Multiple myeloma and related disorders compose a spectrum of diseases that are characterized by the autonomous proliferation of differentiated lymphoid cells and plasma cells whose physiologic function is to secrete immunoglobulins. Plasma cell dyscrasias account for 1% of all cancers in the United States, and approximately 10% of hematologic malignancies. Multiple myeloma is the most common of the dyscrasias, representing about 75% of cases and affecting approximately 15,000 individuals each year. Waldenstrom’s macroglobulinemia accounts for about 20% of cases, with the remainder consisting of other types of heavy-chain disease.
Multiple Myeloma
Multiple myeloma (MM) is a malignancy of clonal plasma cells involving primarily the bone and bone marrow (BM). The median age at diagnosis is approximately 65 years, and fewer than 3% of patients are younger than 40 years. MM remains incurable, with a median survival of approximately 3.5 years. In 2009 an estimated 20,580 patients will be diagnosed, and 10,580 patients will die from the disease ( ).
DISEASE BIOLOGY
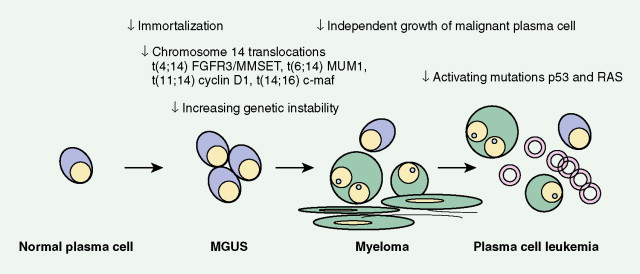
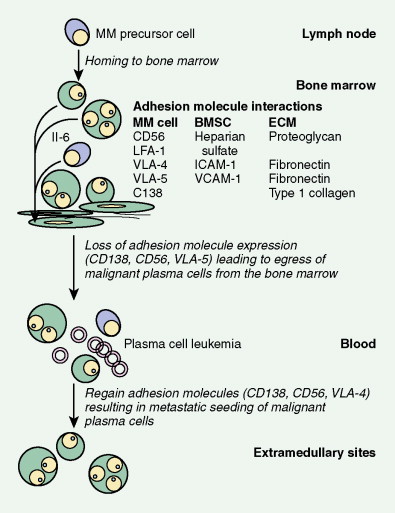
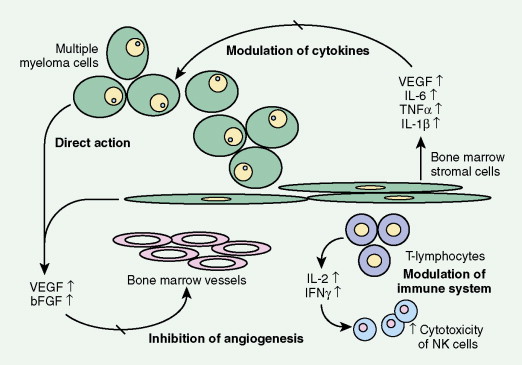
The available evidence suggests that the clonal precursor cell in MM is a B cell that has undergone somatic hypermutation and passed through the germinal center. Within the lymph node illegitimate immunoglobulin (Ig) class switching occurs, resulting in chromosome 14q32 translocations and the dysregulation of a number of oncogenes ( FGFR3/MMSET , cyclin D1, c- MAF ) ( ). Chemokines then mediate homing of the immortalized MM cells from the lymph node to the BM, where binding of MM cells to BM stromal cells (BMSCs) occurs, localizing the MM cells within the BM microenvironment. Adhesion of MM cells results in an increase in the transcription and secretion of a number of cytokines involved in MM cell growth and survival, including interleukin-6, insulin-like growth factor-1, vascular endothelial growth factor, stromal cell–derived growth factor-1α, tumor necrosis factor-α, transforming growth factor-β, and B-cell activating factor, which augment MM cell growth, survival, drug resistance, and migration in the BM milieu. Besides localizing tumor cells in the BM microenvironment, our studies demonstrate that adhesion of MM cells to BMSCs also triggers the paracrine nuclear factor-κB-dependent transcription and secretion in BMSCs of interleukin-6, the major cytokine mediating MM cell growth, survival, and resistance to drug (dexamethasone)-induced apoptosis via mitogen-activated protein kinase and phosphoinositide 3-kinase/Akt, Jak/signal transduction and transcription activator, and phosphoinositide 3-kinase/Akt signaling cascades, respectively. Most importantly, adhesion of MM cells to the BM induces changes in gene profile—that is, upregulation of growth, survival, and drug resistance genes in tumor cells; upregulation of adhesion molecules on MM cells and BMSCs; and changes in cytokines in BMSCs