EPIDEMIOLOGY
PMF is a rare disease. The annual incidence of MF has been estimated to be 0.41 to 1.46 cases per 100,000 individuals.
21,22 The prevalence of MF in the United States has been estimated as 16,000 to 18,500 patients. MF is slightly more common in males and more common in older patients.
21 The median age of patients at the time of diagnosis is 67 years.
21 Among the Philadelphia chromosome negative MPNs (e.g., MF, PV, ET), MF is the most symptomatic and carries the worst prognosis.
A higher incidence of PMF and related MPN has been suggested for persons of Jewish Ashkenazi ancestry.
23 Benzene, industrial solvents, and radiation exposure have been reported to be associated with increased risk of PMF.
24, 25 and 26
PATHOGENESIS
Current understanding suggests that PMF occurs secondary to acquired mutations that target the hematopoietic stem cell.
1,12,27 As a result, ineffective hematopoeisis and proliferation of dysfunctional megakaryocytes are commonly seen in MF. The hallmark of the disease pathologically is bone marrow reticulin and collagen fibrosis, ineffective extramedullary hematopoiesis (EMH), and deregulated cytokine production.
The disease-initiating mutations remain unknown. Cytogenetic abnormalities originating on the progenitor cell level are well described in MF. In general, approximately half of patients with PMF display cytogenetic abnormalities that include del(20) (q11;q13), del(13)(q12;q22), trisomy 8, trisomy 9, del(12) (p11;p13), monosomy or deletions chromosome 7, and partial trisomy 1q. None of these abnormalities is specific to PMF, although the presence of either del(13)(q12;q22) or der(6)t(1;6)(q21-23;p21-23) is strongly suggestive of PMF diagnosis. Molecular cytogenetics using FISH did not reveal additional karyotypically occult cytogenetic lesions, but comparative genomic hybridization (CGH) studies have disclosed gains of chromosome 9p as the most frequent abnormality occurring in 50% of patients.
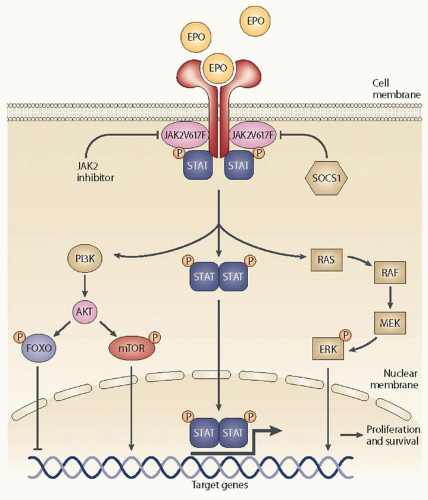
28,29, 30 and 31Deregulation of the Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway is the key contributor to the clinical phenotype of the disease regardless of the presence or absence of the
JAK2 V617F mutation. The mutation described is not a disease initiating abnormality. The JAK-STAT pathway plays a pivotal role in the differentiation and development of hematopoietic cells and the functioning of the immune system.
32 The JAK family comprises JAK1, JAK2, JAK3, and TYK2.
33 Cytokines and growth factors activate the extracellular portion of their cognate receptors,
33,34 which promote the recruitment of JAK proteins to associate closely with the intracellular portion of these receptors and the activation of
the JAK proteins via phosphorylation (
Fig. 83.1). Phosphorylation of JAK proteins, in turn, leads to the phosphorylation and activation of several intracellular downstream signaling proteins, such as STAT proteins.
35 Phosphorylated STATs translocate to the nucleus and act as inducible nuclear transcription factors, which lead to transcriptional modulation and eventual expression of cellular, molecular, and (patho) physiologic actions that were promoted by the initial signal (ligand).
The dysregulation of the JAK-STAT signaling pathway in hematopoietic progenitor cells has been implicated in the pathogenesis of MF. JAK-STAT signaling in the pathogenesis of MF also relates to its role in mediating signaling from proinflammatory cytokines. These cytokines are elevated in environments of myeloproliferative disease and contribute to the debilitating symptoms of MPNs.
36 Somatic mutations that contribute to this dysregulated JAK-STAT activity include gain-of-function mutations directly in
JAK2, or upstream signaling mutations such as the mutation of the thrombopoietin receptor (
MPL W515L),
18 and loss of JAK regulation by mutations in the gene for the lymphocyte-specific adaptor protein (
LNK exon 2 mutations).
37 Mutations in
JAK2 that lead to the constitutive activation of the JAK-STAT pathway, namely the
JAK2 V617F
14, 15 and 16 and
JAK2 exon 12 mutations, have been identified, although this latter mutation was primarily identified in patients with PV.
38 JAK2 V617F is the most common mutation occurring in a large proportion (50% to 60%) of patients with PMF.
14,15 With JAK2 activating mutations, the pathway becomes cytokine- and growth-factor-independent; therefore, even in the absence of these ligands, the intracellular signaling proteins are constitutively active. Clonal expansion leads to an increased
JAK2 V617F allele burden and homozygosity influencing disease phenotype and differentiation of PV and ET.
