Cybèle A. Renault, Joel D. Ernst
Mycobacterium leprae (Leprosy)
Leprosy has a rich history dating to biblical times.1 Even with the development of modern antibiotic therapy, leprosy continues to place a significant burden on individuals and society, and patients continue to be stigmatized by the diagnosis, leading some to avoid proper diagnosis and therapy. Peripheral nerve damage, the most common complication of leprosy, leads to the characteristic deformities of the disease, but early detection and therapy can prevent significant morbidity and disability.
Of the chronic infectious diseases whose clinical and pathologic manifestations arise in a distinct and well-characterized spectrum, leprosy is among the best understood. At one end of the clinical spectrum, tuberculoid leprosy is characterized by a small number of skin lesions, few bacilli in lesions, and development and recruitment of T lymphocytes that contribute to control of the infection.2,3 At the other extreme, lepromatous leprosy is characterized by a larger number of skin lesions, clinically apparent infiltration of peripheral nerves and skin lesions by a large number of bacilli, and the presence of fewer T lymphocytes in lesions whose effector mechanisms are unable to control the infection.2,3
The discovery of Mycobacterium leprae has historical significance: Gerhard Henrik Armauer Hansen discovered the microbe in 1873, before Koch’s discovery of Mycobacterium tuberculosis, but the inability to cultivate M. leprae in vitro (a problem that remains unsolved) allowed Koch to be credited with the discovery and the germ theory of disease.
Before the advent of effective antibiotics, treatment consisted of isolating patients in leprosaria. It was in the 1940s when dapsone was recognized as an effective antimycobacterial agent, and in the past several decades there have been major changes in both the management and the understanding of leprosy. In the 1920s, the U.S. Public Health Service (USPHS) established a program for patients with leprosy that dramatically changed the management of this disease. The United States formally adopted a multidrug treatment policy and an ambulatory care program in 1981, and under USPHS guidance the World Health Organization (WHO) adopted a similar program in 1982. Despite these advances, leprosy is still a disease that is far from being eradicated.
Epidemiology
Because detection of asymptomatic leprosy is difficult and because of the stigma associated with the diagnosis, estimates of leprosy incidence and prevalence are underestimated compared with those of other diseases. Because M. leprae cannot be cultured in vitro and because there is no sensitive and specific diagnostic test for the detection of individuals who are infected without clinical disease, transmission of leprosy is still poorly understood. The predominant mode of transmission is likely to be through respiratory droplets or nasal secretions, because up to 107 viable bacilli per day can be shed in respiratory secretions of people with multibacillary leprosy.4 Modes of nonrespiratory transmission, such as transplacentally5 or via breast milk,6 are also possible. Because M. leprae has been detected in skin,7–9 sebaceous gland secretions, and eccrine sweat glands,10 skin-to-skin transmission has not been excluded. Other modes of transmission, such as soil, insect vectors, and exposure to infected animals, remain speculative. Regarding zoonotic transmission, there is strong evidence supporting armadillos as a natural reservoir but chimpanzees and monkeys are also possible sources.11–13
The likelihood of developing disease is determined by several variables. Age is an important factor, because clinical disease has a bimodal distribution: adolescents aged 10 to 19 years of age are the most susceptible, followed by a second peak at the age of 30 years or older.14 Gender is relevant in adult patients: in contrast to children, where there is no difference in prevalence between the sexes, adult men are twice as likely to develop disease as adult women.15 The risk for disease is associated with contact with infected individuals; individuals who are in contact with multibacillary patients are at highest risk for developing disease.15