both in vitro and in our in vivo models of human MM in mice. Interaction of MM cells with BMSCs activates Notch signaling, which induces melphalan resistance. Others have shown that MM cell adhesion to fibronectin confers cell adhesion–mediated drug resistance to conventional chemotherapy, with induction of p27 and G1 growth arrest. Excitingly, novel agents including thalidomide and derivatives including lenalidomide, as well as proteasome inhibitor bortezomib (Velcade; PS-341, Millenium Takeda Oncology) can target both the tumor cell and its BM microenvironment, thereby overcoming cell adhesion–mediated drug resistance. Induction of proteasome activity when MM cells bind to BMSCs may sensitize them to therapy. Other cytokines are produced that promote bone resorption, tumor migration, and invasion, including matrix metalloproteinase-1, interleukin-1β, and vascular endothelial growth factor ( ). As disease progresses, the development of plasma cell leukemia (PCL) is characterized by increasing genetic instability of the MM cell associated with a high frequency of RAS, TP53, and c- MYC mutations ( ). This is accompanied by a decreased expression of certain adhesion molecules, which facilitates tumor cell mobilization into the peripheral blood. The acquisition of other adhesion molecules on the MM cell surface leads to the metastasis of MM cells to sites outside the BM and the development of extramedullary plasmacytoma.
DIAGNOSIS AND MORPHOLOGY
The Durie and Salmon major and minor diagnostic criteria have been conventionally used for the diagnosis of MM. These include the presence of excess monotypic marrow plasma cells, monoclonal Ig in either the serum or urine, decreased normal Ig levels, and lytic bone lesions ( ). These diagnostic criteria are now being replaced by the . They help in distinguishing active MM from other disorders characterized by monoclonal gammopathies, both malignant and otherwise, in particular monoclonal gammopathy of undetermined significance (MGUS) and smoldering MM. Other conditions such as Waldenstrom’s macroglobulinemia, non-Hodgkin lymphoma, light-chain amyloid, idiopathic cold agglutinin disease, essential cryoglobulinemia, and heavy-chain disease should also be differentiated from MM.
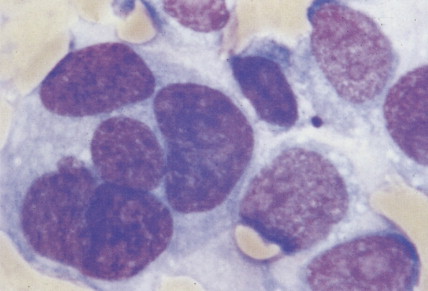
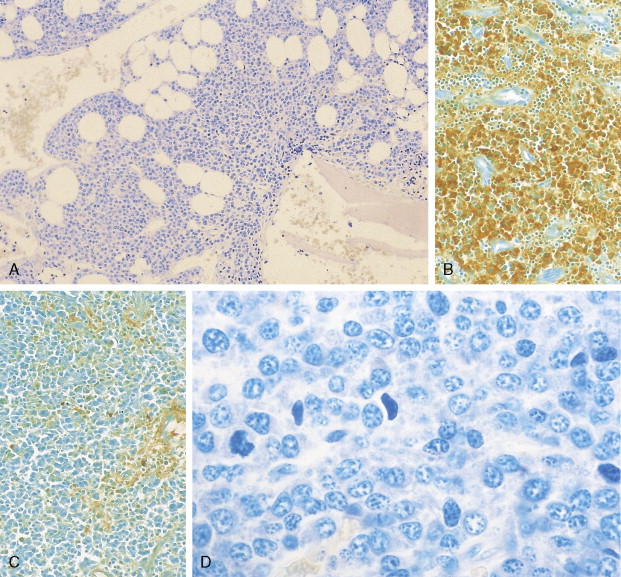
The morphology of plasma cells in MM varies from typical small, mature cells that appear normal to larger cells with immature nuclei containing prominent clear nucleoli, little or no perinuclear clear area (hof), and a rim of basophilic cytoplasm. In between these extremes are medium-sized cells, often characterized by an eccentric nucleus with one or several small nucleoli, a diffuse chromatin pattern, perinuclear hof, and a variable amount of blue cytoplasm. On biopsy normal BM architecture is lost, and the plasma cells are found as single cells or small clusters between adipocytes. As the disease progresses, diffuse marrow replacement occurs, resulting in a packed marrow.
