female carriers are sometimes identified through neonatal jaundice screens because these babies also occasionally develop severe neonatal jaundice.97 The blood film is usually completely normal with no irregularly contracted cells, reticulocytosis or erythroblastosis. However, in the small proportion of neonates who develop anaemia due to G6PD deficiency, the blood film shows typical changes of oxidative haemolysis (Fig. 2.17). It is not clear why some, but not all, G6PD‐deficient neonates develop neonatal jaundice as this does not appear to correlate with the level of G6PD enzyme activity in the red cells, although this is controversial.97 The pathogenesis of the jaundice is also unclear as most babies with G6PD deficiency have no evidence of haemolysis. Current views are that the jaundice is likely to be of hepatic origin and to reflect inefficient bilirubin conjugation rather than haemolysis.98 The diagnosis of G6PD deficiency is made by assaying G6PD on a peripheral blood sample, the result being interpreted in the light of any elevation of the reticulocyte count above normal neonatal levels and the higher normal range in neonates. It is essential to perform the G6PD assay before any red cell transfusions are given (or once any transfused cells have been cleared from the circulation, which may take up to 8 weeks). Mutation analysis of the G6PD gene should be considered for atypical cases – for example those presenting with severe anaemia where there is no evidence of either HDFN or red cell membranopathy. Some mutations are associated with chronic non‐spherocytic hemolytic anaemia and mutation analysis is especially useful for establishing the diagnosis in these cases. Fig. 2.17 Blood film of a neonate with oxidative haemolysis due to glucose‐6‐phosphate dehydrogenase (G6PD) deficiency showing irregularly contracted cells and a bite cell. MGG, ×40. Management of neonatal G6PD deficiency requires close monitoring of the bilirubin to prevent kernicterus, particularly when interactions with other risk factors for neonatal hyperbilirubinaemia, such as Gilbert syndrome, HDFN or hereditary spherocytosis are present. If exchange transfusion is required for severe hyperbilirubinaemia, conventional guidelines for exchange transfusion can be followed.61 It is important to provide information to the parents/carers of affected neonates as to which specific medicines, chemicals and foods may precipitate haemolysis (these are listed in Table 2.10). In countries where malaria is endemic, G6PD deficiency should be excluded before giving primaquine for malaria eradication due to the high risk of drug‐induced haemolysis in affected G6PD‐deficient individuals. It is also useful to be aware that acute haemolysis due to exposure of neonates to naphthalene‐containing mothballs has been reported99 and that consumption of fava beans or quinine‐containing drinks by breastfeeding mothers may cause haemolysis in G6PD‐deficient neonates.100,101 Folate supplementation is not routinely indicated in G6PD‐deficient neonates because, in the vast majority of cases, G6PD deficiency does not cause chronic haemolysis. However, for those infants with one of the uncommon variants of G6PD deficiency associated with chronic non‐spherocytic haemolytic anaemia, such as G6PD Volendam,102 folic acid supplements should be given as described above for the red cell membrane disorders. Table 2.10 Drugs and chemicals that may precipitate haemolysis in neonates and infants with G6PD deficiency * Avoid in breastfeeding mothers. † Not absolutely contraindicated but use with caution. In contrast to G6PD deficiency, pyruvate kinase (PK) deficiency is fairly rare, with an estimated incidence of 1 in 20 000 in Caucasians.92 As a result, even large neonatal centres are only likely to identify one case of PK deficiency every 5 years. PK deficiency is transmitted in an autosomal recessive fashion and is due to bi‐allelic mutations in the PKLR gene that encodes the PK enzyme.103 More than 120 different PKLR variants have been described and the frequency of novel pathogenic variants that have not previously been reported is high (approximately 20%).104 PK deficiency is clinically heterogeneous, varying from anaemia severe enough to cause hydrops fetalis to a mild unconjugated hyperbilirubinaemia (Case 2.5, see page 98). However, the PKLR genotype does not seem to correlate with the frequency of complications in utero or in the newborn period.104 A recent study of the natural history of PK deficiency in over 250 patients found that perinatal complications were common in PK deficiency, including anaemia that required transfusion and hyperbilirubinemia as well as hydrops fetalis and prematurity.103 This study clearly shows that jaundice is a common finding in neonates with PK deficiency as the majority of neonates (93%) were treated with phototherapy and almost half (46%) required exchange transfusion.103 In severe cases, extramedullary haemopoiesis, presenting as a typical violaceous, papular facial rash, has been reported.105 The diagnosis of PK deficiency is made by measuring pretransfusion red cell PK activity. In mild cases enzyme activity may be only modestly reduced and in such cases it can be useful to assay PK levels in the parents. However, mutation analysis of the PKLR gene is increasingly used as the definitive diagnostic tool either by directly sequencing the gene or as part of a targeted next generation sequencing gene panel.3,104 The blood film is sometimes distinctive, with small numbers of markedly echinocytic cells (Fig. 2.18), but more often shows non‐specific changes of non‐spherocytic haemolysis. Occasional cases of PK deficiency present with extreme and persistent neonatal hyperbilirubinemia and hepatic failure with or without evidence of haemolysis,106 making the diagnosis extremely difficult.103 Fig. 2.18 Blood film of a neonate with pyruvate kinase (PK) deficiency showing marked erythroblastosis and polychromasia and occasional echinocytes. There is also left shift. MGG, ×100. Management of PK deficiency in the neonatal period depends on the severity of the jaundice and anaemia. As mentioned above, the majority of affected neonates will require phototherapy for jaundice and almost half will require exchange transfusion. Just over 50% of children remain transfusion‐dependent.107 For those that are transfusion‐independent, folic acid supplements should be given to prevent deficiency due to chronic haemolysis.103 Splenectomy later in childhood reduces the transfusion requirement in 90% of cases. However, management of PK deficiency in children is likely to change in the near future with the promising results of a new small molecule allosteric activator of the PK enzyme (mitapivat), which has been shown to increase red cell enzyme activity and Hb in PK‐deficient adults.108 This poorly understood condition causes acute haemolysis confined to the neonatal period and accounts for about 10% of cases due to red cell enzyme deficiencies.109 In the original report of this disorder, infantile pyknocytosis was defined as the presence of ‘distorted, irregular, densely stained erythrocytes, presenting with several to many spiny projections’ in a neonate with self‐limiting acute haemolytic anaemia.110 These morphological features, and the natural history, strongly point to a red cell enzyme defect. However, the genetic basis of infantile pyknocytosis remains unknown. Recent targeted next generation sequencing has excluded mutations in most of the red cell enzyme genes associated with neonatal haemolytic anaemia, including PKLR, G6PD, hexokinase (HK1), phosphoglycerate kinase (PGK1), phosphofructokinase (PFKL), glucose phosphate isomerase (GPI), triosephosphate isomerase (TPI1) and NT5C3A, the gene encoding pyrimidine‐5′‐nucleotidase.111 In many cases affected neonates are found to have reduced levels of glutathione peroxidase (reference 112 and unpublished data) but no mutations in the GPX1 gene encoding the enzyme have been reported, even when specifically tested.111 Infantile pyknocytosis is more common in preterm neonates and when it occurs in twins, both are usually affected. The anaemia associated with infantile pyknocytosis is often moderately severe and may require one or two red cell transfusions; it then gradually resolves by the age of 2–3 months. The blood film typically shows changes of oxidative haemolysis during the neonatal period but returns to normal as the haemolysis resolves (Case 2.6, see page 100). Measurement of glutathione peroxidase levels in affected neonates may be useful (parental levels are normal) but the diagnosis is usually easily made from the blood film and natural history. There are no reports of recurrence of haemolysis beyond the neonatal period in babies with infantile pyknocytosis and care should be taken to exclude other causes of haemolytic anaemia in any cases where the haemolysis does not resolve, or recurs, after the age of 3 months. The other red cell enzymopathies reported to present in the neonatal period are all rare and are shown in Table 2.9. Several of these disorders cause very severe fetal and/or neonatal haemolytic anaemia. Glucose phosphate isomerase (GPI) and hexokinase (HK) deficiencies, which are autosomal recessive conditions, have both been reported to present with severe neonatal haemolytic anaemia including hydrops fetalis.113–116 Deficiency of pyrimidine‐5′‐nucleotidase due to mutations in the NT5C3A gene usually causes mild chronic haemolytic anaemia and does not present until adolescence; however, about 15% of cases have neonatal jaundice and this diagnosis should be considered in unexplained neonatal haemolysis where the blood film shows prominent basophilic stippling.117 As phosphofructokinase (PFK) deficiency can involve red cells, muscle or both, depending on the PFK subunit affected, the disease may manifest as haemolytic anaemia and/or myopathy. In severe cases the myopathy causes death during infancy as a result of respiratory insufficiency and other complications.118 Phosphoglycerate kinase (PGK) deficiency is a sex‐linked disorder that largely affects males and occasionally presents in the neonatal period with jaundice and non‐spherocytic haemolytic anaemia. More often PGK deficiency presents in older children and clinically has a variable combination of chronic haemolytic anaemia, developmental delay and myopathy.119 Triosephosphate isomerase (TPI) deficiency causes neonatal haemolytic anaemia in around one‐third of patients. This is important to recognise because the haemolytic anaemia often presents many months before the devastating neurological features of this disorder become apparent.120 Other causes of neonatal haemolytic