Cases of leprosy exhibit geographic (e.g., village) and family clustering. Although clustering of leprosy in families can be due at least in part to shared environments and exposure, there is strong evidence for genetic determinants of susceptibility to leprosy.16 Moreover, there are clearly genetic variations that influence susceptibility to the presence of infection (leprosy per se), whereas other genetic loci influence the clinical form of the disease (paucibacillary or multibacillary). Most early efforts to define the genetic determinants of susceptibility to leprosy were association studies that examined the frequency of human leukocyte antigen (HLA) types with leprosy per se or the clinical form. Although these studies demonstrated associations between HLA-DR2 (now subtyped as DRB1*1501 and DRB1*1502) and tuberculoid leprosy, they also revealed that HLA genes did not fully account for the genetic effects on susceptibility, and other genes have been identified whose variants contribute to leprosy susceptibility, including SLC11A1 (formerly NRAMP1),17,18 vitamin D receptor (VDR),19 TNF,20,21 TLR1,22,23 and lymphotoxin (LTA).24
A genome-wide linkage scan identified strong linkage between susceptibility to leprosy per se and a region on chromosome 6q25-6q26 in a Vietnamese population.25 Subsequent analyses of that chromosomal region revealed specific single-nucleotide polymorphisms (SNPs) that formed haplotypes in the shared promoter region of the PARK2 and PACRG genes; heterozygosity and homozygosity for the susceptible haplotype were associated with very high odds ratios of 3.23 and 5.28, respectively, compared with homozygosity for the resistant haplotype; subsequent analysis in a Brazilian population confirmed the association of variation at the PARK2/PACRG locus with leprosy.26 PARK2 encodes parkin, an E3 ubiquitin ligase, which indicates that regulated protein degradation or autophagy or both in mononuclear phagocytes or Schwann cells or both is important for resistance to leprosy; the function of the Parkin coregulated gene protein (PACRG) is unknown, but it is also thought to be related to proteasome function.
Recently, results of studies of genetic polymorphisms in leprosy have converged with studies of Crohn’s disease susceptibility to reveal strong evidence that similar pathways operate in both diseases. In particular, NOD2 and RIPK2 interact directly in a proinflammatory signaling pathway implicated in Crohn’s disease, and these loci are implicated in susceptibility to leprosy in studies of subjects in China, Vietnam, and Nepal.27–29 Other loci implicated in both leprosy and Crohn’s disease include IL23R,30 LRRK2, LACC1, and TNFSF15.29 Studies of the functions of these genes and their pathways in leprosy and Crohn’s disease are likely to be mutually beneficial in understanding the pathogenesis of both diseases.
In contrast to these findings, genetic studies have revealed a lack of association of leprosy susceptibility with the chromosomal region (5p) that contains the T-helper type 2 (Th2) cytokine gene cluster.31 Because a Th2 response is characteristic of lepromatous leprosy, this finding suggests that genetic variations in this region are not primary determinants of polarization of the immune response to M. leprae. Additional studies are necessary to understand more fully the genetic basis of susceptibility to leprosy and to define precisely the polymorphisms in the chromosomal regions linked to susceptibility.
Global Epidemiology
In 2011, the WHO reported detection of 219,075 new cases of leprosy,32 but active case finding has established that there is an additional hidden case load: for example, a study found that there were six times the number of leprosy cases in Bangladesh than that number that was reported by the WHO.33 Of the 219,075 new cases detected in 2011, 160,132 were detected in South and East Asia, followed by 36,832 cases in the Americas and 12,673 cases in Africa.32 In 2011, 83% of the incident cases were reported from three countries: India (58%), Brazil (16%), and Indonesia (9%).32
In 1991, the WHO first embarked on a campaign for the elimination of leprosy, defined as prevalence of less than 1 case per 10,000 people. The program has successfully increased the percentage of leprosy patients receiving multiple drug therapy, reduced the global prevalence of leprosy from 5.2 million in 1985 to 181,941 cases at the end of 2011, and eliminated leprosy from 119 of the 122 countries considered to be endemic for leprosy in 1985.34
United States
The epidemiology of leprosy in the United States reflects immigration patterns. There are currently approximately 6500 cases registered with the National Hansen’s Disease Program (NHDP) (www.hrsa.gov/hansensdisease; tel: 1-800-642-2477), with 213 new cases reported in 2009. California, Texas, Louisiana, New York, Hawaii, Florida, and Massachusetts reported the largest number of cases (65%) in the 2009 surveillance report, but cases were reported from 33 states. Approximately 75% of the patients were born outside the United States. The largest proportion (35%) of U.S. cases self-identified as Asian or Western Pacific Islanders, almost 70% of the patients were men, and patient age ranged from 10 to 94 years. With regard to the history of leprosy in the United States, a marked increase in cases occurred with the emigration of Southeast Asian refugees during 1978 to 1988, but this was not accompanied by an increase in cases in people born in the United States, indicating that transmission of leprosy within the United States is rare.35 However, endemic cases have been identified in the New York City metropolitan area, Mississippi, Louisiana, and Texas.11,36