The diagnosis of MM can be confirmed by immunoperoxidase staining that demonstrates the presence of monoclonal cytoplasmic-staining light chains (κ or λ) or monoclonal heavy chains (IgG, IgA, or IgD). Since plasma cells are terminally differentiated B cells, they express a number of B-cell antigens as well as myeloma-associated antigens including CD38, CD138 (syndecan-1), Muc-1, and PCA-1. They lack CD10, CD20, CD23, CD34, and CD45RO.
STAGING OF MULTIPLE MYELOMA
The Durie-Salmon staging system was conventionally used to separate patients into prognostic groups using laboratory measurements. These stages are now being replaced by the International Staging System ( ). This is simple and correlates well with survival. It relies on serum albumin and β 2 -microglobulin (β2m) levels at diagnosis. In addition to a full physical examination, a complete blood count with blood smear, and blood chemistries including renal studies, are required. Serum electrophoresis with quantification of protein, as well as a 24-hour urine electrophoresis and quantification of Bence-Jones protein (light chains), should also be performed. The serum-free light-chain assay is a new test that allows diagnosis of patients with light-chain disease. BM aspirate and biopsy are indicated. An estimation of the extent of bone disease using a skeletal bone survey is advisable, although in many centers this has now been superceded by magnetic resonance imaging.
CLINICAL MANIFESTATIONS
The clinical picture of MM involves a combination of bone destruction leading to pain or fracture with hypercalcemia; infection due to immune deficiency; BM failure leading to anemia and, less commonly, thrombocytopenia; and renal failure due to hypercalcemia, direct damage from paraprotein, or precipitation of light chain in renal tubules. A hyperviscosity syndrome may occur in some patients, and neurologic complications such as spinal cord compression and peripheral neuropathy may be seen.
A number of studies have evaluated various parameters for prognostic significance. β2m, a polypeptide that forms the extracellular portion of the light chain of the class I major histocompatibility complex, is the single most important variable. Levels correlate with a high tumor burden and decreasing renal function. The extent and type of BM infiltration are also important: patients with a plasmablastic morphology have a much shorter median survival compared with patients with other morphologic subtypes ( ). Measures of tumor cell proliferation such as the plasma cell labeling index (PCLI) are also useful; a high PCLI correlates with a shorter survival time independent of tumor cell mass. The presence of certain cytogenetic abnormalities also has prognostic significance: patients with partial or complete deletions of chromosome 13 or abnormalities of 11q have an adverse outcome.
Three recent prognostic staging systems have been proposed to better predict patient outcome. First, an international staging system based upon serum β2m and albumin has provided a new three-stage International Staging System ( ). Second, cyclin D dysregulation has been identified as an early and unifying event in MM. Using gene expression profiling to identify five recurrent translocations, specific trisomies, and expression of cyclin D2 genes, MM can prognostically be divided into eight translocation/cyclin D groups ( ). Additional molecular classifications have been proposed, with high-risk myeloma defined by deregulated expression of genes mapping to chromosome 1. Most recently, the first DNA-based classification scheme has been proposed to predict outcome to high-dose therapy ( ).