anaemia are shown in Table 2.5. These include infections, rare extrinsic causes of haemolysis, such as thrombotic thrombocytopenic purpura (TTP) and cavernous haemangiomas (Kasabach–Merritt syndrome). Exposure to oxidant drugs and chemicals can cause haemolysis even in babies without G6PD deficiency. It has been reported, for example, in a neonate exposed to higher than recommended levels of methylene blue during creation of a gastrointestinal anastomosis (Case 2.7, see page 103).121 As well as perinatally acquired bacterial infection, a number of congenital infections have been reported to cause neonatal anaemia via a haemolytic mechanism, including CMV, herpes simplex and rubella viruses, syphilis, malaria and toxoplasmosis (see Table 2.5). While there is good evidence for significant neonatal haemolytic anaemia due to syphilis, malaria and toxoplasmosis, solid evidence of haemolysis due to CMV infection is scanty. Perinatally acquired syphilis remains a significant clinical problem in many countries, affecting an estimated 713 600–1 575 000 pregnancies worldwide (reviewed in reference 122). Just over half of all neonates born with congenital syphilis will have Coombs‐negative haemolytic anaemia, which may persist for weeks after treatment.122 Although fatal cases are uncommon, severe cases may present with hydrops fetalis.123 Congenital toxoplasma infection may also present with severe neonatal haemolytic anaemia and/or hydrops fetalis.124,125 Despite the prevalence of malaria in pregnant women, congenital malaria is surprisingly rare, even in countries where malaria is endemic, and it is exceptionally rare in non‐endemic countries,126,127 perhaps because of protective IgG antibodies in the mother.128 Congenital malaria presents with anaemia, jaundice, hepatosplenomegaly and non‐specific signs such as poor feeding, lethargy and fever.126,127,129 Typically, the first signs of malaria arise at 10–28 days of age, although presentation within the first few hours or as late as 8 weeks of age has been reported.130 CMV has been shown to cause haemolytic anaemia in infants131 but this seems to be very rare in neonates. In most case series in neonates where anaemia and/or an increased transfusion requirement is reported, there is insufficient evidence to indicate the mechanism of the anaemia and other causes, such as the side effects of anti‐viral drugs, may be responsible.132 In one well‐documented case, concomitant CMV infection increased the extent of haemolysis in a neonate with immune haemolytic anaemia secondary to maternal lupus.133 Similarly, although acute haemolysis has been reported in children with herpes simplex infection134 and a small proportion of neonates with congenital herpes simplex have anaemia, there is no definite evidence that this is caused by haemolysis.135 This is also the case for congenital rubella. Neonates with congenital rubella syndrome may have hepatosplenomegaly and jaundice but anaemia is not usually a feature,136 although older children may develop autoimmune haemolytic anaemia in response to acute rubella infection.137 Neonates may also develop microangiopathic haemolytic anaemia (MAHA) secondary to extrinsic damage to neonatal red cells. The three major types of disorder associated with MAHA in neonates are congenital TTP, atypical haemolytic uraemic syndrome (aHUS) and congenital haemangiomas. Hereditary TTP typically presents in the neonatal period with severe haemolysis and hyperbilirubinaemia although, as in many very rare disorders, the diagnosis is often delayed by several years (reviewed in reference 138). Hereditary TTP is caused by bi‐allelic mutations in the ADAMTS13 gene.139 In neonates, severe haemolytic anaemia is the most common presentation together with severe thrombocytopenia and red cell fragmentation seen in the blood film. Over one‐third of the cases reported in a recent series required exchange transfusion in the neonatal period due to severe haemolysis in the first 2 days of life.140 In most of these cases there was a single acute episode of haemolysis and thrombocytopenia; in at least one case the apparent trigger was the altered pattern of blood flow (high sheer stress) through a persistent patent ductus arteriosus, although the trigger for the other cases could not be identified. The diagnosis of inherited TTP should therefore be considered in any neonate with unexplained Coombs‐negative severe hyperbilirubinaemia and thrombocytopenia. Infusions of fresh frozen plasma (FFP) are the mainstay of treatment of recurrent episodes of haemolysis and thrombocytopenia in childhood and beyond.138 Atypical haemolytic uraemic syndrome is another rare disorder that may present in the neonatal period with MAHA and thrombocytopenia but with the additional problem of acute kidney injury. Most forms of aHUS are associated with dysregulation of the alternative complement pathway and about two‐thirds of patients have mutations or likely‐pathogenic variants in genes encoding complement pathway proteins, including complement factor H (CFH), complement factor I (CFI), membrane cofactor protein (encoded by CD46), complement factor B (CFB) or complement factor 3 (C3).141 As for TTP, the blood film shows red cell fragmentation together with severe thrombocytopenia with no evidence of disseminated intravascular coagulation (DIC) on the coagulation screen. Early diagnosis is important because affected neonates respond well to treatment with the C5 inhibitor ecluzimab.141–143 A recent report suggests that measurement of the Fragmented Red Cell (FRC) count now available on some blood count analysers may be useful to alert clinicians to a potential diagnosis of aHUS in neonates.144 Microangiopathic haemolytic anaemia may also be part of the presentation of a group of disorders known as kaposiform haemangioendotheliomas (KHE) and tufted angiomas, which are rare vascular tumours derived from capillary and lymphatic endothelium.145 The majority of such tumours affect the extremities or the torso.146 In 50% of cases these tumours present in the neonatal period and around two‐thirds of these will have associated consumptive coagulopathy and thrombocytopenia (known as the Kasabach–Merritt phenomenon [KMP]).146,147 Although the coagulopathy and thrombocytopenia are the main haematological findings in KMP, some affected neonates do have a microangiopathic anaemia.148 It is now clear that KMP is confined to cases of KHE and tufted angiomas and is not seen in the more common benign infantile haemangiomas.147 KHE with KMP rarely, if ever, regresses spontaneously and treatment in the neonatal period is usually with a combination of prednisolone and vincristine.147 Platelet transfusion should be avoided unless there is active bleeding or a surgical procedure is required.147 Clinically significant haemoglobinopathies that present with anaemia or jaundice in the newborn are summarised in Table 2.11. These are predominantly α thalassemia syndromes and structural defects of the α globin chain or γ thalassaemias and γ globin structural variants. The mechanism of anaemia varies between different haemoglobinopathies and is often multifactorial. The most clinically severe haemoglobinopathies that present in the fetal or neonatal period are α thalassaemia major, in which all four α globin genes are deleted and, uncommonly, haemoglobin H disease, in which three α globin genes are deleted or mutated.149 Alpha thalassaemia trait, where two α globin genes are deleted, and which is much more common, is often easy to suspect in the neonatal period since the MCV is reduced below the lower limit of normal and other causes of microcytosis in neonates (see Table 2.12) are very rare. Non‐thalassaemic structural α and γ globin gene mutations, which are clinically silent in adults and children, can cause transient neonatal haemolytic anaemia because the variant haemoglobin is unstable, whereas the β globin haemoglobinopathies (sickle cell disease and β thalassaemia major) do not cause haemolysis or anaemia in neonates and rarely manifest clinically until after 6–8 weeks of age. Methaemoglobinaemia due to rare inherited mutations in an α or a γ globin gene causing the production of haemoglobin M (see also Table 2.16) can also present in the neonatal period and is discussed at the end of this chapter. Alpha thalassaemia is one of the most common inherited disorders, with a worldwide carrier rate of approximately 5%.150 The most significant form predominantly affects families of Southeast Asian origin, where the carrier rate approaches 40%, although it is also common in parts of India and the Mediterranean (Greece, Turkey, Cyprus and Sardinia).151 Alpha thalassemia is most commonly caused by one of more than 50 deletions affecting either one (referred to as α+ thalassaemia) or both (referred to as α0 thalassaemia) of the α globin genes on one copy of chromosome 16.152 The severity of α thalassaemia depends on the number of affected α globin genes. Where both parents are carriers of α0 thalassaemia, their offspring may be homozygous for α0 and inherit no normal α globin genes; deletion of all four α globin genes results in α thalassaemia major, which has the most severe presentation and nearly always manifests in utero when it typically presents with severe second‐trimester fetal anaemia and hydrops fetalis with an average Hb of 64 g/l.153 As the principal haemoglobin produced by affected fetuses is haemoglobin Bart’s (tetramers of fetal γ globin chains, γ4), this condition is often known as haemoglobin Bart’s hydrops fetalis syndrome (BHFS) to distinguish it from other causes of hydrops fetalis with severe anaemia.154 The main clinical features of this syndrome are severe pallor with jaundice, cardiac failure, pleural and pericardial effusions, ascites, hepatosplenomegaly and respiratory distress.154 Table 2.11 Haemoglobinopathies presenting with anaemia and/or jaundice in the neonatal period* * Beta globin disorders can be identified by screening in the neonatal period but almost never have clinical features at this age; presentation of haemoglobin H disease in the neonatal period is not common. HPLC, high performance liquid chromatography. The cause of the severe anaemia in BHFS is multifactorial and includes reduced haemoglobin synthesis, reduced haemoglobin stability (haemoglobin Bart’s), dyserythropoiesis due to precipitation of γ and β chains within the erythroid cells and hypersplenism, which together cause a dramatic anaemia that is partly haemolytic.149 Furthermore, oxygen delivery to the tissues is severely reduced, not only by the low Hb, but also by the high oxygen affinity of haemoglobin Bart’s.155 Infants with BHFS almost always die either in utero (23–38 