Microbiology and Genome Sequence
Despite many generations of effort, M. leprae cannot be cultured in vitro. Consequently, knowledge of the biology of M. leprae has been restricted to biochemical and physiologic characterization of bacteria isolated from experimentally infected nine-banded armadillos. M. leprae is a straight or slightly curved rod-shaped organism that is 1 to 8 µm long and 0.3 µm in diameter. It is Gram stain positive and acid fast, although staining with carbol fuchsin can be irregular. On the basis of assays in footpads of immunodeficient mice, the doubling time of M. leprae has been estimated to be 11 to 13 days.1 Like other mycobacteria, M. leprae possesses a cell wall that contains diverse lipids and glycolipids, including an abundant antigenic glycolipid termed phenolic glycolipid 1 (PGL-1).
The availability of the genome sequence of M. leprae has provided substantial information on the biology of M. leprae, its relationship to other mycobacteria, potential explanations for the inability to culture it in vitro, and potential new targets for drug therapy. The genome contains approximately 3.3 million base pairs, with an average G + C content of 57.8%. It compares to the genome of M. tuberculosis, which contains approximately 4.4 million base pairs, with an average G + C content greater than 65%.37 In addition to the reduction in the size of the M. leprae genome compared with that of M. tuberculosis, only 49.5% of the M. leprae genome is predicted to contain protein-coding genes, because of a high frequency of pseudogenes and noncoding DNA. Therefore, the functional genome of M. leprae appears to be less than 40% of the size of the genome of M. tuberculosis. Because it is generally believed that M. leprae and M. tuberculosis evolved from a common mycobacterial ancestor, M. leprae appears to have lost approximately 2000 genes since the divergence, which has left it dependent on highly specialized ecologic niches for its survival.
Analysis of the residual functional genes of M. leprae in comparison with those of M. tuberculosis and the sequenced genomes of other bacteria has provided considerable insight into its biology. On the basis of the presence of intact operons, it appears that M. leprae has retained nearly all essential anabolic pathways, including synthesis of amino acids, purines, pyrimidines, nucleosides, nucleotides, and many vitamins and enzyme cofactors. In contrast, it lacks the diversity of the apparatus for lipid synthesis and modification characteristic of M. tuberculosis. In particular, M. leprae lacks methoxymycolates, probably because of the absence of the mmaA2 and mmaA3 genes whose products are responsible for methoxy modification of mycolic acids in M. tuberculosis.38 In addition, M. leprae has only six genes encoding polyketide synthases, compared with 18 in M. tuberculosis. Because specific polyketides contribute to pathogenesis of M. tuberculosis39and Mycobacterium ulcerans,40 the differential polyketide synthase gene content may account for some of the differences in pathogenesis of M. leprae compared with other virulent mycobacteria.
One of the polyketide synthases absent from the M. leprae genome is encoded by mbtB, which is essential for synthesis of salicylate-derived mycobactin siderophores in M. tuberculosis and is necessary for optimal acquisition of iron and growth in iron-poor media or macrophages.39 The absence of the mbt operon from M. leprae implies that this species depends on other mechanisms for iron acquisition and retention, which may contribute to the narrow ecologic niche occupied by M. leprae. A glycolipid that is important in M. leprae pathogenesis and that is absent from M. tuberculosis is PGL-1. PGL-1 is derived from the precursor phthiocerol dimycocerosate, a lipid that is common in other mycobacteria. Reconstitution of M. bovis bacillus Calmette-Guérin (BCG) with a complex of six genes from two loci of M. leprae has enabled study of the biosynthetic steps and functional roles of PGL-1 in the context of culturable bacteria.41 This revealed that PGL-1 enhances uptake and intracellular replication of bacteria by human macrophages and dendritic cells and reduces activation of nuclear factor kappa B and secretion of tumor necrosis factor (TNF). In contrast to evidence obtained from experiments with purified PGL-1, expression of PGL-1 in BCG did not confer resistance to reactive oxygen or reactive nitrogen metabolites.
