INTRODUCTION
SUMMARY
Myelodysplastic syndromes (MDS) are a heterogenous group of clonal hematopoietic neoplasms defined by morphologic dysmorphia, one or more blood cytopenias, and an increased risk of clonal evolution to acute myelogenous leukemia (AML).* These disorders can occur at any age but have an incidence that rises exponentially after age 40 years with a median age at diagnosis of 72 years. Most cases are acquired de novo through the accumulation of somatic mutations, although a small fraction arises after exposure to DNA damaging agents, such as chemotherapy and radiation. A minority of cases are the result of inherited mutations that predispose to the development of MDS and related myeloid disorders. Subtypes of MDS are largely defined by clinical features and range from refractory cytopenias (typically including anemia) to oligoblastic myelogenous leukemia with an increase in marrow myeloblasts (5 to 19 percent). Cases with 20 percent or more myeloblasts in the marrow (an arbitrary boundary) or specific chromosomal translocations are defined as AML. The diagnostic criteria for MDS include dysmorphogenesis in one or more blood cell lineages, often resulting in exaggerated apoptosis during later stages of maturation. Poikilocytosis, anisocytosis, anisochromia, and basophilic stippling are features of the abnormal red cells. The marrow usually contains increased erythroid precursors with dysmorphic features, including nuclear distortions and scanty, poorly hemoglobinized cytoplasm or macroerythroblasts. Pathologic ring sideroblasts are a common feature used to define particular subtypes of MDS. Neutrophils may have bilobed or hypersegmented nuclei and hypogranulated cytoplasm in association with increased marrow granulocyte precursors. Giant and microcytic platelets, sometimes with abnormal or absent granulation, in the blood are associated with megakaryocytic hyperplasia and atypical lobulation of the nucleus, megakaryocyte clustering, and decreased marrow megakaryocyte size. Clonal cytogenetic abnormalities occur in approximately 50 percent of patients, typically as recurrent deletions of entire chromosomes or chromosomal segments. Trisomy 8 is the only frequent copy number gain and recurrent translocations are rare. Various prognostic models for MDS incorporate cytogenetic abnormalities along with marrow blast proportion and blood cytopenias to predict the mortality and risk of clonal evolution to AML. The selection and timing of therapy for MDS is largely driven by risk stratification. Newer prognostic scoring systems have begun to consider somatic mutations as markers of disease-associated risk as pathogenic driver mutations can be identified in almost all cases of MDS and several lesions have a prognostic significance that is independent of other known risk factors. As detectable somatic events, driver mutations are markers of clonal hematopoiesis and could help establish the diagnosis in some cases. Recurrent somatic mutations identify the heterogenous molecular pathways frequently disordered in MDS. These include mutations in multiple components of the RNA splicing machinery, several epigenetic regulators of DNA methylation and histone modifications, various hematopoietic transcription factors, and growth factor signaling pathway members among others. Certain mutations are tightly associated with clinical features including ring sideroblasts, chromosomal instability, and severe cytopenias. Current treatment guidelines for MDS are based on clinical risk assessments and generally do not consider genetic abnormalities. The exception are cases with del(5q) and noncomplex karyotypes that have a high rate of deep and sustained responses to the immunomodulator drug lenalidomide. Many patients with lower-risk MDS may not require treatment. Others may benefit from hematopoietic growth factor support or immune suppression with antithymocyte globulin and a calcineurin inhibitor, depending on specific clinical features. Higher-risk MDS is typically treated with one of the hypomethylating agents, azacitidine or decitabine, and eligible patients are evaluated for hematopoietic allogeneic hematopoietic stem cell transplantation, the only potentially curative treatment for MDS. Despite these treatment options, outcomes for persons with MDS remain poor overall. Novel therapies targeting recently identified molecular pathways have been developed and are being pursued in clinical trials.
Acronyms and Abbreviations
AHSCT, allogeneic hematopoietic stem cell transplantation; ALIP, abnormal localized immature precursor; ALL, acute lymphocytic leukemia; AML, acute myelogenous leukemia; ATG, antithymocyte globulin; ATRA, all-trans-retinoic acid; aUPD, acquired uniparental disomy; CALGB, Cancer and Leukemia Group B; CDR, commonly deleted region; CLL, chronic lymphocytic leukemia; CMML, chronic myelomonocytic leukemia; ESA, erythropoiesis-stimulating agent; FAB, French-American-British; FPD, familial platelet disorder; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; HLA, human leukocyte antigens; IDH, isocitrate dehydrogenase; IL, interleukin; IPSS, International Prognosis Scoring System; IPSS-R, International Prognosis Scoring System Revised; LGL, large granular lymphocyte; MAP, mitogen-activated protein; M-CSF, monocyte colony-stimulating factor; MDS, myelodysplastic syndrome; MDS/MPN, myelodysplastic syndrome/myeloproliferative neoplasm; miRNA, microRNA; NCI, National Cancer Institute; NK, natural killer; PLK, polo-like kinase; PRC, protein-repressive complex; RA, refractory anemia; RAEB, refractory anemia with excess blasts; RAEB-t, refractory anemia with excess blasts in transformation; RARS, refractory anemia with ring sideroblasts; RARS-t, refractory anemia with ring sideroblasts and thrombocytosis; RBC, red blood cell; SEER, Surveillance, Epidemiology and End Results; snRNP, small nuclear riboprotein complex; TET, ten-eleven translocation; TGF, transforming growth factor; TNF, tumor necrosis factor; WT, Wilms tumor; WHO, World Health Organization.
*We acknowledge Drs. Marshall Lichtman and Jane Liesveld who wrote this chapter in previous editions of this text. We have retained sections of their previous chapter.
DEFINITION
Myelodysplastic syndromes (MDS) represent a collection of hemapoietic neoplasms characterized by abnormal differentiation, dysmorphology, and resultant blood cytopenias. The hallmarks of these clonal disorders include exaggerated apoptosis of hematopoietic precursors in the marrow, common chromosomal abnormalities, frequent somatic gene mutations, and a variable predilection to undergo clonal evolution to acute myelogenous leukemia (AML). The clinical course of patients with MDS is variable and ranges from relatively indolent clonal cytopenias (e.g., refractory anemia) with a low rate of AML transformation, to more aggressive disease defined by an increased proportion of marrow myeloblasts, oligoblastic myelogenous leukemia (refractory anemia with excess blasts), and a greater risk of progression to AML.
Despite their phenotypic variability, MDS share a common pathophysiology. They are neoplasms derived from the clonal expansion of a somatically mutated, multipotent hematopoietic progenitor cell. A wide range of genetic aberrations can contribute to the development and progression of MDS and these somatic mutations can exist in an even greater variety of combinations. The specific profile of genetic lesions present in a given case contributes to the eventual disease phenotype.
MDS are closely related to other myeloid neoplasms such as AML and some myeloproliferative neoplasms. The dysmorphia of neoplasia (commonly referred to as dysplasia in describing MDS) refers to the abnormal morphology than can be observed in neoplastic mature blood cells and maturing marrow erythroid, granulocytic, and megakaryocytic precursor cells, and is one of the distinguishing characteristics of MDS. The dysmorphia is essential in diagnosis, but is an epiphenomenon. The essential abnormality is the neoplastic transformation of a primitive multipotential myeloid cell. A lower fraction of myeloblasts in the marrow is a reflection of its being at the less-severe end of the spectrum of myelogenous leukemia and is a feature of the diagnosis, creating an arbitrary division between MDS and AML. The diagnostic criteria that separate various myeloid neoplasms (and MDS subtypes) from each other are somewhat ambiguous because they are so closely related. This is evident at the molecular level where no particular genetic abnormality or mutation profile is entirely unique to MDS. Both somatic mutations and chromosomal abnormalities found in MDS are observed in related disorders, albeit often with different frequency. RNA splicing factors and epigenetic regulators are the most common classes of genes affected by mutations in MDS followed by mutations in transcription factors, tyrosine kinase signaling genes, and TP53. MDS should be thought of as a minimal to moderately deviated neoplasm in the spectrum of myelogenous leukemias (Chap. 83).
A minority of MDS cases are attributable to prior exposure to DNA-damaging agents, including chemotherapy, high-dose ionizing radiation, and benzene-containing compounds. The latter external factor requires an exposure of sufficient duration and magnitude to be considered causal, rare now in countries with regulations regarding the content of benzene in products such as paints and solvents. Cigarette smoking is considered a risk factor for AML by the U.S. Public Health Service and, presumably, this should apply to MDS, although it has not been studied as extensively. Other chemical exposures have not been established as causative agents by the International Agency for Research on Cancer. A small number of patients come from families with a high penetrance of MDS and related myeloid disorders or have an inherited or congenital syndrome predisposing to MDS. Although rare, identification of the genetic abnormalities in many of these cases has informed our understanding of the molecular pathophysiology of MDS in general. Yet, the vast majority of MDS cases are age-related without a clear precipitating factor. Although MDS can occur at any age, including rare pediatric forms, its incidence increases exponentially after 40 years of age, making it one of the most common myeloid neoplasms of older adults. This pattern is related to the evidence that somatic mutations in primitive hematopoietic cells increase significantly with age.1
HISTORY
MDS have historically been subject to confusing and shifting terminology and definitions related to incomplete biologic understanding of disease.2,3 With the advent of routine molecular analysis of primary patient samples and increasing insight into disease pathobiology, classification of MDS is likely to evolve from the current systems based on cytologic morphology and enumeration of marrow and blood cell subsets to nosology based on DNA mutation patterns, clonal architecture, and predicted evolution to AML.4
Although anemia had been recognized since the early 19th century as a specific deficiency of red cells (also known as “colored corpuscles,” a term that distinguished these cells from leukocytes in the era before histological stains), marrow biopsies were not regularly performed on living patients until after the 1920s.5 Therefore, conditions such as MDS that are associated with and defined by specific marrow findings could not be described as distinct syndromes until the first half of the 20th century. Still, early suggestive reports can be found in the medical literature: a 1907 report by Luzzatto of megaloblastic “pseudo-aplastic anemia,”6 for example, could have included MDS cases.