weeks) or shortly after birth154 (Case 2.8, see page 105). This condition also carries a serious risk to the mother of the affected fetus, including mortality, with up to 50% developing severe pre‐eclampsia, at least in part because of placentomegaly.156 Table 2.12 Causes of neonatal microcytic anaemia MCV, mean cell volume. Affected fetuses are usually identified on routine ultrasound scanning at 20–21 weeks’ gestation by the clinical findings of fetal ascites, pleural effusion, pericardial effusion or hydrops fetalis.154 These findings in a pregnancy at risk of α thalassaemia should at once prompt fetal blood sampling to determine the Hb and findings on high performance liquid chromatography (HPLC). The diagnosis of α thalassaemia major is confirmed when HPLC shows predominantly haemoglobin Bart’s and haemoglobins F and A are absent. Depending on the extent of the α globin cluster deletion, some affected fetuses (e.g. with the homozygous Southeast Asian variant, which is the commonest cause of BHFS) will also have up to 20% embryonic haemoglobin Portland (ζ2γ2), which is the only functional oxygen‐carrying haemoglobin in these fetuses,155 and sometimes haemoglobin H. The blood film shows hypochromic, often microcytic red cells with vast numbers of circulating NRBC (Fig. 2.19). The mean cell haemoglobin (MCH) and mean cell haemoglobin concentration (MCHC) are reduced but the MCV is not always reduced. Checking the blood counts of the parents will immediately identify whether they are at risk of having a child with α thalassaemia major; both parents will have microcytosis, with the MCV usually below 74 fl and MCH usually less than 24 pg (Fig. 2.20). Fig. 2.19 Blood film of a preterm neonate with haemoglobin Bart’s hydrops fetalis on day 1 of life showing markedly hypochromic red cells, many of which are microcytic, as well as polychromasia and four NRBC. Two of the NRBC show clearly defective haemoglobinisation. MGG, ×100. Fig. 2.20 Blood film of the mother of the neonate whose film is shown in Fig. 2.19, showing microcytosis and mild hypochromia. The mother had deletion of two of four α genes on one chromosome 16 (αα/−−SEA). MGG, ×100. The only long‐term survivors of α thalassaemia major are those who have received IUT followed by regular postnatal transfusions and/or a bone marrow transplant.154 Affected children also have a high incidence of hypospadias, limb defects and atrial septal defects and others have severe neurological problems. If IUT is delayed until anaemia is severe, neonatal pulmonary hypoplasia is a cause of early mortality.154 The diagnosis of α thalassaemia major should be suspected in any case of severe fetal anaemia that presents in the second trimester, and any case of hydrops fetalis with severe anaemia in which the parents come from high‐prevalence areas, particularly Southeast Asia. In addition to the typical presentation, some neonates have had severe anaemia without hydrops fetalis. Antenatal screening of pregnant women and, when relevant, their partners should lead to prediction of the possibility of haemoglobin Bart’s hydrops fetalis, with termination of pregnancy usually being offered when the diagnosis is confirmed. Increasing awareness of BHFS, and the increase in cases in countries such as the USA where this condition used to be extremely rare, has led to major changes in the treatment options available and to the development of consensus guidelines for the perinatal management of patients with BHFS.157 These note that lack of awareness of the improving outcome has meant that parents of affected fetuses may not be offered IUT. A recent international conference (https://conference.globalcastmd.com/ucsf‐alpha‐thalassemia‐major/archive) reviewed existing knowledge regarding the prenatal screening, perinatal care, and maternal and childhood outcomes of patients with α thalassaemia major, and summarised their recommendations for management of the condition.157 These include recommendations for preconceptual (or early antenatal) screening of couples at risk and the offer of fetal blood sampling in all cases of hydrops fetalis in pregnancies at risk in order to make a rapid diagnosis on the basis of blood count and HPLC, with IUT at the same time if the family wishes to pursue intervention in order to boost oxygen delivery to the tissues and minimise complications. These guidelines also suggested that couples at risk should be counselled about the options for a future pregnancy, including the possibility of preimplantation genetic diagnosis.157 This disorder is usually caused by deletion and/or mutation of three α globin genes. In most cases affected individuals are compound heterozygotes for two different mutations rather than homozygotes for a moderately severe molecular defect. Haemoglobin H disease varies considerably in severity. In general, the phenotype correlates very well with the degree of α globin chain deficiency and non‐deletional types of haemoglobin H disease are more severe than the more common deletional types.158,159 The most severe cases present with severe neonatal haemolytic anaemia and, occasionally, with hydrops fetalis.160–163 This includes haemoglobin H/Constant Spring (−−/αCSα); haemoglobin Constant Spring is the most common non‐deletion α‐thalassaemia in Southeast Asia and Southern China and this compound heterozygous state is an important cause of hydrops fetalis.164 A diagnosis of haemoglobin H disease in the newborn period is suggested by the presence of hypochromic, microcytic anaemia together with jaundice and the presence of large amounts of haemoglobin Bart’s (γ4) and as well as some haemoglobin H (β4). The heterogeneity of haemoglobin H disease makes genetic counselling particularly difficult. Most cases resulting from simple deletion of the α globin genes are mildly affected and prenatal diagnosis may not be appropriate; virtually all severe cases have at least one non‐deletional allele.161–163 Awareness of the potential severity of coinheritance of α0 thalassaemia alongside non‐deletional α thalassaemia alleles, such as in haemoglobin H/Constant Spring, in at‐risk populations should lead to careful in utero monitoring of affected pregnancies for signs of hydrops fetalis as prompt IUT has been shown to prevent fetal morbidity and mortality.164 While most α chain mutations do not cause clinically significant disorders either in the newborn or in older children, occasional variants do cause problems specifically in the newborn. In particular, they are a rare cause of unexplained neonatal jaundice and anaemia.165 The best known example is haemoglobin Hasharon (p.Asp>HisHBA2:c.142G>C), which is mildly unstable (due to a higher propensity for dissociation into subunits) and is found mainly in Ashkenazi Jewish families. The interaction between Hasharon α chains and γ chains in the fetal form of haemoglobin Hasharon results in an unstable haemoglobin that manifests as neonatal haemolytic anaemia.166 The haemolysis is transient, resolving with the transition from the fetal to the adult form of haemoglobin Hasharon trait at several months of age. The haemolysis is usually mild and, in practice, most infants with α chain mutations are detected only as part of routine neonatal screening. However, the diagnosis may be difficult to establish because of the instability of the abnormal haemoglobin produced; mass spectrometry or molecular analysis is usually more reliable than HPLC. At least 130 different γ chain variants have been described, many of which either cause transient neonatal haemolytic anaemia or are picked up as a result of neonatal haemoglobinopathy screening programmes.167 The most well‐known unstable γ chain variants are haemoglobin F Poole168 and haemoglobin F Bonheiden,169 which both cause haemolytic anaemia with Heinz bodies (red cell inclusions composed of denatured haemoglobin) confined to the first few weeks of life. The anaemia completely resolves as γ chains are replaced by the production of normal β chains. Hydrops fetalis has also been reported in a unique family where triplets, who were homozygous for haemoglobin Taybe, were born at 27 weeks’ gestation with microcytic anaemia (Hb 71–95 g/l), generalised oedema and ascites.170 Although rare, the diagnosis of heterozygous εγβδ thalassaemia (often referred to as γδβ thalassaemia) should be considered in neonates with unexplained haemolytic anaemia associated with microcytic hypochromic red cells and erythroblastosis.171,172 Sometimes there is hepatosplenomegaly and the blood film may show basophilic stippling. These conditions result either from large deletions that remove the entire β globin gene cluster172 or from deletions in which the structural genes are left intact but important regulatory elements are removed.173 Homozygosity has not been reported and would be incompatible with life. Heterozygous neonates often require red cell transfusion but generally become transfusion‐independent after the first few months of life.171 IUT has also occasionally been required.174 Beta globin disorders usually present in the neonatal period as a result of neonatal haemoglobinopathy screening results rather than with clinical problems, which usually do not develop until after the first few months of life. Beta thalassemia major, even when caused by homozygous β0 mutations, typically has no overt clinical or haematological abnormalities at birth. As the Hb falls over the second and third months of life, infants feed more slowly and the rate of weight gain slows. Increasing anaemia is accompanied by noticeable pallor and the development of splenomegaly together with the appearance of polychromasia and circulating erythroblasts. Although there are no consistent or reliable abnormalities in the red cell indices or blood film in neonates with this condition, an interesting report published more than 60 years ago does suggest that subtle features, including very mild anaemia, may be present in some affected neonates,175 although possibly because of concomitant causes of anaemia in those cases. A diagnosis of β thalassaemia major is strongly suggested by the absence of haemoglobin A on HPLC, especially in a term neonate, and where both parents have been shown to have β thalassaemia heterozygosity. Neonates with β thalassemia trait have no abnormal haematological findings either in the red cell indices or in the blood film; microcytosis and hypochromia normally become evident by 6 months of age. Antenatal (or preconceptual) screening should be offered to all couples at risk of having a child affected by β thalassaemia major. Babies with sickle cell disease (sickle cell anaemia and compound heterozygous states causing a sickling disorder) are generally