An additional application of genome sequence data is the use of sequence variations to study evolution and transmission history. A recent study determined the genome sequences of four isolates from distinct global regions to identify polymorphic sites, mainly single nucleotide polymorphisms (SNPs), which were then confirmed in a total of 400 independent strains from 28 regions.42 This revealed that the four previously discovered lineages of M. leprae could be subdivided into a total of 16 subtypes and provided evidence for the origin of M. leprae in East Africa (SNP type 2), which gave rise to SNP type 1, which was carried by human migration eastward to Asia, and gave rise to SNP type 3, which was transported westward to the Middle East and Europe. SNP type 4, which is prevalent in West Africa and countries with contact with West Africa through the slave trade, arose from SNP type 3. The evidence also suggests that M. leprae entered Asia by a southern route and also by a northern route that corresponded to the Silk Road. In contrast to SNPs, whose rates of change in M. leprae are too slow to be valuable in studies of transmission dynamics and epidemiology, studies using variable numbers of tandem repeat typing have so far implied that their rate of variation may be excessively high for studies of human-human transmission, because they have revealed variable results from samples of different sites from individual patients.43,44
Immunology
Clinical Manifestations and Immunologic Spectrum of Leprosy
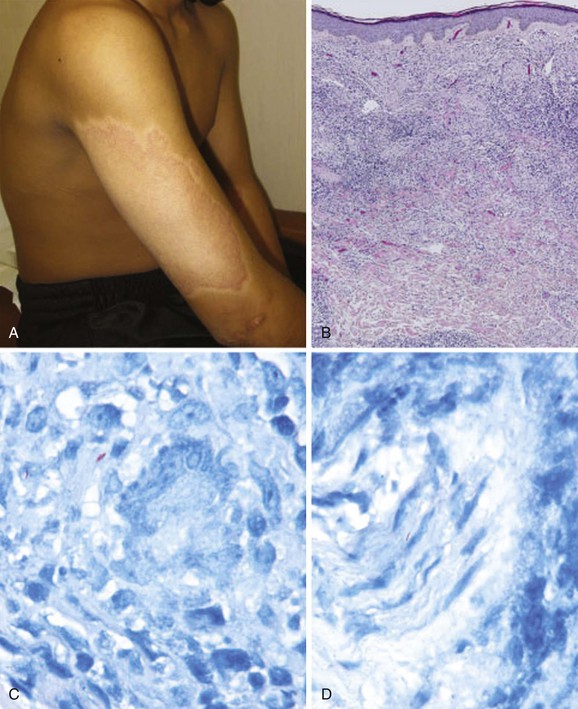
In endemic countries, clinical leprosy has long been categorized by two WHO-classified subtypes: paucibacillary, referring to five or fewer skin lesions and negative acid-fast bacilli (AFB) skin slit smears, and multibacillary, referring to more than five skin lesions and positive AFB skin slit smears. In resource-constrained regions where skin slit smears are not available, the advantage of the WHO classification system is that patients can be grouped on the basis of the number of skin lesions alone. The Ridley-Jopling classification system is more commonly used to describe the subtype of disease. This system classifies patients into five types—tuberculoid (TT), borderline tuberculoid (BT), mid-borderline (BB), borderline lepromatous (BL), and lepromatous leprosy (LL)—and is based on a combination of clinical manifestations (number and appearance of skin lesions, peripheral nerve thickening/impairment and systemic or mucosal involvement), the bacillary load, and histopathology.2 Patients with tuberculoid and lepromatous leprosy have stable cell-mediated immunity; their disease manifestations do not change over time. In contrast, patients with borderline disease have unstable cell-mediated immunity and their clinical manifestations may change over time (e.g., manifestations can “upgrade” or “downgrade” toward tuberculoid or lepromatous presentations, respectively). Some patients develop an indeterminate form of the disease that is distinct from the subtypes included within this spectrum; this very early form of leprosy is seen most frequently in young children, is associated with a short duration of symptoms and a lack of neurologic involvement, and can resolve without treatment.