In the mid-1920s, Di Guglielmo in Naples described a group of marrow disorders associated with bizarrely shaped erythrocytes and cytopenias, which in some cases ultimately proved fatal.7 For many years thereafter, a heterogeneous group of marrow disorders associated with anemia and erythroid dysmorphology were called “Di Guglielmo syndrome” by hematologists, a term that was used variably and is still employed occasionally as an eponymous description of erythroleukemia (AML M6). Some cases of Di Guglielmo syndrome would be called MDS today.
The first MDS-specific term familiar to contemporary hematologists, “refractory anemia,” was coined in the 1930s to describe patients who had unexplained anemia as a consequence of marrow underproduction and who failed to respond to treatment with the available hematinics: iron salts and the liver extract found by Minot and Murphy in the early 1920s to cure pernicious anemia.8,9 It is not clear how many of the 100 cases of refractory anemia in the classic 1938 series of Rhoads and Barker in New York City actually had MDS—many appear to have had anemia of chronic disease from infections or associated malignancies, such as Hodgkin lymphoma—but this term and its cognate, “refractory cytopenias,” are still used today to describe some of the lower-risk forms of MDS with blast proportions less than 5 percent.9,10
In 1942, Chevallier and colleagues in France discussed syndromes they labeled as “odo-leukemias.”11 The French investigators chose the Greek word odo, meaning threshold, to highlight disorders on the threshold of leukemia, and proposed leucoses as a generic term for the leukemias so that marked variations in white cell counts and other highly variable presenting features would not engender inappropriate terminology. However, this proposal was neglected, in part because the paper was published in French and there was no international network of hematologists with which to discuss such concepts in the 1940s.
Despite such provincialism, the idea that what would later be known as MDS could precede and terminate in AML soon made its way in the English medical literature. In 1949, Hamilton-Paterson in London used the term preleukaemic anemia to describe patients with refractory anemia antecedent to AML.12 In 1953, Block and coworkers in Chicago expanded the concept to include cytopenias of all lineages and described cases that closely fit with our current concepts of a clonal myeloid hemopathy prior to evolution to overt AML.13 In 1956, Björkman in Malmö, Sweden described four cases of idiopathic refractory sideroblastic anemia, one of which terminated in AML.14 Descriptive terms, such as herald state of leukemia, refractory anemia, sideroachrestic anemia, pancytopenia with hyperplastic marrow, and others, were coined to describe the various hematopoietic derangements that preceded the onset of florid AML. By the 1970s, the relationship of acquired idiopathic cytopenias to the subsequent onset of AML had become broadly appreciated, although such cases were still thought to be rare. In a 1973 review, Saarni and Linman found only 143 cases of “preleukemic anemia” in the medical literature.15
In 1970, Dreyfus proposed the designation “les anémies réfractaires avec excès de myeloblasts” (i.e., refractory anemia with excess myeloblasts) and in 1976 he and colleagues proposed a preliminary classification of these syndromes. Dreyfus published a paper in which refractory anemia with an excess of myeloblasts was amplified, parenthetically, as smoldering acute leukemia.16,17 The synonym, oligoblastic leukemias, had been used also to describe those cases with low proportions of leukemic myeloblasts and relatively protracted courses.18,19
In 1975, at a conference held in Paris, Bessis and Bernard used the term hematopoietic dysplasia, later shortened to myelodysplasia, for the group of disorders having a more indolent course than AML. The concept that neoplasia is a tissue abnormality defined by its origin in the mutation(s) within a single cell (monoclonality) and that dysplasia is a polyclonal tissue change, not neoplasia, was ignored and took a back seat to the participants’ primary interest in the dysmorphia of cells that characterized most of these syndromes, hence the application of the term dysplasia, which has become entrenched.
In the year following the Paris conference, a group of seven hematopathologists from France, the United States, and England—the “French-American-British (FAB) Co-Operative Group”—instituted the classification of acute leukemia,19 developed by Dalton and Dacie, and called it the FAB classification.20 In the FAB classification, acute leukemia was defined by the presence of 30 percent blasts in the marrow; the original report also included two preleukemic syndromes that should be distinguished from acute leukemia, refractory anemia with excess blasts (RAEB), and chronic myelomonocytic leukemia (CMML), defined by greater than 1000/μL of blood monocytes. Because most patients with preleukemia do not go on to develop leukemia, the FAB group proposed the term “dysmyelopoietic syndrome” as an alternative to preleukemia.20 A few years later, “dysmyelopoietic syndrome” was revised to become the “myelodysplastic syndrome(s)” still used today.
The 1976 FAB classification included only two subtypes of dysmyelopoietic syndromes, but in 1982 the FAB proposed a specific MDS classification that included five entities: refractory anemia (RA), refractory anemia with ring sideroblasts (RARS), RAEB (defined by 5 to 19 percent marrow blasts), refractory anemia with excess blasts in transformation (RAEB-t, defined by 20 to 29 percent marrow blasts), and CMML.21 The 1982 FAB classification, while not without limitations, proved influential and was the basis of subsequent classifications of MDS and related disorders by the World Health Organization (WHO). Subsequent developments in prognostic scoring systems and biologic understanding of MDS are described below.
CLASSIFICATION
A classification of MDS and related disorders was defined by the WHO in 2001 and revised in 2008. It includes six major subtypes of disease distinguished by the type and number of dysplastic lineages, the proportion of marrow blasts, and in one subtype, the presence of a specific chromosomal abnormality, del(5q). These subtypes include (1) refractory cytopenia with unilineage dysplasia (RCUD), which is typically RA, (2) RARS, (3) RA with ringed sideroblasts and thrombocytosis (RARS-t), (4) MDS with isolated del(5q), (5) refractory cytopenia with multilineage dysplasia, (6) RAEB (type 1 or type 2 depending in the proportion of marrow or blood blasts), and (7) unclassifiable MDS (Table 87–1). MDS patients with 20 to 29 percent marrow blasts, previously defined as RAEB-t, are considered to have AML in the WHO classification. However, this sharp cutoff is arbitrarily defined. In practice, patients with blast proportions in this range have comparable outcomes to patients with RAEB-2, including similar rates of benefit from MDS-directed therapy.
1. Refractory cytopenia with unilineage dysplasia (RCUD)
Most often is refractory anemia (RA) but can be refractory neutropenia (RN) or refractory thrombocytopenia (RT) in rare cases 2. Refractory anemia with ring sideroblasts (RARS)
The cutoff for ring sideroblasts is arbitrary and does not reflect the clinical behavior of this subtype as accurately as the frequently associated mutations of SF3B1 3. Myelodysplastic syndromes (MDS) associated with isolated del(5q)
This subtype overlaps with, but is not entirely synonymous with the “5q-minus syndrome” recognized prior to the establishment of the WHO classification system for MDS 4. Refractory cytopenia with multilineage dysplasia (RCMD)
5. Refractory anemia with excess blasts (RAEB)
6. Unclassifiable MDS (MDS-U)
|
The boundaries between subtypes involving percentages of myeloblasts or ring sideroblasts are also arbitrarily defined. Patients may see their disease classification change over time as a result of clinical progression or response to therapy. Clonal cytopenias with dysmorphia may also be present in patients with myeloproliferative features such as thrombocytosis or monocytosis. CMML, for example, was previously considered an MDS subtype in the FAB classification, but is now recognized as a myelodysplastic/myeloproliferative neoplasm (MDS/MPN) overlap syndrome. Some patients diagnosed with MDS may have their diagnosis change to CMML once their monocyte count exceeds 1 × 109/L. Similarly, RARS may be reclassified as RARS-t, if the platelet count rises above 450 × 109/L. CMML, RARS-t, and related MDS/MPN subtypes are discussed separately in Chap. 89 with the chronic myelogenous leukemias.
Classification systems for MDS have descriptive merit, but do not capture many disease variables with clinical significance. The WHO subtypes of MDS identify patients with similar prognoses because patients with multilineage dysplasia and increasing blast percentages are at higher risk of AML transformation and death. However, dedicated prognostic models that include features not considered by the WHO classification system are better suited for the estimation of disease risk. Similarly, WHO-defined subtypes do not necessarily share common pathogenic elements or identify groups of patients most likely to respond to particular therapies. Our greater understanding about the underlying molecular abnormalities that occur in MDS demonstrate how clinically defined subtypes are genetically very heterogenous. It is likely that future classification systems will consider somatic mutations in MDS driver genes, genes responsible for the clonal outgrowth and the eventual development of disease, as a basis for defining disease subtypes.
EPIDEMIOLOGY
The incidence and prevalence of MDS have been difficult to establish, in part because patients are not consistently reported to central cancer registries.22,23,24 Only since 2001 has the United States (U.S.) National Cancer Institute (NCI) Surveillance, Epidemiology and End Results (SEER) database included MDS cases (Fig. 87–1).25 In 2003, approximately 10,000 new cases were reported to NCI SEER, most “unclassified” (i.e., not distinguished as lower- or higher-risk).25 Improved methods of case ascertainment using claims-based data suggest a high rate of unreported cases and also indicate that MDS is one of the most common hematological malignancies.26
Figure 87–1.