Megakaryocyte-derived transforming growth factor-β1 (TGF-β1) has been identified as the primary cytokine that mediates many of the bone marrow stromal changes (i.e., collagen fibrosis, osteosclerosis, angiogenesis) in PMF.
39,40 In mice, the PMF phenotype has been induced either by systemic overexpression of thrombopoietin (TPOhigh mice) or by megakaryocyte lineage-restricted underexpression of the transcription factor GATA-1 (GATA-1low mice). In both instances, the megakaryocytes display abnormal distribution of P-selectin that is believed to promote a pathologic interaction between megakaryocytes and neutrophils, resulting in the release of both fibrogenic and angiogenic cytokines including TGF-β, platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF), tissue inhibitors of matrix metalloproteinases, and neutrophilderived elastase and other proteases.
41, 42Another characteristic feature in PMF that accompanies the aberrant bone marrow stromal reaction involves peripheral blood expansion of both CD34-positive myeloid progenitors and endothelial cells.
43,44 Current consensus implicates both circulating progenitor cell trapping and abnormal cytokine stimulation of embryonic hematopoietic sites as mechanisms of hepatosplenic EMH in PMF.
45,46 Such a contention is supported by the high concordance between bone marrow and splenic tissue cytogenetic findings in PMF.
47Disease progression to blast phase (acute myeloid leukemia [AML]) is probably driven by additional clonal evolution and acquired mutations (
see section “Disease Course”).
CLINICAL FEATURES
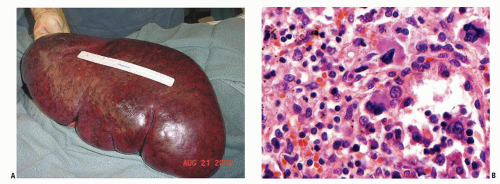
Splenomegaly is a well-established clinical feature of MF with 85% or more of MF patients presenting with palpable splenomegaly at the time of diagnosis. One third of patients will have marked splenomegaly (
Fig. 83.2) and 50% of patients will also have hepatomegaly.
48,49 Spleen-related symptoms include abdominal discomfort, early satiety, and pain under the left ribs. Portal hypertension and variceal bleeding can be morbid complications of splenomegaly. Some patients may experience severe pain secondary to splenic infarcts.
50Constitutional symptoms are common and often debilitating. Disease burden due to those symptoms often interfere with daily quality of life. These symptoms include fatigue, pruritus, night sweats, fever and bone/muscle pain, and cachexia. Presence of those symptoms is associated with worse outcome.
Cytopenias can dominate the course of the disease especially at the advanced stages. Two thirds of patients may have anemia at diagnosis and 20% are transfusion-dependent.
51,52 Thrombocytopenia may be present in 21% to 37% and leukopenia in 7% to 22%.
53Some patients present with leukocytosis (41% to 49% incidence), and/or thrombocytosis (13% to 31%). Furthermore, >10% of patients may present with extreme thrombocytosis (platelet count >1,000 × 10
9/L), whereas >20% present with marked leukocytosis (leukocyte count >20 × 10
9/L).
53 Patients with MF are also at risk for developing thrombo-hemorrhagic complications secondary to leukocytosis and/or thrombocytosis.
54Other potential symptoms and complications include ascites, portal hypertension, lymphadenopathy, pleural effusions, and nerve or cord compression secondary to EMH.
55Characteristic laboratory findings in MF may include peripheral blood leukoerythroblastosis, dacryocytosis, teardrop-shaped red blood cells (see
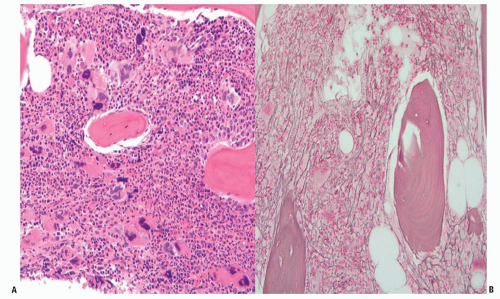
Fig. 80.9), circulating immature myeloid cells, increased serum lactate dehydrogenase, increased vitamin B12 levels, and hyperuricemia. The characteristic bone marrow aspirate and biopsy findings may be limited by the inability to collect an adequate bone marrow aspirate (so-called dry tap). The clustering of atypical megakaryocytes, which may often be mistaken as dysplasia by an inexperienced pathologist, is a pathologic hallmark of myeloproliferative syndromes.
56 Reticulin staining demonstrates an increased deposition of reticulin fibers. Collagen fibrosis can be appreciated and may be more disease-specific compared to reticulin staining. In some early cases of MF, the bone marrow could only be hypercellular with no evidence of fibrosis.
7 Figure 83.3 illustrates typical bone marrow findings in MF. Circulating levels of CD34
+, hematopoietic stem cells and progenitors are high in patients with MF compared with healthy patients as well as patients with other Philadelphia chromosome negative MPNs.
42