The Ridley-Jopling classification system has been widely used, at least in part because of the clear correlation between the histologic appearance of the local immune response (abundant lymphocytes and well-formed granulomas in the tuberculoid type; fewer lymphocytes without well-formed granulomas in the lepromatous type) and the number of bacteria (few in the tuberculoid type; numerous in the lepromatous type). From these observations, a correlation between the nature of the immune response, control of bacterial growth, and clinical appearance in leprosy has provided a basis for studies of the protective cellular immune response to M. leprae.
Mechanisms of Immunity
The early observation that tuberculoid leprosy lesions contain large numbers of CD4+ T lymphocytes and low numbers of M. leprae45 supported the concept that adaptive cellular immunity contributes to control of the infection. This concept was refined by the discovery that tuberculoid leprosy was associated with lymphocyte expression of interleukin (IL)-2 (IL-2), lymphotoxin, and interferon-γ, whereas lepromatous leprosy was associated with expression of IL-4, IL-5, and IL-10, and not interferon-γ, in lesions.3 This pattern of cytokine production fits the paradigm of helper T cell type 1 (Th1) or Th2 differentiation of T lymphocytes and has been the basis of further studies to understand the determinants of an effective immune response to M. leprae.
Given the evidence that a Th1 CD4+ T-cell response to M. leprae is associated with control of infection and a Th2 response is not, identifying the factors that determine the polarity of the immune response in leprosy is of crucial importance. Although individuals with lepromatous leprosy exhibit little, if any, Th1 immune responses to M. leprae antigens, their CD4+ T lymphocytes are capable of responding to other antigens with a Th1 (i.e., interferon-γ) response, which demonstrates that lepromatous leprosy is not the result of a global inability to generate a Th1 response.46 This finding has focused attention on the interactions of M. leprae and antigen-presenting cells (monocytes and dendritic cells), because the outcome of these encounters determines the polarity of CD4+ T-cell differentiation (i.e., Th1 vs. Th2).
As described in Chapter 6, antigen stimulation of CD4+ T lymphocytes is mediated by presentation of pathogen-derived peptide (bound to major histocompatibility complex class II) or glycolipid (bound to CD1a, b, c, or d) antigens on monocytes or dendritic cells for recognition by specific T-cell antigen receptors. The determinants of the polarity of antigen-stimulated T lymphocytes are imposed by antigen-presenting cells. A major determinant of Th1 differentiation of naïve T lymphocytes is IL-12, whose expression is approximately 10-fold higher in tuberculoid lesions than in lepromatous lesions,47 although monocytes and monocyte-derived dendritic cells from patients with lepromatous leprosy and tuberculoid leprosy produce similar amounts of IL-12 in response to a triacylated lipopeptide derived from M. leprae.46 Lipopeptide stimulation of antigen-presenting cell IL-12 production is mediated by Toll-like receptors 1 (TLR1) and 2 (TLR2), and expression of TLR1 and TLR2 is higher in lesions of tuberculoid compared with lepromatous leprosy.48
In addition to Toll-like receptor–stimulated production of IL-12, stimulation of antigen-presenting cells through CD40 by activated T cells expressing CD40L contributes to production of IL-12. Expression of CD40 and CD40L has also been found to be deficient in lepromatous compared with tuberculoid lesions.49 The low expression of TLR1, TLR2, and CD40 in lepromatous lesions may contribute to deficient induction of IL-12, and the resulting deficiency of IL-12 in lepromatous lesions is consistent with failure to generate Th1, interferon-γ–producing CD4+ T cells that respond to M. leprae antigens. However, evidence that a polymorphism in TLR1 that disrupts trafficking of TLR2 to the cell surface is associated with protection from leprosy per se22 and from reversal reactions23 indicates that the role of TLR2 is more complex than the in vitro studies imply.