The annual incidence of myelodysplastic syndrome shown by age. There is an exponential (approximately linear on semilogarithmic plot) increase in incidence from age 40 years on. In persons younger than age 40 years, the incidence is so low that it is aggregated as <40 years. (Data from the United States National Cancer Institute, Surveillance, Epidemiology, and End-Results Program.)
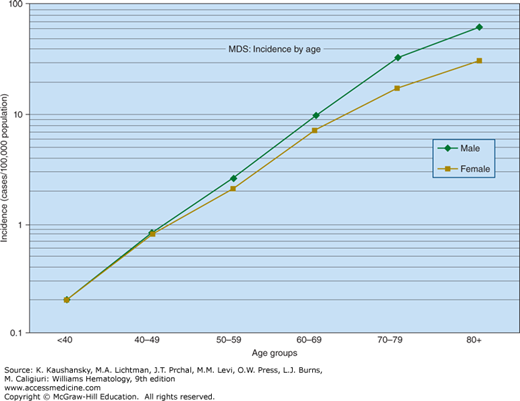
Claims-based data suggest that conservatively, at least 30,000 new cases annually are diagnosed in the United States.27 It is likely that many additional elderly patients with unexplained cytopenias have MDS, but are incompletely evaluated because of severe comorbid conditions limiting life expectancy, clinician oversight of blood test results, or a sense of nihilism. Rates of MDS are similar in Western Europe and the United States.28
In certain Asian countries and Eastern Europe, MDS is diagnosed at a younger age on average than in the United States and Western Europe.29,30,31 The subtypes of MDS that are diagnosed in different regions of the world are also distinct32,33; for instance, for unclear reasons RARS is rare in Japan compared to the West.34 Exposure to ionizing radiation from the 1945 Hiroshima and Nagasaki atomic bomb explosions continues to be associated with an increased MDS risk among exposed Japanese more than 50 years after the events.35
The median age at MDS diagnosis in the United States is approximately 71 years.36 Onset of MDS before the age of 50 years is uncommon, except in cases preceded by irradiation or cytotoxic chemotherapy given for another malignancy.37,38,39 MDS, as defined by the WHO classification, occurs in children ages 5 months to 15 years at a rate of approximately one per 1 million children per year. In contrast to adults, most pediatric cases are oligoblastic myelogenous leukemia (RAEB); clonal sideroblastic anemia is rare.40,41 A proportion of childhood cases evolve from inherited predisposing diseases, such as Down syndrome and Fanconi anemia, or are associated with germline GATA2 or RUNX1 mutations.42,43,44 Some cases of Fanconi anemia first present in adulthood as MDS, often in the absence of typical dysmorphology.45 Dyskeratosis congenita and other telomeropathies can also end in MDS, but many congenital marrow failure syndromes such as Diamond-Blackfan anemia or Shwachman-Diamond syndrome do not carry an increased MDS risk.46
With the exception of the 5q-minus syndrome, males are affected with MDS up to 1.5 times as often as females.47 Case-control studies of possible occupational or environmental associations have provided many possible candidates as contributors to MDS, but none other than benzene (exposure of ≥40 parts per million [ppm]-years) has been observed consistently.48,49,50,51 Cigarette smoking, and a family history of hematologic malignancy also seem plausible risk factors.52 Chemicals other than benzene have not been established as causative factors by epidemiologic studies that fully meet the guidelines proposed by Bradford Hill for causation by an external factor. Moreover, given the requirement for biologic plausibility, such chemicals should be shown to induce the specific driver mutations required to cause MDS.
ETIOLOGY AND PATHOGENESIS
The etiologic factors that increase the incidence of MDS are similar to the factors affecting the incidence of AML (Chap. 88). Exposure to prolonged or high levels of benzene,34,35 chemotherapeutic agents, particularly alkylating agents and topoisomerase inhibitors,36,37,38,39,40,41,42,43 and radiation44,45 increases the risk of these clonal hemopathies. These agents may cause DNA damage, impair DNA repair enzymes, and induce loss of chromosome integrity. Most cases of secondary or posttreatment MDS occur in patients treated for a lymphoma or a solid tumor. Increasing reports of MDS as a complication of treatment of myeloid diseases, such as acute promyelocytic leukemia, may reflect a second clonal myeloid disease from another primitive hematopoietic cell injured during therapy.43 The increased life span of patients with acute promyelocytic leukemia and other cancers after effective therapy may make these events more common. More common environmental exposures, such as cigarette smoke, may contribute to the likelihood of developing MDS.
Inherited diseases, such as Fanconi anemia, known to predispose to AML development occasionally evolve instead into a clonal myeloid hemopathy (see Chap. 88, Table 88–1).46 Other syndromes, of either a familial (inherited) or spontaneous nature, have been associated with a high risk of developing myeloid neoplasms. Germline mutations of the hematopoietic transcription factor RUNX1 are associated with a familial platelet disorder with predisposition at AML (FPD-AML). Affected individuals often have qualitative and quantitative platelet abnormalities that precede the development of a more aggressive myeloid neoplasm such as MDS or AML. Transformation typically occurs in the third decade of life, but penetrance is variable between individuals and kinships. The long latency prior to progression suggests that the acquisition of additional cooperating mutations is required for transformation. Somatic RUNX1 mutations are also common in de novo and therapy-related MDS cases highlighting the oncogenic driver nature of these abnormalities. In contrast, somatic mutations of Fanconi anemia genes are extremely rare in MDS. Congenital FANC mutations may instead cause DNA damage and accelerated exhaustion of normal stem cells allowing mutant clones to expand more readily. Similarly, inherited CCAAT/enhancer binding protein alpha (C/EBPA) mutations, often associated with eosinophilia, typically predispose to AML without an MDS-like clinical phase and are only rarely found as somatic mutations in MDS.
Congenital mutations of another hematopoietic transcription factor, GATA2, have been linked to familial MDS.53 The syndromic manifestations of germline GATA2 mutations are highly varied and can include lymphedema, cutaneous warts, sensorineural hearing loss, pulmonary alveolar proteinosis, and disseminated nontuberculous mycobacterial infections.54,55 Several distinct clinical syndromes that include subsets of these features are now known to be caused by germline GATA2 mutations. These include Emberger syndrome comprising MDS, verrucae, and congenital lymphedema, as well as the MonoMAC syndrome comprising monocytopenia and nontuberculous mycobacterial infections.55,56,57,58 Not all patients show overt syndromic features prior to developing MDS, even as adults, and several pediatric marrow failure syndromes can be associated with germlinel GATA2 mutation in the absence of syndromic features.59
The combined incidence of familial (<2 percent) and therapy-related MDS (~5 to 10 percent) pales in comparison to the frequency of de novo MDS that has age as its dominant predisposing factor. This may be simply a matter of probability, with aged stem cells being more likely to have acquired somatic driver mutations. It may also reflect age-related changes in the microenvironment or stem cell epigenetic state as hematopoietic stem cells from elderly persons without disease are known to have an exaggerated myeloid differentiation bias.60 In concert, age-related drop out of normal hematopoietic stem cells could lead to oligoclonal, or even monoclonal, hematopoiesis derived from stem cells with weakly selective abnormalities that then serve as fertile ground for cooperating MDS-related somatic mutations.61
MDS arise from the clonal expansion of a mutated multipotential hematopoietic cell. For patients without excess blasts, the cell of origin is presumed to be a lymphohematopoietic pluripotential stem cell based on the presence of disease-associated driver mutations in cells that share the surface protein immunophenotype of functionally defined stem cells.62 Subsequent evolution measured by the acquisition of additional mutations takes place in this cellular compartment and can occur in more differentiated progenitors, if they confer the capacity for sustained self-renewal. Evidence for the clonal nature of MDS is supported by studies of skewed X-chromosome inactivation in female patients heterozygous for glucose-6-phosphate dehydrogenase isoenzymes. The hematopoietic progenitors,63,64 and sometimes B lymphocytes,65 of such patients had only one isoenzyme present, supporting the concept of clonal expansion of a neoplastic early progenitor cell. Subsequent studies confirm the presence of acquired chromosomal abnormalities and somatic mutations in hematopoietic progenitors as well as in B and T lymphocytes in some, but not all, cases.62,66,67,68,69,70,71,72
This process of clonal expansion takes place in the context of the marrow microenvironment and host immune response (Chap. 5). These features extrinsic to the cells in the neoplastic clone generate the selection pressures that drive disease evolution and can significantly influence the clinical manifestations of MDS.