not anaemic at birth and have no consistent abnormalities of red cell morphology in blood films examined in the first few weeks of life. Target cells are occasionally present in neonates with sickle cell disease, particularly those with haemoglobin S/haemoglobin C compound heterozygosity. HPLC in neonates with sickle cell anaemia (βSβS) typically shows 15–20% haemoglobin S and the absence of haemoglobin A (Fig. 2.21), in contrast to neonates with sickle cell trait (ββS) where there is approximately 10% haemoglobin A together with a similar amount of haemoglobin S (Fig. 2.22). In cases where clinically evident sickle cell disease has been diagnosed during the neonatal period, the most frequent findings are jaundice, fever, pallor, respiratory distress and abdominal distention.176 The differential diagnosis of neonatal microcytosis and microcytic anaemias is shown in Table 2.12). This includes specific red cell membrane disorders, such as HPP, discussed earlier, most of the haemoglobinopathies that present in the neonatal period, and chronic in utero blood loss as discussed later. A small number of rare sideroblastic anaemias present with hypochromic microcytic anaemia in the neonatal period. Congenital sideroblastic anaemia is a rare disease caused by mutations of genes involved in haem and iron–sulphur cluster formation, and mitochondrial protein biosynthesis. While the majority of these disorders present later in childhood or in adulthood, a number present in the neonatal period with microcytic anaemia with or without syndromic features.177 A recent European survey of congenital sideroblastic anaemia identified neonatal presentations in some cases of X‐linked sideroblastic anaemia (XLSA) due to mutations in the ALAS2 gene, in some cases with autosomal recessive sideroblastic anaemia caused by bi‐allelic mutations in the SLC25A38 gene and in cases with mutation of the TRNT1 gene.177 The same report noted that a molecular defect could not be identified in almost one‐third of patients with congenital sideroblastic anaemia, including several that presented in the neonatal period. Of note, several series have demonstrated that congenital sideroblastic anaemia caused by SLC25A38 mutations is nearly always associated with severe microcytic anaemia in the neonatal period.177–179 Infants with anaemia due to SLC25A38 mutations usually require red cell transfusion during the neonatal period and remain transfusion‐dependent thereafter. Congenital sideroblastic anaemia due to TRNT1 mutations is also associated with severe neonatal microcytic anaemia in virtually all reported cases. This syndromic disorder, first termed SIFD syndrome, is characterised by a tetrad of congenital sideroblastic anaemia, B cell immunodeficiency, periodic fevers and developmental delay.180 Severe cases of SIFD due to mutations in TRNT1 may present with hydrops fetalis due to progressive fetal anaemia.181 Affected infants remain transfusion‐dependent and haemopoietic stem cell transplantation is usually recommended as a curative approach for both the anaemia and immunodeficiency.181 Fig. 2.21 High performance liquid chromatography (HPLC) on a BioRad Variant II instrument of a neonate with sickle cell anaemia (SS) showing 84.7% haemoglobin F, no haemoglobin A and an estimated 17.9% in the haemoglobin S window. The complex peaks to the left are post‐translationally modified haemoglobin F. Note the very low percentage of haemoglobin A2, which is expected in a neonate. Fig. 2.22 HPLC on a BioRad Variant II instrument of a neonate with sickle cell trait (AS) showing 75.3% haemoglobin F, 15.3% haemoglobin A and 9.9% in the haemoglobin S window. Note that in sickle cell trait the S percentage is lower than the A percentage. Table 2.13 Useful diagnostic tests in neonatal anaemia due to blood loss Blood loss as a cause of neonatal anaemia may be very obvious (for example, a cephalohaematoma or rupture of the umbilical cord) but more often the blood loss is concealed and easy to miss unless specifically sought (for example a fetomaternal bleed). Usually, the most serious blood loss occurs prior to delivery and important causes of this are twin‐to‐twin transfusion and fetomaternal haemorrhage. Acute perinatal blood loss as a cause of unexpected severe neonatal anaemia and severe hypovolaemic shock is often missed initially, leading to a delay in treatment and a significant risk of neonatal death or severe anoxic brain damage leading to neurological damage.182,183 Recent data suggest that up to 40% of cases of severe fetomaternal haemorrhage are missed184 and awareness of this complication amongst obstetricians is often low.185 Although much less dramatic, chronic neonatal blood loss secondary to iatrogenic blood letting is also important to consider as this is by far the most common cause of anaemia in hospitalised neonates and strategies are now available to minimise this.186 The main causes of neonatal anaemia due to blood loss are shown in Table 2.13. This occurs in monochorionic twins with monochorial placentas where there are two amniotic sacs (diamniotic).187 In this situation, the twins share a placenta within which there are multiple vascular anastomoses that allow blood to flow between the twins either in fetal life or during delivery.188 Although blood flow between the twins is balanced in most cases, in up to 20% of cases net blood flow is towards one of the twins. When this occurs before birth this is known as twin‐to‐twin transfusion syndrome (TTTS) or twin anaemia–polycythaemia sequence (TAPS), depending upon the size of the vascular anastomoses and the associated clinical sequelae; when bleeding between the twins occurs during delivery, it is known as peripartum TTTS (reviewed in reference 189). Twin‐to‐twin transfusion affects around 10% of monochorionic twin pregnancies.190 Bleeding in TTTS usually occurs during the second trimester and carries a very high mortality rate if left untreated.191 The diagnosis is based on the ultrasound findings, which predominantly show oligohydramnios in the donor twin and reciprocal polyhydramnios in the recipient twin. The presence of haematological problems in neonates affected by TTTS depends on the type of antenatal treatment. While twins treated with laser coagulation surgery generally have no significant difference in Hb levels, for TTTS twins treated conservatively (expectant management or serial amnioreduction), donor twins typically have a significantly lower Hb at birth than recipient twins; a recent study reported a median inter‐twin difference of 36 (16–60) g/l.192 In conservatively managed cases, around one‐third of donor twins will require red cell transfusion for anaemia soon after delivery, while recipient twins may develop severe hyperviscosity–polycythaemia, leading to ischaemic limb necrosis.193 A recent retrospective study showed that even after fetal laser coagulation surgery, residual patent anastomoses mean that around 50% of donor twins will require red cell transfusion at birth, and one‐third of recipient twins will have severe hyperviscosity/polycythaemia and require exchange transfusion.193 Nevertheless, recent data show that treatment with fetoscopic laser coagulation has significantly improved the outcome from a mortality rate of up to 80% to 30–40%, although about 10% of survivors will have impaired neurodevelopmental outcome, including cerebral palsy.193 Twin anaemia–polycythaemia sequence is a newly described form of chronic inter‐twin blood transfusion.194,195 This also leads to anaemia in the donor twin and polycythaemia in the recipient but without signs of oligo‐ or polyhydramnios, respectively. In contrast to TTTS, TAPS is caused by bleeding through very small placental vascular anastomoses (diameter <1 mm), allowing slow transfusion of blood from one twin to the other, without the major haemodynamic imbalance seen in TTTS. In some cases TAPS can occur with, or following treatment of, TTTS, when it typically manifests during the second and third trimesters of pregnancy. The diagnosis may be made antenatally by identifying differences in the MCA peak systolic velocity, with increased velocity predicting anaemia in the donor twin while reduced velocity in the recipient is associated with polycythaemia.196 The diagnosis of TAPS after birth is based on the extent of differences in Hb at birth in monochorionic twins. Generally accepted postnatal criteria are an inter‐twin Hb difference >80 g/l and either a donor/recipient reticulocyte count ratio of >1.7 and/or the presence of minuscule placental anastomoses (diameter <1 mm).197 In this situation, the donor twin is smaller, pale and lethargic and may have overt cardiac failure. By contrast, the recipient twin is often plethoric, with polycythaemia, hyperviscosity, hyperbilirubinaemia and, in the most severe cases, DIC.193 The majority of donor twins will require short‐term red cell transfusion by the end of the first day of life but, unlike TTTS, emergency transfusion at birth is not usually needed as the anaemia is chronic. Recipient twins, on the other hand, will frequently require exchange transfusion for severe polycythaemia to reduce the risk of limb ischaemia, skin necrosis and brain injury.198–201 Bleeding may also occur acutely during labour with signs similar to those of fetomaternal haemorrhage. However, acute peripartum TTTS is very uncommon, affecting around 1.5–2.5% of monochorionic twins and little is known about the pathogenesis.202 Most significant fetomaternal bleeds occur during the third trimester, either spontaneously or secondary to trauma, and involve very small quantities of blood (0.5 ml or less).185 The risk of fetomaternal haemorrhage is increased by invasive procedures, including caesarean section, fetal blood sampling, amniocentesis and chorionic villus sampling, as well as by obstetric complications such as placental abruption, placenta praevia, vasa praevia, choriocarcinoma and chorioangioma.203,204 Fetomaternal haemorrhage may also follow falls or major trauma, such as road traffic accidents, where haemorrhage can be massive and rapid leading to a high rate of fetal loss.205 Massive fetomaternal haemorrhage, usually defined as the loss of more than 20% of the blood volume or >30 ml fetal blood transfused into the maternal circulation (Case 2.9, see page 107), may cause intrauterine death or the baby may be born with hydrops fetalis or circulatory shock.206,207 In some cases the baby appears well, but pale, at birth and collapses a few hours later. Careful questioning of the mother may elicit a history of reduced or absent fetal movements, bleeding or trauma. As fetomaternal haemorrhage can have a devastating outcome, both in the acute