A recent study reported that M. leprae or a synthetic agonist of NOD2 promotes differentiation of human monocytes into dendritic cells with the capacity for efficient activation of antigen-specific CD4 and CD8 T cells, and that activation of NOD2 drives this cellular differentiation program by inducing IL-32.50 This finding, together with the results of genetic studies implicating NOD2 polymorphisms in susceptibility to leprosy, may provide useful insight into an early step in responses to M. leprae. Evidence that IL-32 also induces expression of NOD2 implies that these may be components of a positive feedback response that amplifies responses to NOD2 agonists.51
Although understanding of the differences in the immune response in lepromatous compared with tuberculoid leprosy has advanced considerably, and recent studies indicate a role for disproportionately abundant M. leprae antigen-specific regulatory T cells in lepromatous leprosy,51a the essential initial determinant of the distinct responses remains to be identified. The polarity of the cellular immune response (i.e., Th1 vs. Th2) is likely to be determined during the initial encounter of a naïve host with M. leprae, but it is not yet clear whether the essential determinant of that outcome is influenced by the route of infection, the precise phenotype of the antigen-presenting cells that present M. leprae antigens to naïve T cells, or other host cofactors such as infection with other pathogens or altered endogenous microbiota. The association of leprosy with polymorphisms of NOD2 and genes whose products function in the same pathway provide a logical entry point for further investigation.
Pathogenesis of Nerve Damage
Peripheral sensory nerve damage is the leading cause of functional morbidity in people with leprosy and is characteristic of both paucibacillary and multibacillary disease.52,53 Peripheral nerve damage in leprosy can be mediated directly by M. leprae, as well as by the immune response to the organism.54,55
Direct Nerve Damage by Mycobacterium leprae
M. leprae invades Schwann cells, the glial cells of the peripheral nervous system. Because Schwann cells form a functional unit with peripheral nerve axons surrounded by a basal lamina, studies have focused on the interaction of M. leprae with proteins of the basal lamina. These studies revealed that M. leprae specifically interacts with the G domain of the α2-subunit of laminin-2, a neural-specific extracellular matrix protein.56 This domain of laminin-2 can bind simultaneously to M. leprae and to the Schwann cell laminin receptor α-dystroglycan, allowing high-affinity binding of M. leprae to Schwann cells by using laminin-2 as a bridging molecule.57 Laminin-2 interacts with two distinct molecules on the surface of M. leprae, a 21-kDa protein and the abundant glycolipid PGL-1. The 21-kDa protein, termed M. leprae laminin-binding protein (ML-LBP21) (also known as histone-like protein/Hlp58), interacts with the G4 module of the α-subunit of laminin-2, and ML-LBP21 is sufficient to mediate invasion of Schwann cells.59 PGL-1, which is a highly abundant component of M. leprae, also binds to the α2-subunit of laminin-2, through the G4 and G5 modules of the G domain.60 PGL-1, like ML-LBP21/Hlp, is sufficient to mediate invasion of Schwann cells and interacts with laminin-2 through the unique trisaccharide of PGL-1 (3,6-di-O-methylglucose linked α-1→4 to 2,3-di-O-methylrhamnose linked β-1→2 to 3-O-methylrhamnose).60,61 These studies provide considerable insight into the molecular mechanism of the direct interaction between M. leprae and Schwann cells of peripheral nerves and illustrate that M. leprae uses a neural-specific target for its apparently redundant bacterial molecules (ML-LBP21/Hlp and PGL-1) to achieve its unique tropism for peripheral nerves.