The hallmark of clonal hematopoiesis is the presence of a somatic genetic abnormality. Approximately 50 percent of patients with MDS will have a grossly abnormal karyotype, typically in the form of a partial or total chromosomal deletion. A fraction of the remaining cases with a “normal” karyotype will have cryptic cytogenetic abnormalities that can include small microdeletions and areas of copy number neutral loss of heterozygosity. This latter phenomenon occurs by mitotic recombination during cell division and results in acquired uniparental disomy (aUPD) where both copies of a large chromosomal segment appear to be derived from a single parent. The most common somatic genetic lesions in MDS are mutations of individual genes. More than 50 recurrently mutated genes have been identified with nearly all patients harboring one or more such mutations (Table 87–2).
| Mutated Gene | Frequency in MDS (%) | Prognostic Value | Additional Information | |
|---|---|---|---|---|
| Splicing | SF3B1 | 20–30 | Favorable | Strongly associated with ring sideroblasts |
| SRSF2 | 10–15 | Adverse | More frequent in CMML | |
| U2AF1 | 8–12 | Adverse | Associated with del(20q) | |
| ZRSR2 | 5–10 | ? | ||
| Epigenetic regulators | TET2 | 20–25 | Neutral | More frequent in CMML |
| DNMT3A | 12–18 | Adverse | ||
| IDH1/IDH2 | <5 | ? | ||
| ASXL1 | 15–25 | Adverse | More frequent in CMML | |
| EZH2 | 5–10 | Adverse | More frequent in CMML | |
| ATRX | <2 | ? | Associated with ATMDS | |
| KMD6A | <2 | ? | ||
| Transcription | RUNX1 | 10–15 | Adverse | Familial in rare cases |
| GATA2 | <2 | ? | Commonly familial, rarely somatic | |
| ETV6 | <5 | Adverse | Rarely translocated in MDS | |
| PHF6 | <2 | ? | ||
| TP53 | 8–12 | Adverse | Associated with complex karyotypes | |
| Cohesins | STAG2 | 5–10 | ? | |
| RAD21 | <5 | ? | ||
| SMC3 | <2 | ? | ||
| SMC1A | <2 | ? | ||
| Signaling | NRAS/KRAS | 5–10 | Adverse | More frequent in CMML |
| JAK2 | <5 | Neutral | Enriched in RARS-t | |
| CBL/CBLB | <5 | Adverse | More frequent in CMML | |
| PTPN11 | <2 | Adverse | More frequent in JMML, can be germline | |
| Others | GNAS/GNB1 | <2 | ? | G-protein signalling pathway |
| BRCC3 | <2 | ? | DNA repair pathway | |
| PIGA | <2 | ? | Cause of PNH clones | |
| TERT/TERC | <2 | ? | Can be germline | |
| FANC genes | <2 | ? | Typically germline |
The recurrent nature of many of these genetic events has helped identify molecular mechanisms associated with the development and progression of MDS. Many of these lesions are associated with particular clinical phenotypes, including differences in disease manifestation, response to therapy, risk of AML transformation and overall survival. However, the use of genetic features to classify disease subtypes or personalize the care of individual patients is still rudimentary.
Chromosomal amplifications and translocations are relatively rare events in MDS compared with other hematologic malignancies. The most frequent chromosomal abnormalities seen in MDS, present in nearly half of all cases, are deletions of chromosomal segments or loss of entire chromosomes (monosomies). Several such lesions are recurrent and typically involve commonly deleted regions that are presumed to harbor one or more tumor-suppressor genes. Identifying individual gene drivers from these regions has been challenging because of the large amount of genomic territory that they encompass and the likelihood that multiple gene losses cooperate to generate a disease-related phenotype.73 However, MDS-related chromosomal abnormalities do have important clinical significance. For example, they can establish the presence of clonal hematopoiesis and in the appropriate context, can serve as presumptive evidence of MDS. Chromosomal abnormalities are key elements in the determination of prognosis and in the case of del(5q), can predict response to a particular treatment.
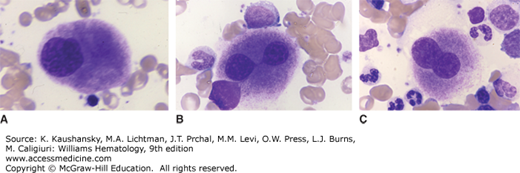
Del(5q) Deletion of the long arm of chromosome 5 is the most common karyotypic abnormality observed in MDS, occurring in 15 percent of cases, half of which have several other karyotype abnormalities. Studies examining the breakpoints of deletions across multiple patients have identified two commonly deleted regions (CDRs), one on 5q31.1 and the other at 5q32–33.3. Patients with del(5q) often have deletions that encompass both CDRs and can include proximal and distal chromosomal regions as well. Larger deletions that include more proximal genes, like APC, and more distal genes, like NPM1, are much more common in higher-risk MDS and AML where del(5q) is considered an adverse cytogenetic abnormality.74 In MDS, smaller deletions that include the 5q32–33.3 region are associated with a more favorable prognosis and a marked sensitivity to treatment with lenalidomide. Such patients with a sole del(5q) abnormality and no excess blasts represent the only genetically defined MDS subtype in the WHO classification system. Some of these patients have characteristics of the “5q-minus syndrome,” which is characterized by dyserythropoietic anemia, micromegakaryocytes with a preserved or elevated platelet count, female predominance, and lower risk of transformation to AML (Fig. 87–2).
Figure 87–2.
Composite from marrow films of patient with the 5q– syndrome. Characteristic hypolobulated megakaryocytes. A. Monolobed megakaryocyte. B. Bilobed megakaryocyte. Lobes connected by a nuclear bridge. C. Bilobed megakaryocyte. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
The pathogenic mechanisms associated with del(5q) are not completely understood. Patients with del(5q) do not routinely carry point mutations on the remaining intact 5q arm, suggesting that the inactivation of classic tumor-suppressor genes is not responsible for the selective advantage associated with this lesion.75 Instead, haploinsufficiency of genes lost in the deleted regions of chromosome 5q is largely responsible for the disease phenotype. For example, deletion of the ribosomal subunit gene RPS14 creates dyserythropoiesis mediated by TP53 activation in differentiating erythroid cells analogous to that seen in congenital haploinsufficient ribosomopathies such as Diamond-Blackfan anemia.76,77,78,79 Isolated mutations or deletions of RPS14 are not known to occur in MDS, suggesting that loss of this gene may influence the clinical presentation of MDS, but is not directly responsible for its development. Instead, codeleted genes must be drivers of transformation and several candidates have been proposed. These include microRNA (miRNA) genes 145 and 146a involved in the regulation of innate immune signaling and megakaryocyte differentiation,80,81,82 a mitochondrial heat shock protein HSPA9,83,84 and the zinc finger transcription factor EGR1.85,86 Several other genes, both in and out of the CDRs, have been implicated in the pathogenesis of MDS, including MAML1, a coactivator of the Notch signaling pathway, and the casein kinase gene, CSNK1A1.74,87
A del(5q) is frequently found as one of several chromosomal abnormalities in patients with complex disease karyotypes (defined as three or more chromosomal abnormalities).88 In this context, it is associated with an adverse prognosis, a poor response to lenalidomide, and frequently cooccurs with mutations of TP53 or abnormalities of 17p where TP53 resides.89,90,91,92 The association between del(5q) and TP53 lesions occur more often than predicted by their independent incidences alone, suggesting pathogenic cooperation between these abnormalities.86 Even in cases of isolated del(5q), small subclonal TP53 mutations can be found in 15 to 20 percent of cases. These patients appear to have a greater than predicted risk of AML transformation and inferior responses to treatment with lenalidomide.93,94,95
Monosomy 7 and Del(7q) Abnormalities of chromosome 7 are prognostically adverse lesions found in approximately 5 percent of MDS patients, often as part of a complex karyotype. Studies indicate that isolated monosomy 7 is a more adverse abnormality than isolated del(7q), and both occur more frequently (~50 percent) in patients with prior exposure to alkylating agents.88,89,96 Several distinct CDRs have been reported, including regions 7q22, 7q32–34, and 7q36.97,98,99 The relative pathogenic contribution of deletions in each of these regions is not well understood.
Several recurrently mutated genes reside on chromosome 7q. The histone methyltransferase gene, EZH2, is located on 7q36 and is mutated in approximately 6 percent of MDS cases.100,101,102 In some patients, an EZH2 mutation is accompanied by aUPD of 7q, but most EZH2 mutant patients do not have –7 or del(7q) and most patients with these chromosomal lesions do not harbor EZH2 mutations. In AML, the MLL3 gene, also located at 7q36, has been proposed as a haploinsufficient driven of disease.103 More proximal lies CUX1, a 7q22 gene implicated in MDS pathogenesis, which, like EZH2, is associated with a poor prognosis when mutated.104 Inactivating mutations of CUX1 tend to be heterozygous, suggesting that haploinsufficiency of this gene found in 7q22 might be a disease driver.104 However, deletion in mice of the region syngeneic to 7q22 produced no discernable phenotype.105
Trisomy 8 This is the only large-scale amplification frequently encountered in MDS, present in approximately 5 percent of cases. It is also highly nonspecific as it can occur in patients with myeloproliferative neoplasms, acute myeloid leukemia, and even aplastic anemia. Trisomy 8 is associated with an intermediate prognosis and is often acquired late in the disease course.106 In some cases, it may be acquired in myeloid progenitors as opposed to more pluripotential CD34+CD38–CD90+ stem cells where the MDS-initiating clone is presumed to have developed.107 How trisomy 8 leads to a selective growth advantage is not well understood. Progenitor cells with trisomy 8 express high levels of apoptosis-related genes and demonstrate dysregulation of immune response genes. Patients can harbor T cells that preferentially suppress trisomy 8 progenitor cells, particularly in response to overexpression of Wilms tumor 1 (WT1), which is upregulated in trisomy 8 cells.108,109 These findings may indicate selective pressure from the immune system on the disease clone with potential for collateral autoimmune suppression of normal hematopoiesis. Patients with trisomy 8 may benefit from immune suppression even if the aneuploid clone expands after response to treatment.110 Other autoimmune phenomena, such as Behçet disease, have been associated with trisomy 8 MDS.111,112,113
Del(20q) This abnormality is another nonspecific, yet recurrent, chromosomal abnormality found in approximately 2 percent of MDS cases. As an isolated lesion it is associated with disease risk comparable to that of MDS patients with normal karyotypes.114,115 However, del(20q) may be acquired late in the course of disease, indicating clonal progression and a more adverse prognosis.116 A CDR on 20q has been defined, but no single gene has been identified as the pathogenic driver responsible for the recurrent selection of del(20q) clones in MDS.117,118 Candidate disease genes on 20q include MYBL2,119,120 which lies within the CDR, and ASXL1,121,122 which lies outside the CDR and is mutated in a large fraction of MDS patients with or without del(20q). Patients with del(20q) appear more likely to have thrombocytopenia and are enriched in mutations of the splicing factor gene U2AF1.123,124
Loss of Y The isolated loss of the Y chromosome is a rare recurrent abnormality observed in just over 2 percent of males with MDS. Like the del(20q) abnormality, –Y is associated with the same cytogenetic risk as patients with normal karyotypes.114 It has been argued that –Y is not a pathogenic lesion in MDS, but instead an age-related event that can occur in men without cytopenias.125,126 However, the presence of –Y as a somatic change is indicative of oligoclonal, if not monoclonal hematopoiesis and is therefore consistent with the diagnosis of MDS in a cytopenic patient. The larger the –Y clone, the more likely that evidence of an underlying neoplasm like MDS will be found.127 Other genetic abnormalities, including mutations in ten-eleven translocation 2 (TET2) and DNMT3A have also been identified in elderly patients without cytopenias or other evidence of hematologic disease, yet these are considered pathogenic lesions in MDS. It is unclear if cytopenic patients with these markers of clonality, such as –Y, and insufficient evidence for an MDS diagnosis have comparable prognoses to patients with MDS.