THERAPY
Treatment for MM has evolved significantly in the last 3–4 years. Although the use of conventional low-dose chemotherapy or high-dose chemotherapy with autologous or allogeneic stem cell transplantation is able to reduce tumor burden, complete molecular remission of disease is rare, and all patients eventually relapse. Novel pharmacologic agents including thalidomide, lenalidomide, and proteasome inhibitors like bortezomib are now approved by the U.S. Food and Drug Administration for the treatment of MM ( ). These agents have been used, either alone or in combination with conventional and high-dose chemotherapy, to improve response and outcome.
Other novel single agents of great promise include new proteasome inhibitors NPI-0052 and PR-171, fibroblast growth factor receptor-3 (FGFR3) inhibitors, humanized antibodies to CD40, antibody to CS1, mitogen-activated protein/extracellular signal–regulated kinase kinase (MEK) inhibitor AZD6244, cyclin-dependent kinase inhibitor P276.00, Hsp90 inhibitors, and histone deacetylase (HDAC) inhibitors. NPI-0052 and PR-171 are next-generation proteasome inhibitors that are active against bortezomib-resistant MM, are nontoxic in preclinical models, and are already in clinical trials in relapsed MM. FGFR3 inhibitors specifically target those 15% to 20% of patients with t(4;14) translocation. CD40 humanized antibodies have demonstrated early activity against MM, and Hsp90 inhibitors as single agents can achieve responses in relapsed refractory MM. HDAC inhibitors block aggresomal breakdown of ubiquinated proteins; combined with proteasome inhibitors to inhibit proteasomal degradation of ubiquinated proteins, they mediate significant toxicity. Clinical testing of the HDAC inhibitor panobinostat (LBH589; Novartis) is ongoing, with a combination LBH and bortezomib trial soon to follow. Immunologic-based treatment approaches (e.g., vaccination, antibody therapy) are also being actively evaluated ( ).
Plasma Cell Leukemia
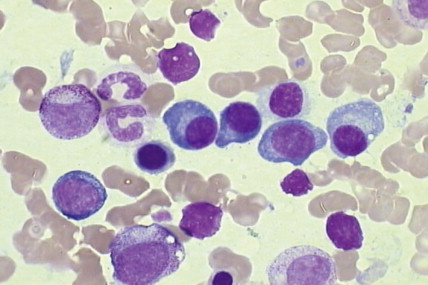
PCL is characterized by the presence of more than 2 × 10 9 plasma cells per liter within the peripheral blood, which constitutes 20% of all circulating cells. The majority (60%) of cases are de novo or primary, in which a leukemic picture develops in the absence of documented preceding myeloma, whereas 40% of cases are secondary and occur in 1% of myeloma patients with advanced myeloma that is refractory to treatment ( ). The symptoms of primary disease are similar to myeloma, although the disease course is often more aggressive with symptoms relating to extramedullary disease (plasmacytomas and hepatosplenomegaly) and BM failure (anemia, infections, and bleeding).
Monoclonal Gammopathy of Undetermined Significance
MGUS describes a condition characterized by the presence of a low level of paraprotein in the absence of other clinical features of MM, Waldenstrom’s macroglobulinemia, or other B-cell lymphoproliferative disorders. MGUS is present in 3.2% of persons 50 years of age or older and 5.3% of persons 70 years of age or older. Independent prognostic factors associated with MGUS transformation to MM include (1) more than 5% BM plasma- cytosis, (2) Bence-Jones proteinuria, (3) decrease in polyclonal serum Ig, and (4) an elevated erythrocyte sedimentation rate. More recently, risk factors for progression include an abnormal serum κ/λ free light-chain ratio, a high serum monoclonal protein level, and non IgG MM. The risk of progression from smoldering MM to symptomatic disease is related to the proportion of BM plasma cells and serum monoclonal protein level at diagnosis ( ). The previous term “benign monoclonal gammopathy” is a misleading description of the disease, since approximately 25% of patients will go on to develop an overt plasma cell disorder.