Once M. leprae or its PGL-1 is bound and internalized by Schwann cells, it can cause demyelination of peripheral nerves in vitro and in vivo in the absence of a cellular immune response.54 Binding and activation of the receptor tyrosine kinase ErbB2 by one or more unidentified molecules on the surface of M. leprae can also lead to rapid demyelination of peripheral nerves in vitro.62 Demyelination by M. leprae can promote further invasion of Schwann cells by the bacteria, because M. leprae preferentially invades nonmyelinated Schwann cell-axon units. M. leprae-mediated demyelination occurs without early cell death or toxicity, although Schwann cells and neurons can die by apoptosis later after infection.54,63 In addition, dead M. leprae or PGL-1 shed from live or dying M. leprae can mediate peripheral nerve demyelination,54 which may contribute to the ongoing nerve damage that can follow initiation of active chemotherapy. Furthermore, prolonged M. leprae infection of Schwann cells in vitro causes marked dedifferentiation to cells with mesodermal characteristics, and this is proposed to play a role in dissemination of the bacteria.64
In addition to PGL-1, an M. leprae 19-kDa lipoprotein can mediate Schwann cell apoptosis as an agonist of TLR2. TLR2 is expressed on Schwann cells in vitro and in vivo, and apoptotic Schwann cells can be found in human leprosy lesions.63 The results of these studies clearly demonstrate that M. leprae is capable of direct peripheral nerve damage, even in the absence of inflammation or a cellular immune response. These mechanisms are likely to be especially responsible for peripheral nerve damage in multibacillary leprosy.
Immunologically Mediated Peripheral Nerve Damage in Leprosy
In addition to direct damage to peripheral nerves by M. leprae, there is abundant evidence that the immune response in leprosy contributes to nerve damage. This contribution is likely to account for much of the nerve damage that occurs in paucibacillary leprosy, in which the bacteria and PGL-1 are present in insufficient quantities to cause widespread nerve damage, and in reversal reactions, in which inflammation is particularly prominent.
Several distinct immunologic mechanisms probably contribute to nerve damage in leprosy.65 Proinflammatory cytokines such as tumor necrosis factor, IL-1β, and interferon-γ are especially prominent in lesions during reversal reactions,66 when marked and irreversible nerve damage can occur. Because these molecules can directly and indirectly contribute to inflammatory tissue damage and can induce apoptosis of Schwann cells in vitro,67 it is likely that these mediators play an active role in nerve damage. Reversal reactions are also characterized by an increase in the number of CD4+ T lymphocytes in lesions, and at least some of these CD4+ cells exhibit a cytotoxic phenotype and kill M. leprae–infected Schwann cells through antigen- and class II–dependent secretion of cytotoxic granule contents.55 Whether similar mechanisms of nerve damage occur in chronic tuberculoid leprosy is not established, but qualitatively similar cytokines and T lymphocytes are found in tuberculoid lesions. Investigation of the potential roles of proinflammatory Th17 cells and the presence68 and activity of regulatory T cells in reversal reactions and nerve damage in chronic tuberculoid leprosy are also warranted.
Clinical Manifestations
The cardinal manifestations of leprosy are hypopigmented, erythematous or infiltrative skin lesions with or without neurologic signs or symptoms (including hypoesthesia, weakness, autonomic dysfunction, or peripheral nerve thickening). The clinical manifestations of leprosy vary significantly, depending on the subtype of disease. Patients with stable tuberculoid or borderline tuberculoid leprosy may present with skin lesions but without subjective complaints (Fig. 252-1). In contrast, patients with advanced tuberculoid leprosy may present with asymmetric peripheral neuropathy. Patients with stable lepromatous or borderline lepromatous leprosy may present with widespread infiltrative skin lesions or prominent peripheral neuropathy with secondary deformities (e.g., claw hand) or nonhealing painless ulcers (Fig. 252-2). Reactions (see later) include reversal reactions, which present most often with increased erythema of preexisting skin lesions and progressive peripheral neuropathy, and erythema nodosum leprosum (ENL), which presents as systemic signs and painful erythematous skin nodules (Fig. 252-3). The history of a patient with suspected leprosy should include whether the person has resided in an area with high prevalence and whether the person has been previously diagnosed or treated for leprosy. Certain patients may deny knowledge of a prior diagnosis or may report that skin lesions or neuropathy or both are acute, because they wish to avoid the stigma of a diagnosis of leprosy; this occurs even in immigrants to developed countries.