Chromosome 17 Abnormalities Including del(17p) A small fraction of MDS patients will have an abnormality of chromosome 17, typically in the context of a complex karyotype. The TP53 gene is located on 17p13.1 in a region that is recurrently deleted in those rare patients with either del(17p) or monosomy 17. One copy of 17p is typically retained in these cases suggesting that total loss of this region is not tolerated. However, the TP53 gene on the remaining chromosome is often mutated leaving no wild-type protein. Patients with chromosome 17 abnormalities typically have a poor prognosis, particularly in the presence of a TP53 mutation. The 17p region can be effectively lost in patients with an isochromosome 17p abnormality [i(17q)], occurring in just under 1 percent of cases. While these lesions predict a high rate of leukemic transformation, they are rarely associated with a coexisting TP53 mutation.128 Instead, they are often found to cooccur with mutations of SETBP1, abnormalities that are found more often in patients with both dysplastic and proliferative disease features.129
Complex and Monosomal Karyotypes MDS karyotypes can be defined by the number of abnormalities present instead of focusing on the specific regions involved. Complex karyotypes are defined as having three or more cytogenetic abnormalities of any sort and are strongly associated with an adverse prognosis. In the International Prognosis Scoring System-Revised (IPSS-R), complex karyotypes are further separated in those with exactly three abnormalities and those with four or more, with the latter group having the highest associated disease risk. Monosomal karyotypes are defined as the loss of two or more entire chromosomes or the deletion of a single chromosome and the presence of another structural cytogenetic abnormality. Monosomal karyotypes are not necessarily complex and complex karyotypes are not necessarily monosomal, but in practice, there is substantial overlap between the two. The most frequent abnormalities seen in both monosomal and complex karyotypes involve chromosomes 5 and 7. The IPSS-R considers complex, but not monosomal karyotype as an independent risk factor and there has been ambiguity about whether complex versus monosomal karyotypes are better predictors of disease risk.130,131,132,133
Approximately 50 percent of patients with complex karyotypes have a concomitant TP53 mutation and account for the majority of patients with mutations of this gene.134 The incidence of TP53 mutation is particularly high when the complex karyotype includes del(5q) as an associated abnormality.90,92,135 The adverse prognostic significance associated with complex karyotypes may be largely driven by their frequent association with TP53 mutations.134,136 Complex karyotype MDS patients with intact TP53 have a median overall survival comparable to that of patients with noncomplex karyotypes, whereas those with a TP53 mutation have a significantly shorter overall survival.134 Patients with complex karyotypes on average have fewer mutations in genes other than TP53.
Acquired mutations of individual genes are significantly more common than karyotypic abnormalities in patients with MDS. Chromosomal rearrangements detected by standard cytogenetic techniqes are present in just under 50 percent of cases and more sensitive techniques with greater resolution can uncover small scale or copy number neutral abnormalities in an additional 25 percent. However, recurrent mutations of single genes can be identified in more than 80 percent of patients with MDS using targeted sequencing techniques and are likely to be found in all cases studied with whole-genome approaches. Unlike most chromosomal abnormalities that span large regions of the genome containing many candidate disease genes, most recurrent mutations affect the coding sequence of single genes, identifying them and their associated molecular pathways as pathogenic drivers. As of this writing, there are more than 50 genes known to be recurrently mutated in patients with MDS. A handful of these genes are mutated in a significant fraction of cases (10 to 35 percent) with several others in the 5 to 10 percent range. But the majority of recurrently mutated genes are found only rarely, encompassing fewer than 5 percent and, generally, less than 1 percent of cases. In some instances, these rare mutations occur in genes, like the splicing factors U2AF2 or SF1 that are in the same family as more commonly mutated genes. In other cases, the infrequently mutated genes, like GNAS and GNB1, represent their own molecular pathways suggesting that clonal myelodysplasia is a phenotypic manifestation that can be caused by a variety of pathogenic mechanisms. The large number of potentially mutated genes and multitude of ways in which they can be combined result in a staggering number of possible genetic profiles. The apparent cooperativity between some lesions and the mutual exclusivity of other limits this variability to some extent, but still allows for tremendous complexity at the genetic level. As a determinant of disease phenotypes, this variation likely explains much of the clinical heterogeneity seen in patients with MDS.137
Not all somatic mutations have equal pathogenic or clinical significance. Any patient with clonal hematopoiesis will harbor a large number of acquired mutations throughout their genome. The vast majority of these are incidental mutations acquired over the lifetime of the particular stem cell that eventually grew to clonally dominate hematopoiesis, most of which occurred prior to its expansion. These preceding mutations are not related to the development of disease and are distributed randomly, typically in noncoding and nonconserved areas of the genome, suggesting that they have no positive or negative selective significance. Collectively, these nonrecurrent mutations without pathogenic significance are described as passenger mutations because their presence in the expanded clone is only because they happen to coexist with much rarer driver mutations, responsible for the clonal outgrowth and the eventual development of disease. Driver mutations are typically recurrent and predicted to have pathogenic consequences like the alteration of a protein coding sequence or changes in the expression of one or more disease-related genes. Driver mutations can be early transformative events, in which case they would be found in every subsequent disease cell derived from that clone, or they can be late events, often associated with progressive disease. Some genes, like splicing factors, are predominantly mutated early and are typically found as part of the dominant disease clone. Other genes, like NRAS and SETBP1, are typically secondary mutations acquired later in the disease course and are often found as emergent subclones that may expand in size during progression.90,138,139
Somatic mutations can have important clinical implications for patients with MDS and the physicians who treat them, but their interpretation can be challenging. Several factors may influence the clinical significance or recurrent gene mutations. These include the type of mutation present (e.g., missense, nonsense, splicing, or frameshift), the configuration of the mutations (e.g., homozygous, heterozygous, hemizygous, or compound heterozygous), the fraction of disease cells that contain the mutations (i.e., presence in the dominant clone versus a subclone), the mutations that coexist in that patient and if these mutations are in the same clone or a sister clone, and whether mutations add information above and beyond what can be learned by looking at more readily accessible clinical variables like age, blast proportion, and blood counts. Despite these complexities, there are several scenarios in which specific gene mutations can inform the clinical care of patients with clonal cytopenias like MDS.
Splicing Factors The most frequently mutated class of genes in patients with MDS encode splicing factor proteins involved in the excision of introns and the ligation of exons from maturing pre-mRNA strands. There are at least eight recurrently mutated splicing factor genes identified in MDS and nearly two-thirds of patients will carry at mutation of a member of this gene family.140 Splicing factor mutations are largely mutually exclusive. Patients with one splicing factor mutation rarely have a mutation in another suggesting that these lesions are either not tolerated in combination or, more likely, have a common mechanism of action. Thus, a disease stem cell that acquires a second splicing factor mutation would gain no selective advantage and may well develop a selective disadvantage as a consequence of that second mutation. Despite a presumed common pathogenic role, patients with mutations in different splicing factor genes often have very disparate clinical phenotypes.137 This is partially because disease manifestations are often determined by the effect of driver mutations in the differentiating progeny of disease stem cells. Some may induce profound dysplasia while others promote lineage-specific proliferation, for example. Different splicing factor mutations may also be associated with certain clinical phenotypes because of their distinct patterns of comutation with other MDS-related genes.90,135,141 These comutations may, for example, drive the disease manifestations or prognosis. If there is a common mechanism by which splicing factor mutations drive the development of MDS, it is not yet well understood. These mutations are not unique to MDS as they can be found in other malignancies, including some solid tumors, where they occur at low frequency. Several studies have identified changes in splicing efficiency and gene expression associated with splicing factor mutations, but these effects are subtle and do not readily identify a downstream pathogenic mechanism.