Waldenstrom’s Macroglobulinemia
Waldenstrom’s macroglobulinemia (WM) is a chronic B-cell lymphoproliferative disorder in which most of the clinical manifestations are due to the presence of a high level of serum IgM paraprotein ( ). It is predominantly a disease of the elderly, with a median age at presentation of 65 years, and remains incurable with a median survival of 5 years. The disorder is characterized by BM infiltration with small lymphocytes, plasma cells, and characteristic lymphoplasmacytoid cells, which have the nucleus of a lymphocyte and cytoplasm of a plasma cell. The immunophenotype of WM cells is between that of well-differentiated small lymphocytes and plasma cells, since cells express monoclonal surface and cytoplasmic IgM pan-B-cell markers CD19, CD20, and CD22, and late B-cell differentiation markers such as CD38 and FMC7, but lack CD5, CD10, and CD23. Most patients develop symptoms due to tumor cell infiltration of BM resulting in anemia, increased infections, and bleeding. Splenomegaly, hepatomegaly, and lymphadenopathy are common. Many patients present with hyperviscosity syndrome accompanied by headache, confusion, and blurred vision. Cryoglobulinemia, cold agglutinin disease, and neurologic manifestations may also occur. In contrast to other plasma cell disorders, there is an absence of bony changes and lytic lesions.
LIGHT CHAIN–ASSOCIATED AMYLOIDOSIS
Light-chain amyloid, previously called primary amyloid, is characterized by the extracellular deposition of fibrillar protein derived from monoclonal light chains ( ). The fragments form β-pleated sheets, which become insoluble and resistant to degradation following the deposition of glycosaminoglycans and the normal protein serum amyloid P component. The clinical features depend on the spectrum of organ involvement, with the most commonly affected organs being the heart, kidneys, and peripheral nerves. Other features include macroglossia (infrequent but pathognomonic), gastrointestinal malabsorption, hepatosplenomegaly, and skin involvement including papular and nodular lesions and characteristic purpura around the eyes. A monoclonal component is usually present in the serum or urine, although immunofixation is often required to demonstrate its presence, since the peak may be small. Amyloid may occur as a long-term complication of most clonal B-cell disorders, especially myeloma and, less commonly, WM.
Heavy-Chain Disorders
This group of rare plasma cell disorders is characterized by the production of a monoclonal Ig that is formed from truncated heavy chains with no associated light chains ( ). α-Heavy-chain disease, a subtype of immunoproliferative small intestine disease, is diagnosed by the identification of monoclonal Ig α-heavy-chain fragments in the plasma or urine. The pathologic process consists of a lymphoma-like proliferation of lymphoid cells and plasma cells in the lamina propria of the small intestine and in mesenteric nodes, accompanied by diarrhea and malabsorption. Occasionally the disease may present with respiratory symptoms due to infiltration in the respiratory tract. It was previously referred to as Mediterranean abdominal lymphoma, but it may also be identified in patients of non-Mediterranean ancestry.
Acknowledgments
We are extremely grateful to Dr. Andrew Jack (Haematological Malignancy Diagnostic Service, Leeds General Infirmary, Leeds, UK) and Professor Gareth Morgan and Dr. Faith Davies (Royal Marsden Hospital, Sutton, UK) for allowing us to use some of their figures.



| Types | Frequency (%) |
|---|---|
| IgG | 52 |
| IgA | 22 |
| κ only | 9 |
| λ only | 7 |
| IgD | 2 |
| Biclonal | 1 |
| IgM * | 0.5 |
| Negative | 6.5 |
* IgM rarely occurs, since it usually indicates Waldenstrom’s monoglobulinemia. Light-chain disease is defined by circulating light chains only with Bence-Jones proteinuria and hypogammaglobulinemia. As the result of imbalanced immunoglobulin production, most patients with an M component also have monoclonal light chains that vary from barely detectable levels to grams per day.
| Parameter | MGUS | Smoldering myeloma | MM |
|---|---|---|---|
| M component | <30 g/L | >30 g/L | >30 g/L |
| % BM plasma cells | <10% | >10%, <30% | >30% |
| Symptoms | Nil | Nil | Yes |
| Bone lesions | Nil | Nil | Present |
| Anemia, hypercalcemia, or renal impairment | Nil | Nil | Present |
| Follow-up | Stable | Stable initially, once progressed similar to MM | Median survival 3.5 years |
| Cytokine | Role |
|---|---|
|
|
Related posts:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