setting and secondary to long‐term neurological damage, prompt diagnosis is crucial. The diagnosis should be considered in any neonate with unexpected severe neonatal anaemia with or without signs of hypovolaemic shock. In fact, the presence of moderate or severe anaemia at birth in an otherwise well baby with no or minimal jaundice is a key diagnostic clue. The most useful diagnostic tests are a Coombs test to exclude immune haemolysis, a reticulocyte count to exclude red cell aplasia, a Kleihauer test or flow cytometry on maternal blood to quantify the number of haemoglobin F‐containing fetal red cells in the maternal circulation, and a blood film,207 which shows large numbers of NRBC, particularly where there has been a large acute bleed (Fig. 2.23a,b). The Kleihauer (or Kleihauer–Betke) test has been the mainstay of laboratory methods for estimating the volume of fetal haemorrhage for more than 50 years. This test relies on the principle that adult (maternal) red blood cells normally contain almost entirely haemoglobin A, which is sensitive to acid elution in vitro, giving rise to colourless ‘ghost’ cells (Fig. 2.24a); by contrast, fetal red cells contain mostly haemoglobin F, which is resistant to acid elution and stains a deep pink colour (Fig. 2.24b). The Kleihauer test remains very widely used as it is rapid and inexpensive and can be performed without expensive equipment.208 However, careful attention to detail and controls are essential as systematic evaluation has shown both over‐ and under‐estimation of fetomaternal haemorrhage.209 In addition, the results are difficult to interpret where the mother herself has increased haemoglobin F‐containing cells (‘F‐cells’), for example due to maternal hereditary persistence of fetal haemoglobin (HPFH) or HPFH/sickle trait.210,211 Newer, more accurate methods, such as anti‐haemoglobin F flow cytometry are a promising alternative, although their use is limited by equipment and staffing costs.212 Fig. 2.23 Blood film of two term neonates with anaemia secondary to fetomaternal haemorrhage showing (a) a very marked increase in erythroblasts and marked polychromasia on the first day of life and (b) three erythroblasts as well as an occasional spherocyte and mild polychromasia on day 3 of life. Note that the dyserythropoiesis shown by several erythroblasts is a feature of ‘stress erythropoiesis’. MGG, (a) ×40; (b) ×100. Fig. 2.24 Kleihauer–Betke test. (a) This image shows maternal red blood cells containing almost entirely haemoglobin A after acid elution in vitro causes the appearance of colourless ‘ghost’ cells. (b) This image shows the positive control in which fetal red blood cells have been added prior to acid elution and, as these contain largely haemoglobin F which is resistant to acid elution, these are shown here stained deep pink. ×100. Where chronic fetal blood loss has occurred, the baby is often well at birth but may present with cardiac failure. In this situation, the blood film shows hypochromic, microcytic red cells as well as circulating erythroblasts (Fig. 2.25) and the Kleihauer result may be difficult to interpret. In some cases, however, chronic fetomaternal blood loss is followed by an acute bleed, with resultant severe anaemia (Hb <50 g/l) and a high risk of long‐term brain injury.213 Another potential confounding factor is when there is ABO incompatibility between the mother and baby and the baby is disproportionately anaemic.214,215 In such cases, the fetal cells may be rapidly destroyed within the maternal circulation and a high index of suspicion of fetomaternal haemorrhage as a cause of neonatal anaemia is needed, in order to perform the Kleihauer test as soon as possible. Choriocarcinoma is a very rare cause of massive fetomaternal haemorrhage, which it is important to consider in all cases as the presence of unexplained severe neonatal anaemia may provide the first clue to the diagnosis.216 It may also present as fetal hydrops, IUGR or intrauterine fetal death.217 The presence of choriocarcinoma is often not recognised at delivery because the tumour is not visible macroscopically.218 Despite the risk to the fetus and newborn, recent data show a cure rate in affected women of almost 100% even in the presence of metastases.219 Fig. 2.25 Blood film of a term neonate born with severe anaemia (Hb 50 g/l) showing three normoblasts, an erythroblast containing a mitotic figure, polychromasia and a mixture of normochromic and hypochromic red cells as well as some spherocytes. The Kleihauer–Betke test estimated the fetal blood loss at less than 30 ml and a diagnosis of acute fetomaternal haemorrhage on a background of chronic fetomaternal haemorrhage was suspected. MGG, ×100. Anaemia due to blood loss around the time of delivery is usually due to obstetric complications, including placenta praevia, placental abruption or incision of the placenta during caesarean section or, alternatively, is due to bleeding from the umbilical cord caused by trauma or inadequate cord clamping. In these circumstances, the babies are often extremely ill, with circulatory shock, anaemia worsening rapidly after birth, large numbers of circulating NRBC and DIC. One of the most serious causes of acute fetal blood loss is vasa praevia, an anomaly of placentation which results in aberrant fetal blood vessels running across the internal os of the cervix. These vessels are at risk of rupture during labour when the supporting membranes rupture because they are unsupported by the umbilical cord or placental tissue.220 The classic presentation is with painless vaginal bleeding in the mother, but the diagnosis can easily be missed because the volume of blood is relatively small and it is impossible to distinguish fetal from maternal blood by appearance. The incidence of vasa praevia is estimated to be around 1 in 2500 pregnancies and the condition carries a fetal mortality of almost 50%.220 The presence of vasa praevia can be detected antenatally using vaginal ultrasound, although this is not routine practice. Nevertheless, recent studies have shown that prenatal diagnosis in centres with prenatal diagnosis expertise can be associated with 100% neonatal survival by performing early caesarean section.221 Acute neonatal anaemia may also be due to internal haemorrhage in the baby, usually in association with a traumatic delivery. This may be immediately evident, for example cephalohaematoma, or less obvious, for example pulmonary or intra‐abdominal haemorrhage. In such babies, particularly if there is no history of trauma, it is important to consider whether there an underlying bleeding diathesis, such as haemophilia or vitamin K deficiency. Inherited and acquired coagulation disorders that may present with bleeding at birth or during the neonatal period are discussed in Chapter 4. Anaemia of prematurity refers to the normal physiological fall in Hb over the first 4–8 weeks of life in a preterm neonate. The pathogenesis is multifactorial and includes the shorter lifespan of fetal red blood cells, the relatively low EPO concentrations in the first few weeks of life and the rapid growth of the baby during this period as well as iatrogenic anaemia due to phlebotomy for diagnostic investigations.1,186 Phlebotomy losses are typically highest in very low birthweight neonates (<1500 g at birth) who have been estimated to lose 11–22 ml/kg per week during the first 6 weeks of life, equivalent to 15–30% of their circulating blood volume.222 Nutritional deficiency no longer plays a significant role, at least in well‐resourced countries, due to routine prophylactic supplementation with iron and folic acid.223 The clinical impact of anaemia of prematurity has been the subject of a large number of studies which, together, suggest a significant association between the extent of anaemia and brain injury, necrotising enterocolitis (NEC) and retinopathy of prematurity.224–227 The relationship between the degree of anaemia and tissue hypoxia‐related damage, as well as the impact of different red cell transfusion strategies and the timing and dose of EPO administration, is described in detail in recent reviews.228–230 The diagnosis of anaemia of prematurity is usually straightforward. Typically, a well preterm baby has a slowly falling Hb with an unremarkable blood film showing normochromic, normocytic red blood cells, a slightly low reticulocyte count (e.g. 20 × 109/l) and no NRBC. However, where top‐up red cell transfusion is being considered, it is important to first exclude other potential causes of anaemia using clinical features and a diagnostic algorithm such as shown in Fig. 2.26. Strategies for the management of this neonatal anaemia vary in different countries, different centres and also between clinicians. Of the three main approaches, there is increased use of the most conservative approach (watchful waiting) as the complications and costs of the other two approaches are increasingly recognised – top‐up red cell transfusion and administration of erythropoiesis‐stimulating agents, such as EPO and darbepoetin.230 Whatever option is chosen, it should be combined with appropriate measures to minimise the severity and duration of anaemia, including limiting iatrogenic blood loss by using blood logs, strict criteria for routine blood tests and miniaturised assays.231–233 There is also increasing interest in delayed clamping of the umbilical cord at delivery to increase the Hb/haematocrit at birth. In addition, all preterm neonates should be given iron supplementation (3 mg/kg/day from 4–6 weeks of age or iron‐fortified formula with 0.5–0.9 mg/dl iron) and folate supplementation (50 μg daily or 500 μg once weekly).234 A recent systematic review of delayed (30 seconds or more) versus early clamping of the cord at delivery in preterm infants included 18 randomised studies.235 Although shown to be a safe option, the effects of this approach on the haematocrit at birth was very small (an increase of 2.7%) while the proportion of neonates who were transfused fell by 10%. Nevertheless, this review also showed that in‐hospital mortality was significantly reduced (risk ratio 0.68).235 Importantly, the outcome of two of the randomised studies suggests that neurodevelopmental outcome is improved, and some countries, including the UK (https://www.resus.org.uk/library/2021‐resuscitation‐guidelines/newborn‐resuscitation‐and‐support‐transition‐infants‐birth) and the USA, now recommend delaying cord clamping for at least 30–60 seconds for uncompromised preterm and term neonates (https://www.acog.org/clinical/clinical‐guidance/committee‐opinion/articles/2020/12/delayed‐umbilical‐cord‐clamping‐after‐birth). The safety and efficacy of delayed cord clamping and