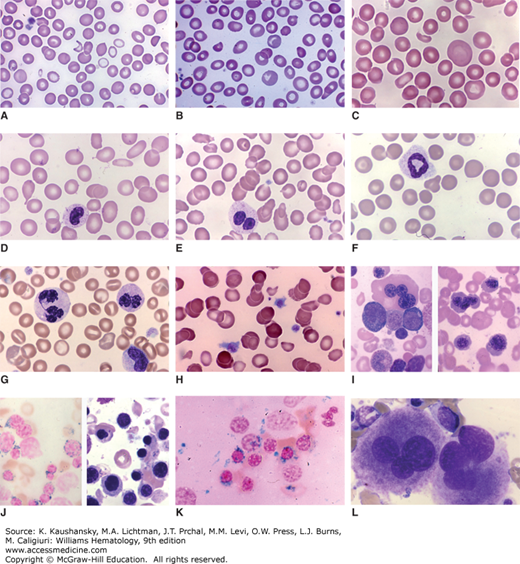
SF3B1 is the most frequently mutated splicing factor gene and encodes the U2 small nuclear riboprotein complex (snRNP) subunit responsible for branch site recognition. Mutations of SF3B1 are present in 20 to 30 percent of patients and are the only somatic mutations associated with a favorable prognosis. The pattern of mutation of SF3B1 suggests an oncogenic gain or change of function for its encoded protein. Mutations are always heterozygous to an intact wild-type allele and are relatively conservative missense substitutions at very specific hotspots.142 These mutations occur in the middle of several consecutive HEAT protein domains of which the K700E substitution is the most common, accounting for more than half of all mutations of SF3B1. Clinically, mutations of SF3B1 are very tightly associated with the presence of ring sideroblasts (Fig. 87–3). More than 85 percent of patients with RARS or RARS-t will have an SF3B1 mutation. These mutations are common in patients with other subtypes of MDS, like refractory cytopenia with multilineage dysplasia, when ring sideroblasts are present. SF3B1 mutations have been associated with a more favorable prognosis but it is unclear if this is independent of other known risk factors. For example, SF3B1 mutant patients are less likely to have cytogenetic abnormalities, including complex karyotypes, and are less likely to have other mutations in genes associated with a poor prognosis. Only mutations in DNMT3A cooccur with SF3B1 mutations more often than would be predicted by chance alone, suggesting a cooperative interaction between these lesions.90,141 SF3B1-mutant MDS may represent its own nosologic entity based on its common clinical features and patterns of comutation regardless of WHO subtype.137 Mutation of SF3B1 is also common in chronic lymphocytic leukemia (CLL) where it occurs in 15 to 20 percent of cases (Chap. 92). However, in contrast with MDS, these mutations in CLL are often subclonal or acquired after initial diagnosis and are associated with treatment resistance and a poor prognosis.143 SF3B1 hotspot mutations are enriched in uveal melanoma and can be found in various other solid tumors at lower frequency, demonstrating that it is not a tissue-specific oncogene.144,145
Figure 87–3.
Blood and marrow films from patients with clonal cytopenias (myelodysplastic syndromes). A. Blood film. Anisocytosis. Poikilocytosis with occasional fragmented cells. Marked anisochromia with marked hypochromia, mild hypochromia and normochromic cells. B. Blood film. Marked anisocytosis. Mild anisochromia. Poikilocytes with occasional fragmented cells and oval and elliptical cells. Two polychromatophilic macrocytes. C. Blood film. Striking anisocytosis with giant macrocytes and microcytes. Poikilocytes with tiny red cell fragment and elliptocyte. D. Blood film. Mild anisocytosis. Ovalocytes and elliptocytes. Dacryocyte. Hyposegmented neutrophil with poor granulation. E. Blood film. Marked anisocytosis (macrocytes and microcytes). Ovalocytes and elliptocytes. Acquired Pelger-Hüet nuclear anomaly (classic pince-nez shape) in neutrophil. F. Blood film. Mild anisocytosis. Abnormal neutrophil with ring nucleus. G. Blood film. Anisochromia. Stomatocytes. Abnormal neutrophil nuclei with hyperlobulation and hyperchromatic staining. Note abnormal elongated nuclear bridge in neutrophil on left. H. Blood film. Atypical platelets. Two macrothrombocytes with excess cytoplasm and atypical central granules. Anisocytosis (conspicuous microcytes). Anisochromia (conspicuous hypochromic cells). Poikilocytosis with occasional fragmented red cells. I. Marrow film. Wright stain. Trilobed megakaryocyte. Wright stain. Macroerythroblasts. J. Marrow films. Prussian blue stain. Ring sideroblasts. Wright stain. Erythroid hyperplasia with macroerythroblasts. K. Marrow film. Prussian blue stain. Ring sideroblasts. L. Marrow film. Wright stain. Trilobed megakaryocytes. (Reproduced with permission from Lichtman’s Atlas of Hematology, www.accessmedicine.com.)
SRSF2 is the second most frequently mutated splicing factor, present in 10 to 15 percent of MDS and 40 percent of CMML cases. It encodes a serine-arginine–rich protein that interacts with the U2 and U1 components of the spliceosome. The predominant mutation in this gene is a missense substitution of the proline at codon 95, although small insertions and deletions at this position that conserve the reading frame have also been reported. As with SF3B1, mutations are heterozygous to a wild-type allele suggesting a very specific gain or change of function. SRSF2 mutations cooccur with mutations of several other genes, such as TET2, ASXL1, CUX1, IDH2, and STAG2, many of which are also enriched in patients with CMML.90 SRSF2 mutations are generally associated with an inferior prognosis.
U2AF1 is the third frequently mutated splicing factor, present in approximately 12 percent of patients. Like SF3B1 and SRSF2, mutations are heterozygous to a wild-type allele and occur as missense mutations at fixed hotspots. The affected amino acids at codons 34 and 157 are in the zinc finger DNA-binding regions of the protein.140,146 U2AF1 encodes an auxiliary factor in the U2 spliceosome responsible for the recognition of the AG splice acceptor dinucleotide at the 3′ end of introns. U2AF1 mutations appear to affect splicing in reporter assays and transgenic mouse models, but how these changes confer a selective advantage to mutant cells is not well understood.140,147 Clinically, U2AF1 mutations are associated with shorter overall survival and increased risk of transformation to acute leukemia.148 Patient with del(20q) may be enriched or U2AF1 mutations.123 Several additional splicing factors can be mutated in MDS, including ZRSR2, SF1, and U2AF2.140 Most of these appear to harbor loss-of-function mutations, but remain largely exclusive of mutations in other splicing factors, suggesting a shared pathogenic mechanism.
Epigenetic changes, defined as heritable covalent modifications of chromatin that do not alter the DNA base sequence, play a role in the development of MDS and other malignancies. Methylation of cytosine residues in DNA represents one form epigenetic modification that can be dysregulated in MDS. Specifically, patients may have global DNA hypomethylation, but will demonstrate hypermethylation in specific regions such as the CpG islands (areas rich in CG dinucleotides) found at or near gene promoters. These epigenetic marks have been associated with a closed chromatin configuration and relative silencing of nearby genes. The simplistic explanation is that aberrant DNA methylation leads to the pathogenic silencing of critical tumor-suppressor genes and is, therefore, oncogenic. Hypomethylating agents, which are inhibitors of the DNA methyltransferases that catalyze cytosine methylation, have been presumed to undo the silencing of these tumor-suppressor genes leading to therapeutic benefit. However, it is not certain that hypomethylating agents work in this way. It is known that several genes involved in the regulation of DNA methylation are mutated in a large proportion of patients with MDS as are genes involved in the regulation of DNA-associated histone modifications. Together these epigenetic regulators form the second largest class of genes mutated in MDS. Unlike the splicing factors, most mutated epigenetic regulator genes are not exclusive of each other and frequently coexist.