cord ‘milking’ in preterm infants less than 29 weeks’ gestation at birth is not yet clear,236 awaiting the outcome of several ongoing trials.237 Fig. 2.26 Diagram showing a suggested approach to the diagnosis of neonatal anaemia. G6PD, glucose‐6‐phosphate dehydrogenase; HDFN, haemolytic disease of the fetus and newborn; MCV, mean cell volume. This remains the mainstay of treatment for anaemia of prematurity. However, increasing recognition of the importance of limiting transfusion to the neonates most likely to benefit from this treatment is leading to more widespread adoption of restrictive transfusion policies. The indications, principles and hazards of neonatal transfusion are briefly summarised below. The two erythropoiesis‐stimulating agents currently available for use in neonates are recombinant human EPO and darbepoetin, a re‐engineered form of EPO with a longer half‐life. There have been many controlled trials of erythropoiesis‐stimulating agents for prevention of early and late neonatal anaemia, which have recently been the subject of Cochrane systematic reviews.228,229 There is no doubt that recombinant EPO is biologically effective in that it stimulates erythropoiesis in virtually all preterm infants and there is no evidence that preterm or term neonates are physiologically insensitive to EPO. EPO is also able to reduce red cell transfusion requirements in preterm infants.228 The recent Cochrane reviews also found that the effects of EPO administration varied depending upon when treatment was started. In particular, early EPO treatment, before the age of 8 days, reduced the number of red blood cell transfusions as well as the total transfusion volume and donor exposure.228 Early administration of EPO also reduced the risk of intraventricular haemorrhage (IVH), periventricular leucomalacia and NEC without a significant increase in the rate of retinopathy of prematurity. However, early administration of EPO is not currently recommended for routine use, largely because it is not clear that the modest reduction in transfusions is clinically meaningful. In addition, a large randomised study (PENUT; NCT01378273) recently reported that there was no neurodevelopmental benefit for high dose EPO administered within the first 24 hours of life until 32 weeks corrected gestational age.238 The results of late administration are also modest and the benefits remain unproven.229 Specifically, EPO started between 8 days and 28 days of life causes a small reduction in the number of red blood cell transfusions but has no significant effect on donor exposure. Given that EPO does have some impact on reducing transfusion requirements, it is possible that future changes in transfusion practice in response to safety concerns or parental acceptance may lead to a more defined role for erythropoiesis‐stimulating agents in neonatal medicine. At present, the main indications for EPO in neonates are to prevent anaemia in those who have received IUT for alloantibody‐mediated anaemia14 and in a non‐emergency situation where red cell transfusions are against the parents’ wishes but are not essential (e.g. preterm babies of Jehovah’s witnesses).239 The recommended dose is 300 μg/kg as a single subcutaneous injection three times per week starting in the first week of life as the Hb does not start to rise until 10–14 days of treatment. Iron supplements (3 mg/kg/day) should be started as soon as possible to prevent the rapid development of iron deficiency in EPO‐treated infants (the dose may need to be increased up to a maximum of 9 mg/kg/day if iron deficiency develops on the standard dose).240 As discussed earlier, red cell disorders associated with neonatal or fetal anaemia present in three main ways: with an isolated low Hb, with jaundice due to haemolysis or with hydrops fetalis. A diagnostic algorithm to help identify which of these causes is most likely, which can be excluded and what further investigations are most appropriate is shown in Figure 2.26. A list of the haematological causes of hydrops fetalis and the most helpful diagnostic clues is shown in Table 2.14. Table 2.14 Haematological causes of hydrops fetalis * G6PD deficiency in neonates most often presents with jaundice with few or no red cell morphological abnormalities and a normal Hb. CDA, congenital dyserythropoietic anaemia; DBA, Diamond–Blackfan anaemia; G6PD, glucose‐6‐phosphate dehydrogenase; Hb, haemoglobin concentration; HLH, haemophagocytic lymphohistiocytosis; PK, pyruvate kinase; TAM, transient abnormal myelopoiesis. Table 2.15 Causes of neonatal polycythaemia CAH, congenital adrenal hyperplasia; Hb, haemoglobin concentration; IUGR, intrauterine growth restriction. For practical purposes, in both term and preterm infants, polycythaemia can be defined as a central venous haematocrit of greater than 0.65, because there is an exponential rise in blood viscosity above this level.242 However, even at haematocrits greater than 0.70, only a minority of neonates have clinical signs of hyperviscosity, such as lethargy, hypotonia, hyperbilirubinaemia and hypoglycaemia.243 In severe cases, skin necrosis has been reported and polycythaemia is also likely to be a contributory factor in the pathogenesis of NEC and in thrombotic disorders such as neonatal seizures, stroke and renal vein thrombosis.244 Causes of polycythaemia are shown in Table 2.15. The most common cause is chronic fetal hypoxia secondary to IUGR, maternal hypertension/pre‐eclampsia or maternal diabetes. Neonates with Down syndrome are also frequently polycythaemic and this is secondary to the effects of trisomy 21 on fetal erythropoiesis rather than congenital heart disease.245 Another important cause of severe polycythaemia is TAPS (see earlier) where there has been chronic inter‐twin transfusion throughout the second and third trimesters of pregnancy.189 Treatment is controversial and is not necessary in infants with very minor signs (for example borderline hypoglycaemia or poor peripheral perfusion). Infants with neurological signs and a haematocrit greater than 0.65 should have a partial exchange transfusion (using a crystalloid solution such as normal saline) to reduce the haematocrit to 0.55. There is no evidence to support the use of FFP or albumin for this procedure.61 Although cyanosis in neonates is nearly always due to underlying cardiac or respiratory compromise, there are several rare and well‐recognised haematological causes of cyanosis (Table 2.16), which may cause diagnostic confusion. Cyanosis (or pseudocyanosis) is present at birth with α and γ chain methaemoglobins.246 The skin colour, brownish/slate, differs slightly from cyanosis due to an increased percentage of deoxyhaemoglobin. In the case of the γ chain variants haemoglobin F‐M‐Osaka, haemoglobin F‐M‐Fort Ripley and F‐M‐Circleville, the cyanosis disappears as β chain synthesis takes over from γ chain synthesis. With α chain variants the cyanosis persists. Haemoglobin M‐Iwate and haemoglobin M‐Boston are not only methaemoglobins but also have a low oxygen affinity so that there is both pseudocyanosis and true cyanosis. In addition, there are two γ chain variants – haemoglobin F‐M‐Fort Ripley and haemoglobin F‐M‐Osaka – that are methaemoglobins and also unstable. Neonatal methaemoglobinaemia can be acquired rather than inherited. Neonates are more susceptible to oxidation of haemoglobin than older infants and adults, both because haemoglobin F is more susceptible to oxidation than haemoglobin A and because methaemoglobin reductase activity is relatively low. The decision to transfuse a neonate with anaemia depends on the cause and natural history of the anaemia as well as the severity, the rate of fall in Hb, the postnatal and gestational age of the neonate and the clinical signs. The majority of red cell transfusions administered to neonates are small ‘top‐ups’ for anaemia of prematurity and/or to compensate for phlebotomy losses. Increasing recognition of the hazards of transfusion in neonates has led to re‐evaluation of transfusion thresholds (see recent reviews).247–249 Many countries have developed expert‐based consensus guidelines for neonatal transfusion which incorporate available evidence, much of which does not yet come from controlled trials. Two large randomised controlled trials of liberal versus restrictive transfusion policies in preterm neonates have recently been designed. The first, the Effect of Transfusion Thresholds on Neurocognitive Outcome of ELBW infants trial (ETTNO trial; NCT01393496), which was completed in October 2018, showed that using a liberal rather than restrictive red cell policy did not reduce the likelihood of death or disability at the age of 24 months.250 The Transfusion of Prematures trial (TOPs trial; NCT01702805) is due to complete in August 2023. In the meantime, the indications for top‐up transfusion and exchange transfusion recommended by the British Committee for Standards in Haematology are summarised in Table 2.17. Table 2.16 Haematological causes of cyanosis Table 2.17 Indications for red cell transfusion in the neonatal period* * Based on the recommendations of the British Committee for Standards in Haematology.61 The main risks of transfusion in neonates include: metabolic complications, such as hyperkalaemia and hypocalcaemia; transfusion‐transmitted infections, such as CMV infection if CMV‐unscreened blood products are used; and immunological complications, such as TA‐GvHD which, though rare in neonates, has a high mortality.230 Many of these risks can be reduced by the use of ‘paedipacks’ of up to six small‐volume red cell transfusion bags prepared from a single donor unit. The use of paedipacks reduces transfusion exposure for multiply transfused preterm neonates; although it may mean using older red blood cells, this has not been shown to adversely affect neonatal outcome.61 In the UK, transfusion of CMV‐seronegative blood products is also recommended for all transfusions in neonates up to 44 weeks corrected gestational age. The optimal transfusion volume for neonatal top‐up transfusions has not been established but most guidelines recommend 10–20 ml/kg because higher volumes may cause clinical problems due to volume overload. IUTs require irradiated blood and, when IUT has been necessary, except in a dire emergency, transfusion of the neonate should continue to be with irradiated components to prevent TA‐GvHD. The frequency of the most serious hazards of transfusion, such as transfusion‐related acute lung injury (TRALI) and transfusion‐associated circulatory overload (TACO), that are well recognised in older patients, is unknown in neonates. (For abbreviations used in the text of the illustrative cases, see page 110.)
2
Red cell disorders: anaemia, jaundice, polycythaemia and cyanosis
Neonatal anaemia
Definition and clinical significance of neonatal anaemia
Neonatal anaemia due to reduced red cell production
Low reticulocytes)
<20 × 109/l
Direct Coombs test
Negative
Bilirubin
Usually normal
Blood film
Red blood cell morphology usually normal
Red cell aplasia and/or dysplasia
Useful diagnostic tests
Parvovirus B19 infection
Blood film: disproportionate lack of polychromasia and NRBC (in setting of low Hb)
Maternal parvovirus B19 serology: IgM antibodies during pregnancy indicates in utero infection
Parvovirus B19 DNA (neonate ± mother)
Diamond–Blackfan anaemia
Family history
Exclude parvovirus B19 infection
Adenosine deaminase assay (pretransfusion), including parents
Molecular analysis (ribosomal protein genes*), including parents
Bone marrow aspirate
Congenital dyserythropoietic anaemia
Family history
Red cell indices (MCV usually increased)
Red cell morphology (macrocytosis and poikilocytosis)
Molecular analysis (known CDA genes)*
Bone marrow aspirate (characteristic dyserythropoiesis)
Electron microscopy (bone marrow cells)
Mitochondrial cytopathy:
Pearson syndrome
Bone marrow aspirate (vacuolation of erythroid and granulocytic precursors)
Molecular analysis of leucocytes (mitochondrial genome sequencing)
Parvovirus B19 and fetal/neonatal anaemia

Genetic red cell aplasia and dysplasia
Diamond–Blackfan anaemia
Pearson syndrome





Congenital dyserythropoietic anaemias

Neonatal anaemia due to increased red cell destruction
Immune haemolysis, including haemolytic disease of the fetus and newborn
Increased reticulocyte count
Usually >150 × 109/l
Direct Coombs test
Positive if immune
Jaundice
Unconjugated hyperbilirubinaemia
Blood film
Red blood cell morphology usually abnormal and provides helpful guide to likely diagnosis
Family history and ethnic origin
Often useful, including history of jaundice, anaemia, splenectomy, transfusions or gallstones
Causes of haemolysis in neonates with anaemia
Useful diagnostic tests
Immune – haemolytic disease of the newborn
Coombs test
Blood group of mother and baby
Neonatal blood film (spherocytes, polychromasia)
Maternal red cell antibody screen
Maternal immune (IgG) anti‐A/anti‐B (if ABO haemolytic disease suspected)
Eluate of antibody from neonatal red cells
Red cell membrane disorders
Neonatal blood film
Parental FBC, reticulocyte count and blood films
EMA dye‐binding test (largely replaced osmotic fragility testing)
Molecular analysis for pathogenic variants in genes known to cause primary red cell membrane disorders (e.g. SPTA1, SPTB, EPB41)
Red cell enzymopathies
Neonatal blood film
Parental FBC, reticulocyte count and blood films
G6PD enzyme activity assay
PK enzyme activity assay (helpful to assay levels in parental samples at the same time)
Molecular analysis for pathogenic variants in genes known to cause red cell enzymopathies (e.g. PKLR, GPI)
Haemoglobinopathies
Neonatal blood film
HPLC
Family studies if results difficult to interpret
Consider mass spectrometry or molecular analysis of globin genes if haemoglobinopathy strongly suspected but no diagnosis on HPLC (e.g. unstable haemoglobin)
Other inherited disorders, e.g. congenital thrombotic thrombocytopenic purpura, congenital haemolytic uraemic syndrome
Neonatal blood film
Plasma ADAMTS13 activity assay
Molecular analysis for pathogenic variants in genes known to cause congenital TTP (ADAMTS13) or HUS (cobalamin C)
Infections associated with neonatal haemolysis
Blood film
Microbiological tests for neonatal bacterial infection or congenital bacterial, viral or parasitic infection
Cytomegalovirus
Disseminated herpesvirus
Adenovirus
Rubella*
Syphilis
Malaria
Toxoplasmosis
Microangiopathic, e.g. Kaposiform haemangioendothelioma with or without Kasabach–Merritt phenomenon; vascular anomalies (renal artery stenosis, coarctation of the aorta)
Blood count and coagulation screen to look for consumptive coagulopathy
Blood film
Imaging
ABO haemolytic disease of the fetus and newborn


ABO
Rh
Haemoglobin concentration
Usually normal
Usually reduced and falls rapidly in first few hours or days
Coombs test
Occasionally negative
Always positive
Reticulocytes
Usually increased
Usually increased
Erythroblastosis
Uncommon
Nearly always
Spherocytes
Easily seen on blood films in very large numbers
Often inconspicuous
Causative antibodies
Anti‐A (usually mild)
Anti‐B (sometimes severe)
Anti‐D (often severe)
Anti‐c (often severe)
Anti‐C
Anti‐E
Rh haemolytic disease of the fetus and newborn



Management of haemolytic disease of the fetus and newborn
Neonatal haemolytic anaemia due to red cell membrane disorders
Hereditary spherocytosis
Condition
Diagnostic clues and useful tests
Hereditary spherocytosis
Autosomal dominant hereditary spherocytosis
Spherocytosis with negative Coombs test and/or maternal blood Group A, B or AB
Usually a strong family history – of HS, gallstones and/or neonatal jaundice or anaemia
Relatively common (1 in 2000–5000 in White Europeans)
Autosomal recessive hereditary spherocytosis
Presents with severe neonatal anaemia and/or hydrops fetalis
Usually no family history of anaemia or jaundice
Some patients with bi‐allelic SLC4A1 mutations have renal tubular acidosis
Hereditary elliptocytosis
Hereditary pyropoikilocytosis
Anaemia usually marked in neonatal period
Due to microspherocytes and other schistocytes, the MCV is typically very low (<70 fl) and platelet counts may be artefactually high
Homozygous/compound heterozygous for α spectrin variants
Often no family history but one or both parents usually have blood film typical of HE
Hereditary elliptocytosis with infantile poikilocytosis
Elliptocytosis with marked poikilocytosis and anaemia (Hb is normal in typical common HE)
Heterozygous for an HE α spectrin variant common in Black Africans
Often no family history but one parent usually has blood films typical of HE
Southeast Asian ovalocytosis
Southeast Asian ovalocytosis
Blood film shows stomatocytosis with a population of macro‐ovalocytes
Caused by heterozygous mutations in SLC4A1
Jaundice common
Mild (or no) anaemia; often have increased MCH
Family history, including origin from areas where the disease is prevalent (e.g. Thailand)
Hereditary stomatocytosis
Overhydrated hereditary stomatocytosis
May present with hydrops fetalis
MCHC may be decreased or normal
Blood film shows stomatocytes
Pseudohyperkalaemia (when sample stored at 4°C overnight)
Gene mutated in HS
Proportion of cases of HS*
Autosomal recessive HS
Autosomal dominant or sporadic HS
Alpha spectrin (SPTA1)
<5%
Yes
No
Beta spectrin (SPTB)
15–30%
Yes
Yes
Ankyrin‐1 (ANK1)
30–60%
Yes
Yes
Band 3 (SLC4A1)
20–30%
Yes
Yes
Band 4.2 (EPB42)
<5%
Yes
No


Hereditary elliptocytosis and hereditary pyropoikilocytosis


Southeast Asian ovalocytosis
Neonatal haemolysis due to red cell enzymopathies
Red cell enzymopathies
Diagnostic clues and useful tests
Glucose‐6‐phosphate dehydrogenase deficiency (G6PD gene)
Neonatal jaundice, often severe, despite normal blood film and Hb, presenting in the first 5 days of life in a male infant
Mother typically of African, Caribbean, Mediterranean, Arabic or Indian subcontinent origin
May also present in neonates a few weeks of age with sudden and profound drop in Hb due to acute oxidative haemolysis, often in association with infection (or drugs/chemicals)
Family history of anaemia usually absent but there may be a history of neonatal jaundice
Pyruvate kinase deficiency (PK gene)
Family history of anaemia, often severe and sometimes transfusion‐dependent
May present with neonatal hepatic failure and severe cholestasis
Males and females equally affected
No ethnic predisposition
Infantile pyknocytosis
Sudden onset of severe anaemia at 2–4 weeks of age, typically with Hb drop of 40–50 g/l over a few days; transfusion often required (usually only 1–2 transfusions)
More common in preterm babies
May occur in twins/siblings but otherwise no family history
Self‐limiting and confined to the neonatal period