DNMT3A This gene encodes a de novo DNA methyltransferase and is the only DNA methyltransferase gene frequently mutated in MDS. It is mutated in approximately 15 percent of cases in a pattern that suggests a resultant loss of function.149,150 Mutations can include frameshifts and premature stop codons, as well as missense mutations spread throughout the length of the gene. The one exception is a high frequency of missense mutations at the hotspot codon 882, which have been shown to impair catalytic activity.151 As with nearly all of the genes mutated in MDS, DNMT3A mutations are not unique to these disorders and can be found in AML, myeloproliferative neoplasms (MPNs), and even lymphoid malignancies. Mouse models of Dnmt3a loss demonstrate hematopoietic stem cell exapansion with impaired differentiation. Dysplasia and leukemic transformation do not occur in this mouse model, suggesting that DNMT3A loss is sufficient to provide a stem cell growth advantage, but insufficient to cause an MDS or AML disease phenotype. This is consistent with the finding that somatic DNMT3A mutations can be found in persons without cytopenias or other elements of disease.152 Therefore, cooperating mutations or microenvironmental changes are likely necessary determinants of disease. Mutations of DNMT3A are found most often in patients with normal karyotypes and cooccur with SF3B1 mutations more often than expected by chance.90,153 DNMT3A mutations in MDS patients appear to confer a poor prognosis.150,154
TET2 The second member of the ten-eleven translocation gene family, TET2, is among the most frequently mutated MDS genes present in 20 to 30 percent of cases, and in more than 40 percent of patients with CMML. It encodes a methylcytosine oxygenase that converts 5-methylcytosine (5-mC) in to 5-hydroxymethylcytosine (5-hmC) using iron and α-ketoglutarate (αKG) as cofactors.155 The TET2 enzyme can further oxidize 5-hmC into 5-formyl- and 5-carobxycytosine (5-fC and 5-caC, respectively).156 This may represent a mechanism for the active demethylation of cytosines as 5-caC can be decarboxylated to form cytosine or treated as a mismatched nucleotide by the base excision DNA repair pathway. Mutations in TET2 are typically truncating or clustered in regions that encode catalytic domains indicating an associated loss of function. Mutations are often compound heterozygous or in areas of aUPD on chromosome 4q, resulting in no viable wild-type allele.157 Patients with TET2 mutations demonstrate increased global DNA methylation, lower levels of 5-hmC and are more likely to have an elevated monocyte count.158,159 Mouse models of Tet2 loss show a similar phenotype with increased stem cell and progenitor numbers, impaired differentiation, and myeloid skewing of hematopoiesis.160,161,162,163 As with DNMT3A, TET2 mutations can also be found in various myeloid and lymphoid malignancies, as well as in persons with clonal hematopoiesis and no hematologic disesase.152 Clinically, TET2 mutations are not likely drivers of MDS prognosis and have variably been associated with favorable, neutral, or poor outcomes.134,164,165
IDH1 and IDH2 Only mutations in the isocitrate dehydrogenase genes 1 and 2 (IDH1 and IDH2, respectively) are exclusive of TET2 mutations, suggesting that they share a common pathogenic mechanism. Mutations of these IDH genes are common in AML and gliomas, but relatively rare in MDS, comprising approximately 5 percent of cases. Oncogenic IDH mutations are always heterozygous missense mutations of specific codons that result in an important change of enzyme function. Instead of converting isocitrate to αKG while generating an nicotinamide adenine dinucleotide phosphate (NADPH) from nicotinamide adenine dinucleotide phosphate–positive (NADP+), the mutant forms of IDH1 and IDH2 catalyze the conversion of αKG to 2-hydroxyglutarate (2-HG) while oxidizing NADPH to NADP+.166 The 2-HG produced is considered an oncometabolite, which can interfere with the activity of αKG-dependent oxygenases, including the TET family of genes, prolyl hydroxylases, collagen synthesis enzymes, and various histone demethylases.167,168,169,170,171 Mouse models of leukemic IDH mutations in the hematopoietic system share several features with Tet2-null mice, including global DNA hypermethylation and increased proportions of early progenitor cells.172 In MDS, the clinical significance of IDH mutations is mixed and may depend on the nature of the mutation.173 Inhibitors of the neomorphic activity of mutant IDH enzymes represent promising therapies as the effects of 2-HG exposure appear to be reversible.174
EZH2 and Other Rare Mutations Several regulators of histone modifications are recurrently mutated in MDS and MDS/MPN disorders. These include the histone methylase EZH2, which encodes the catalytic subunit of the protein-repressive complex 2 (PRC2) responsible for methylating lysine 27 on histone 3 (H3K27). The H3K27 methyl mark is associated with closed chromatic and reduced expression of neighboring genes. Loss-of-function mutations in EZH2, present in 6 percent of MDS, are associated with a poor prognosis in a manner that is independent of common prognostic scoring systems.100,102,134 This is largely because EZH2 mutations are not strongly associated with adverse clinical features such as increased proportions of blasts, complex karyotypes, or severe cytopenias.141 Other members of the PRC2, EED and SUZ12, can also be mutated in very rare cases of MDS.175 Loss of PRC2 activity may, in part, promote the development and progression of MDS through derepression of HOX genes, which are often upregulated or aberrantly expressed in self-renewing leukemic cells.176
ASXL1 ASXL1 is a frequently mutated MDS gene believed to be an epigenetic “reader” capable of binding to specific histone marks through its highly conserved PHD domain. Mutations of ASXL1, present in 20 percent of MDS and 40 percent of CMML, are largely heterozygous truncating mutations in its terminal exon. These lesions are associated with a poorer prognosis than predicted by common clinical assessments alone.134,177,178 ASXL1 interacts directly with the PRC2, directing the activity of EZH2 to specific genomic regions. Loss of ASXL1 is associated with absent H3K27 trimethylation at the HOXA gene cluster.179 ASXL1 mutations may cooperate with mutations in various other genes such as SRSF2, U2AF1, TET2, and NRAS, as mutations of these genes cooccur more often than predicted by chance alone.90 In patients with germline mutations of the transcription factor GATA2, somatic ASXL1 mutation appears to be a common concurrent event at the time of MDS or AML development.180,181
Mutated Transcription Factor Genes Mutations of hematopoietic transcription factors in MDS are typically somatic events, but can be present in the germline either as inherited or spontaneous congenital events in rare cases. RUNX1 is the most frequently mutated transcription factor in MDS. This gene, previously known as AML1, encodes the alpha core binding transcription factor subunit and is altered in many myeloid and lymphoid malignancies. In the acute leukemias, RUNX1 is a frequent translocation partner with other genes such as RUNX1T1 (previously known as ETO) as part of t(8;21) in AML and with ETV6 in (previously known as TEL) as part of t(12;21) in acute lymphocytic leukemia (ALL). RUNX1 is mutated in 10 to 15 percent of MDS where it is associated with a poor prognosis, increased rates of leukemic progression, and thrombocytopenia.134,135 Mutations can affect one or both alleles and often involve the DNA-binding RUNT domain or truncate the more distal protein interaction domain.182,183,184 Persons with congenital mutations of RUNX1 can have an autosomal dominant FPD-AML characterized by numerical and functional platelet abnormalities that precede transformation to AML by many years. Penetrance of FPD-AML is variable and the long latency to transform indicates the need to acquire cooperating mutations.43,185,186 Mutations of C/EBPA can also cause familial propensity for AML in an autosomal dominant fashion, but are very rare mutations in MDS as somatic or inherited abnormalities.187
ETV6 The ets-like transcription factor 6, ETV6, is frequently rearranged, deleted, or mutated in hematologic malignancies. In MDS, ETV6 mutations are present in approximately 5 percent of cases, where they are independently associated with shorter overall survival and progressive disease.134,188
GATA2 Germline GATA2 mutations are responsible for several different congenital syndromes with overlapping features including a predisposition to marrow failure, MDS, and AML.53,58,189,190 Familial GATA2 mutations can manifest as the monoMAC syndrome, characterized by monocytopenia and mycobacterial infections; Emberger syndrome, characterized by congenital lymphedema and risk of developing MDS; or as a deficiency in monocytes, B and natural killer (NK) lymphocytes, and dendritic cells.191 Patients can also have sensorineural hearing loss, alveolar proteinosis, and dermatologic abnormalities. Penetrance is variable and patients with one syndrome often will have features of the others. Several cases of congenital neutropenia or apparently de novo MDS in childhood have been ascribed to germline GATA2 mutations in the absence of other syndromic features.56 However, unlike RUNX1 and ETV6 mutations, mutations of GATA2 are only very rarely somatic events.
TP53 TP53 mutations are present in approximately 10 percent of MDS cases and are strongly associated with a poor prognosis independent of other risk factors.134 Most patients with mutations of TP53 will have a complex karyotype and tend to have fewer point mutations in other typical MDS genes. Patients with del(5q) are more likely to have a TP53 mutation, particularly in the context of a complex karyotype, suggesting pathologic synergy between these abnormalities.90,135 Unfortunately, treatment with either hypomethylating agents or allogeneic hematopoietic stem cell transplantation (AHSCT) fails to rescue the adverse outcomes associated with mutations of TP53.192,193,194
Mutations of Growth Factor Signaling Pathway Genes Mutations of receptor tyrosine kinase genes are common in proliferative myeloid disorders, such as FLT3 in AML, KIT in mast cell neoplasms, and of the receptor gene MPL in MPN, but only rarely present in MDS. Instead, several genes encoding downstream signaling proteins are more frequently mutated in MDS. Many of these mutations are associated with proliferative features and often presage more advance disease or progression to secondary AML. Signaling pathway mutations are typically mutually exclusive, indicating some redundancy in function and are more common in CMML where monocytic proliferation is a defining feature. In MDS, these mutations are frequently present in minor disease subclones demonstrating that they were acquired later in the course of disease. Despite their small abundance, signaling pathway mutations are often predictive of transformation and shorter overall survival.195,196
Activating NRAS mutations are the most common in this category, but found in only 5 to 10 percent of cases. These lesions are associated with excess blasts and thrombocytopenia.134 The E3-ubiquitin ligase CBL regulates tyrosine kinase receptors by marking them for degradation.197 CBL mutations that impair this function are found in 3 to 5 percent of MDS cases and are associated with monocytosis.198,199 Somatic mutations of the tyrosine phosphatase encoded by PTPN11 are seen in very rare cases of MDS and CMML, but are more common in juvenile myelomonocytic leukemia where mutations are often germline lesions and part of a congenital syndrome.200,201 Other genes that are very rarely but recurrently mutated in this pathway include KRAS, BRAF, KIT, and CBLB.
The V617F mutation in JAK2 is found in 3 to 5 percent of patients with MDS and is exclusive of other signaling pathway mutations. However, it does not appear to have prognostic significance and is not associated with an increased red cell mass as it is in polycythemia vera.134 This is likely because JAK2 mutations in MDS are often late events and coexist with other genetic lesions that result in dysplasia, limiting the production of mature red cells. JAK2 mutations are enriched in patients with RARS-t, and MDS/MPN overlap disease.202 Half of these patients will carry a JAK2V617F mutation. This is roughly the same proportion observed in patients with essential thrombocythemia (ET); leading to the speculation that RARS-t is a “frustrated” form of ET with dysplasia caused by the other mutations like those in SF3B1 associated with ring sideroblasts.203 Approximately 5 percent of RARS-t patients will instead have a mutation in MPL, which is similar to the rate at which it occurs in ET as well (Chap. 85).
Cohesin Genes RAD21, STAG2, SMC3, and SMC1A are recurrently mutated members of the cohesin gene family. Collectively, they are mutated in approximately 10 percent of MDS cases, where they may be associated with a poor prognosis.204,205 Cohesins bind to chromatin in a large complex believed to protect chromatid structure and shepherd chromosomes through mitosis. However, mutations of this complex are not associated with chromosomal instability in MDS. Instead, they identify patients more likely to have multilineage dysplasia.137 The pathogenic mechanism of cohesin mutations in myeloid disorders remains poorly understood.
Other Classes of Mutated Genes Several other classes of genes are recurrently mutated in MDS, including DNA repair enzymes, RNA helicases, and members of the G-protein signaling pathway. The long tail of recurrent, but rarely mutated genes in MDS suggest that dysplasia is common final phenotype that can be caused by a variety of different pathogenic abnormalities, each with their own degree of severity, risk of progression, and variation in clinical presentation.
Not all abnormalities identified in the marrow of patients with MDS are intrinsic to the clonal cells that give rise to the disease. There are many microenvironmental alterations that contribute to the distorted hematopoiesis that is characteristic of MDS. Marrow cytokine levels are altered in many cases. Circulating monocyte colony-stimulating factor (M-CSF) is increased in some patients with MDS, AML, and other hematologic malignancies.206 Interleukin (IL)-1α and granulocyte-macrophage colony-stimulating factor (GM-CSF) levels have been undetectable in most patients. IL-6, granulocyte colony-stimulating factor (G-CSF), and erythropoietin concentrations have been variable. Tumor necrosis factor (TNF) concentrations has been inversely related to hematocrit.207 Stem cell factor, a multilineage hematopoietin, can be decreased in some patients.208
The neoplastic clone may induce activation of the innate immune system. Loss of miRNAs 145 and 146a from the CDR of chromosome 5q lead upregulation of toll-interleukin receptor adaptor protein (TIRAP) and tumor receptor-associated factor (TRAF) 6, members of the innate immune signaling pathway downstream of toll-like receptors.80 Toll-like receptors may also be targets of somatic mutations leading to nuclear factor (NF)-κB activation.209 Myeloid-derived suppressor cells, distinct from the clonal disease cells, may contribute to the altered cytokine microenvironment and promote disordered hematopoiesis.210
Dysregulation of the adaptive immune system has also been described. CD40 expression on monocytes is increased, as is CD40L on T lymphocytes, and has been postulated as being a contributing factor to hematopoietic failure in some patients with less-advanced disease.86 Many patients with MDS will demonstrate oligoclonal T-cell expansion with skewing of Vβ-subunits of the T-cell receptor similar to that seen in aplastic anemia.211 In patients that respond to immunosuppressive therapy, this oligoclonality of T cells may be normalized.212 There likely exists substantial pathogenic overlap between aplastic anemia and hypoplastic MDS. Immunosuppression can improve hematopoiesis in both disorders, both can exhibit paroxysmal nocturnal hemoglobinuria (PNH) clones, and both can have somatic mutations typical of MDS (Chap. 35).213 Large granular lymphocyte (LGL) leukemia can cause immune cytopenias and LGL cells are present in some cases of both MDS and aplastic anemia. These lymphocytes can carry somatic mutations of STAT3 indicating their clonal nature, distinct from the MDS clone.214
CLINICAL FEATURES
Patients can be asymptomatic or, if anemia is more severe, can have pallor, weakness, loss of a sense of well-being, and exertional dyspnea.215 Fatigue is a major complaint that is not necessarily related to degree of anemia.216 A small proportion of patients have infections related to severe neutropenia or neutrophil dysfunction, or hemorrhage related to severe thrombocytopenia or platelet dysfunction at the time of diagnosis. Patients with severe depressions of neutrophil and platelet counts at diagnosis usually have more advanced disease. Rarely, patients have fever unrelated to infection. Arthralgia is the initial complaint in some patients. The presentation, infrequently, can mimic a rheumatologic disease. Hepatomegaly or splenomegaly occurs in approximately 5 or 10 percent of patients, respectively.
LABORATORY FEATURES
Anemia is present in greater than 85 percent of patients.217,218,219 In approximately 4 percent of patients, the anemia results from erythroid aplasia.220 Mean cell volume often is increased. Red cell shape abnormalities include oval, elliptical, teardrop, spherical, and fragmented cells. Red cell findings occur in a spectrum. Some patients have only slight anisocytosis. Elliptical red cells sometimes dominate. Basophilic stippling of red cells occurs (see Fig. 87–3). Nucleated red cells are seen in the blood film in approximately 10 percent of cases. Reticulocyte counts are low for the degree of anemia. Other abnormalities of red cells occur, such as an increased proportion of hemoglobin F221 and decreased red cell enzyme activities, especially acquired pyruvate kinase deficiency.222 Hemolysis has occurred in some patients with the latter deficiency. Enhanced sensitivity of membranes to complement223 and modification of red cell blood group antigens may be observed.224 Acquired hemoglobin H disease, a rare superimposition, results in red cell morphology similar to thalassemia (microcytosis, anisocytosis, basophilic stippling, poikilocytosis with target cells, fragmented cells, and teardrop cells). Intracellular precipitates of β-chain tetramers (identified by crystal violet stain) reflect an acquired decrease in the rate of α-chain synthesis in erythroblasts.225,226,227 The decrease in α-globin–chain synthesis is profound, involves each of the four α-chain loci, and results from a transcription abnormality. No gross alterations in genes (e.g., insertions, deletions) are seen in these cases.227 Acquired hemoglobin H disease in this setting has been dubbed the α-thalassemia–myelodysplastic syndrome and is the consequence of acquired mutations in ATRX, the gene associated with the X-linked α-thalassemia/mental retardation (ATR-X) syndrome.225
Neutropenia is present in approximately 50 percent of patients at the time of diagnosis.228 The proportion of monocytes often is increased, and monocytosis per se can be the dominant manifestation of the hematopoietic abnormality for months or years.229,230,231 Morphologic abnormalities of neutrophils can occur, sometimes resulting in the acquired Pelger-Huët anomaly (see Fig. 87–3E). In this condition, neutrophils have very condensed chromatin and unilobed or bilobed nuclei that often have a pince-nez shape. The neutrophils may be in the process of apoptosis.232 Ring-shaped nuclei also can occur in neutrophils (see Fig. 87–3F).233 Neutrophil alkaline phosphatase activity is decreased in some patients.234 Expression of normal surface antigens on neutrophils and monocytes is decreased, and abnormal surface antigen expression occurs in some cases.235 Defective primary granules of abnormal size and shape with decreased myeloperoxidase content can be present.236 Specific neutrophil granules are often decreased in number, producing hypogranular cells.237 Neutrophil granule membranes frequently are deficient in glycoproteins.238 Chemotactic, phagocytic, and bactericidal capability may be impaired.239,240,241 Formyl-leucyl-methionyl-phenylamine receptor signaling and actin polymerization can be abnormal.242,243 Muramidase (lysozyme) activity in blood and urine may be increased, reflecting granulocytic hyperplasia, heightened monocytopoiesis, and monocyte turnover.
Approximately 25 to 50 percent of patients have mild to moderate thrombocytopenia at the time of diagnosis.228,234 Mild thrombocytosis also can occur.228,234 Platelets may be abnormally large, have poor granulation, or have large, fused central granules (see Fig. 87–3H).244,245 Abnormal platelet function can contribute to a prolonged closure time, easy bruising, or exaggerated bleeding. Decreased platelet aggregation in response to collagen or epinephrine is a frequent functional abnormality.246
Patients with clonal hemopathies may have immunologic deficiencies, such as a decrease in NK cells in the blood but no decrease in LGLs,247,248,249,250 a decrease in helper T lymphocytes,248 and a decrease in Epstein-Barr virus receptors on B lymphocytes.248,249,250,251 Antibody-dependent cellular cytotoxicity is normal.248 Thymidine incorporation after mitogenic stimulation252,253 and colony growth of T lymphocytes are decreased.248 Lymphocytes may have an increased sensitivity to irradiation.253 The defects in lymphoid cells could reflect the level of the somatic mutation in a primitive multipotential cell in different cases. Intrinsic, rather than secondary, alterations in lymphocytes are determined by whether no lymphocytes are generated from the clone, B cells are part of the clone, or B and T cells are part of the clone.254 Clonally derived, CD8+CD57+CD244+CD28–CD62L– T lymphocytes are present in marrow and to a lesser extent in blood in approximately 50 percent of patients, independent of type of MDS, age, and sex of the patient,255,256 as are NK and B cells (see “Pathogenesis” above).256
Serum iron, transferrin, and ferritin levels may be elevated as a result of anemia and the shift of erythron iron to plasma and storage compartments. Lactic dehydrogenase and uric acid concentrations can be increased as a result of ineffective hematopoiesis and a high death fraction of maturing marrow precursors. Monoclonal gammopathy, polyclonal hypergammaglobulinemia, and hypogammaglobulinemia each occur with increased frequency.257,258 The frequency of autoantibodies was increased in one report240 but not in another.258 β2-Microglobulin serum levels are increased in proportion to the prognostic category of the disease.259
Marrow cellularity usually is normal or increased.234,260,261 Cellularity is decreased in approximately 15 percent of cases260 and may simulate hypoplastic or aplastic anemia.262 However, islands of dysmorphic cells, especially atypical megakaryocytes, usually are present (see Fig. 87–3L). An increased proportion of blast cells in this setting suggests hypoplastic myelogenous leukemia (Chap. 88).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