Blood film shows features of oxidative haemolysis but G6PD and PK activity are normal
No genetic cause identified; glutathione peroxidase levels are often reduced
Rare enzyme deficiencies (gene mutated):
Chronic non‐spherocytic haemolytic anaemia
Family history of severe, often transfusion‐dependent haemolytic anaemia and/or consanguinity
Associated features, e.g. neurological problems
Glucose‐6‐phosphate dehydrogenase deficiency

Chemicals
Methylene blue
Naphthalene (mothballs)
Fava beans (mother)*
Antibiotics
Nitrofurantoin*
Some sulfonamides, e.g. sulfasalazine*
Quinolones (ciprofloxacin*, nalidixic acid*)
Chloramphenicol†
Trimethoprim*†
Isoniazid
Antimalarials
Primaquine*
Chloroquine†
Quinine†
Other medication
Rasburicase
Dapsone*
Aspirin*†
Pyruvate kinase deficiency

Infantile pyknocytosis
Other red cell enzymopathies
Other causes of neonatal haemolytic anaemia
Neonatal haemolytic anaemia due to infection
Neonatal anaemia due to extrinsic causes of red cell destruction
Neonatal anaemia due to haemoglobinopathies and other microcytic anaemias
Alpha thalassaemia major (haemoglobin Bart’s hydrops fetalis)
Condition
Genetic defect
Alpha globin disorders
Alpha thalassaemia major
Deletion of four α globin genes
Haemoglobin H disease* including that associated with the α globin variants, haemoglobin Constant Spring and haemoglobin Paksé
Deletion and/or mutation of three α globin genes
Unstable α chain variants
Hb Hasharon (α2Asp14→Hisβ2)
Gamma globin disorders
Unstable γ chain variants
Mutation of the Gγ or Aγglobin gene
Gamma beta delta thalassaemia
Deletion of the β globin gene cluster
Red cell disorder
Diagnostic clues and useful tests
Inherited sideroblastic anaemias
Family history
High serum iron, transferrin saturation and ferritin
Low hepcidin
Molecular analysis for mutations in the following genes:
Haemoglobinopathies – α thalassaemias (see Table 2.11)
Mother typically of Far Eastern, African, Mediterranean, Arab, Caribbean or Indian subcontinent origin
Neonatal anaemia (and hydrops) only if α thalassaemia major or haemoglobin H disease
Red cell membrane disorders – hereditary pyropoikilocytosis (see Table 2.7)
Anaemia usually marked
MCV is typically very low (<70 fl)
Homozygous/compound heterozygous for α spectrin variants
Often no family history but parents may have blood films typical of hereditary elliptocytosis
Chronic in utero blood loss (see Table 2.13)
Twin‐to‐twin transfusion (donor twin)
Chronic fetomaternal haemorrhage

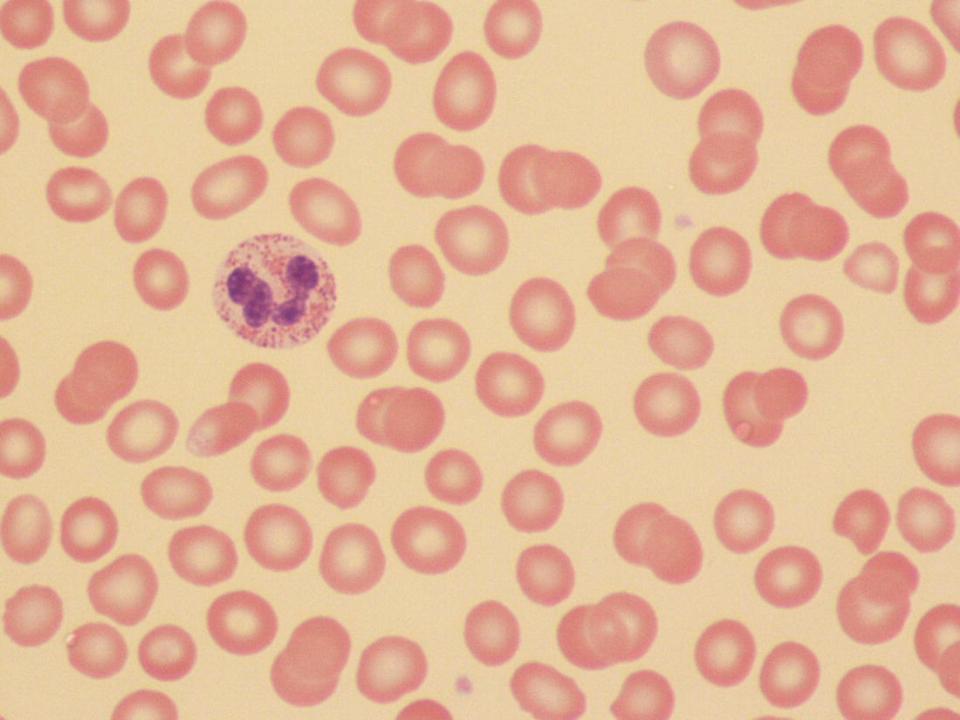
Haemoglobin H disease in the newborn
Neonatal haemolysis due to unstable α chain variants
Neonatal haemolysis due to unstable γ chain variants
Heterozygosity for εγβδ thalassaemia
Beta globin disorders in the neonatal period
Beta thalassaemias
Sickle cell disease
Neonatal microcytic anaemia
Sideroblastic anaemias presenting in the neonatal period


Neonatal anaemia due to blood loss
Diagnostic clues and useful tests
Fetomaternal haemorrhage: acute or chronic
Maternal Kleihauer test or measurement of haemoglobin F‐containing cells in the maternal circulation by flow cytometry
Neonatal blood film (erythroblastosis; dimorphic if acute on chronic blood loss)
Fetofeto haemorrhage (twin‐to‐twin transfusion)
Blood count on both twins
Blood film on both twins – donor twin will have erythroblastosis and, if chronic blood loss, may have hypochromic red cells
Fetal/neonatal haemorrhage: cephalohaematoma, intra‐abdominal bleeding, pulmonary haemorrhage
Blood film: erythroblastosis if large recent haemorrhage
Imaging to identify the site of bleeding
Neonatal anaemia due to blood loss
Twin‐to‐twin transfusion
Fetomaternal haemorrhage



Neonatal anaemia due to blood loss at or after delivery
Anaemia of prematurity
Management of anaemia of prematurity
Delayed clamping of the cord to minimise neonatal anaemia

Top‐up red cell transfusion
Erythropoiesis‐stimulating agents to treat and prevent neonatal anaemia
A simple diagnostic approach to neonatal anaemia
Condition
Diagnostic clues and useful tests
Failure of normal red cell production
Parvovirus B19
Diamond–Blackfan anaemia
Congenital dyserythropoietic anaemia, e.g. CDA type II, CDA type IV (KLF1 mutations)
Rare genetic syndromes, e.g. Southeast Asian ovalocytosis; IKZF1 mutation/deletion
Inappropriately low reticulocyte count
Blood film: characteristic red cell morphology (CDA and Southeast Asian ovalocytosis)
Congenital anomalies (DBA)
Pancytopenia, immune defects (IKZF1 mutation)
Haemolytic disease of the newborn
Rh (mainly anti‐D and anti‐c)
ABO (rare cause of hydrops and mainly due to anti‐B)
Other blood groups: anti‐Kell
Known maternal alloantibodies
Positive Coombs test
Previously affected sibling
Red cell membrane disorders
Hereditary stomatocytosis
Hereditary spherocytosis (rarely)
Hereditary xerocytosis (rarely)241
Family history
Blood film: characteristic red cell morphology
Negative Coombs test
Red cell enzymopathies *
Pyruvate kinase deficiency
Glucose phosphate isomerase deficiency
PK deficiency may present with unexplained neonatal hepatic failure; often difficult to establish the diagnosis from initial enzyme activity assays
Haemoglobinopathies
Alpha thalassaemia major (haemoglobin Bart’s hydrops fetalis)
Haemoglobin H disease, including related to haemoglobin Constant Spring
Typical red cell indices and blood film abnormalities
Family history and ethnic origin
Chronic fetal blood loss
Twin‐to‐twin transfusion
Monochorionic twins
Discrepant Hb between the twins
Storage disorders
Hepatosplenomegaly
Metabolic problems
Family history
Blood film (lymphocyte vacuolation)
Malignancies
Transient abnormal myelopoiesis in Down syndrome
Congenital acute myeloid or acute lymphoblastic leukaemia
Haemophagocytic lymphohistiocytosis
TAM: hepatosplenomegaly, ascites, pleural/pericardial effusions and/or skin rash in severe cases; leucocytosis and increased blast cells are the most reliable haematological feature; anaemia is uncommon
Acute leukaemia: hepatosplenomegaly, skin nodules, leucocytosis, increased blast cells, rapidly progressing anaemia and thrombocytopenia
HLH: hepatosplenomegaly, fever, multiorgan failure, disseminated intravascular coagulation
Condition
Diagnostic clues and useful tests
Chronic intrauterine hypoxia
Intrauterine growth restriction
Maternal hypertension
Maternal diabetes
Clinical evidence of intrauterine growth restriction: low birthweight, reduced end‐diastolic blood flow
Maternal history
Marked erythroblastosis despite high Hb
Associated mild/moderate thrombocytopenia resolving within 7–10 days ± mild, self‐limiting neutropenia
Howell–Jolly bodies
Chromosomal disorders
Trisomy 21
Trisomy 18
Trisomy 13
Typical facies and associated congenital anomalies
Other typical neonatal haematological abnormalities, e.g. thrombocytopenia, dysplastic white cells and increased blast cells
Twin‐to‐twin transfusion
Discrepant Hb concentrations in monochorionic twins (>30 g/l) – one twin plethoric and the other pale in severe cases
Endocrine disorders
Thyrotoxicosis
Congenital adrenal hyperplasia
Thyrotoxicosis: IUGR, goitre, exophthalmos, stare, craniosynostosis, flushing, heart failure, tachycardia, arrhythmias, hypertension, hypoglycaemia
CAH: arrythmias, vomiting, hyponatraemia, hypoglycaemia; ambiguous genitalia (females)
Delayed clamping of the cord
Relevant history of delayed clamping and no evidence of alternative causes
Neonatal polycythaemia
Haematological causes of cyanosis
Principles of red cell transfusion in neonates
Low‐affinity haemoglobin
Methaemoglobinaemia
Variant haemoglobin (Haemoglobin M)
Cytochrome b5 reductase (methaemoglobin reductase) deficiency
Oxidant exposure (high‐dose methylene blue, local anaesthetic)
Postnatal age
Haemoglobin threshold for transfusion
0–7 days
100–120 g/l (if oxygen‐dependent)
<100 g/l (if oxygen‐independent)
8–14 days
95–100 g/l (if oxygen‐dependent)
<75–85 g/l (if oxygen‐independent)
15 days onwards
85 g/l
Illustrative cases
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


