- Monogenic diabetes results from single-gene mutations that cause β-cell dysfunction or, less commonly, insulin resistance.
- Approximately 1–2% of diabetes is monogenic, but this is frequently misdiagnosed.
- Monogenic diabetes should be suspected where: presentation is atypical for type 1 or 2 diabetes; there is an autosomal dominant (or maternally inherited in mitochondrial disorders) family history; there are characteristic associated features such as deafness in mitochondrial diabetes or fat loss in lipodystrophy; or diabetes has been diagnosed within the first 6 months of life.
- Mutations in the glucokinase gene, which is important in “sensing” blood glucose levels in the pancreas, result in resetting of fasting glucose to a higher level (5.5–8.0 mmol/L). Patients have dominantly inherited mild fasting hyperglycemia with only modest changes in glycated hemoglobin. Complications are rare and no treatment is needed.
- Mutations in the transcription factor genes HNF1A and HNF4A result in dominantly inherited progressive hyperglycemia with symptomatic diabetes in adolescence or young adulthood. Patients with HNF1A or HNF4A diabetes are very sensitive to sulfonylurea treatment and may not require insulin until middle or old age.
- Mitochondrial mutations can result in maternally inherited diabetes often with sensorineural hearing loss and a range of other disorders.
- Diabetes diagnosed before 6 months is unlikely to be type 1 diabetes and a genetic cause should be sought even where the patient is now an adult. High dose sulfonylurea treatment is often more effective than insulin where mutations affecting Kir6.2 and SUR1 subunits of the β-cell potassium channel are identified.
- Acanthosis nigricans is the key feature of insulin resistance and a genetic cause should be considered where this is seen in non-obese patients. Partial lipodystrophy results in thin muscular limbs with hypertriglyceridemia and insulin resistance and suggests a mutation in LMNA or PPARG. In the absence of lipodystrophy an insulin receptor mutation is the most common cause.
Introduction
Monogenic diabetes results from inheritance of one or more mutations in a single gene and accounts for 1–2% of diabetes cases. Mutations may be inherited in a dominant or recessive fashion. The majority (90%) of monogenic diabetes cases are initially misdiagnosed as type 1 (T1DM) or type 2 diabetes (T2DM).
Correct genetic diagnosis is important to predict clinical course, explain other associated clinical features, enable genetic counseling and diagnose family members, and most importantly guide appropriate treatment.
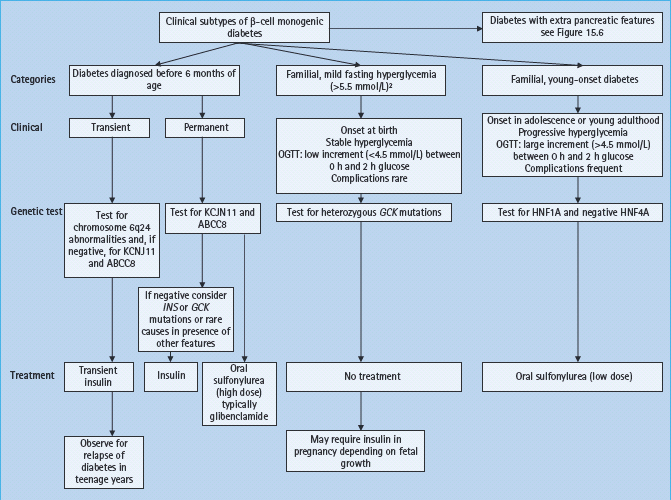
Monogenic diabetes where the primary disorder affects the β-cell has four main clinical presentations: familial mild fasting hyperglycemia (glucokinase maturity-onset diabetes of the young [MODY]), familial young-onset diabetes (transcription factor MODY), neonatal diabetes and diabetes with extrapancreatic features. Clinical and biochemical features that help differentiate the common forms of monogenic diabetes that result in β-cell dysfunction from T1DM and T2DM are summarized in Table 15.1. Clinical features and management of monogenic diabetes without extrapancreatic features are further summarized in Figure 15.1. Single-gene mutations may also cause diabetes through insulin resistance as occurs in the inherited lipodystrophies and insulin receptor mutations. A number of monogenic multisystem diseases (e.g. hemochromatosis and cystic fibrosis) may cause diabetes; these are beyond the scope of this chapter and are discussed elsewhere (see Chapter 18).
Table 15.1 Differentiating β-cell monogenic diabetesfrom type 1 and 2 diabetes.
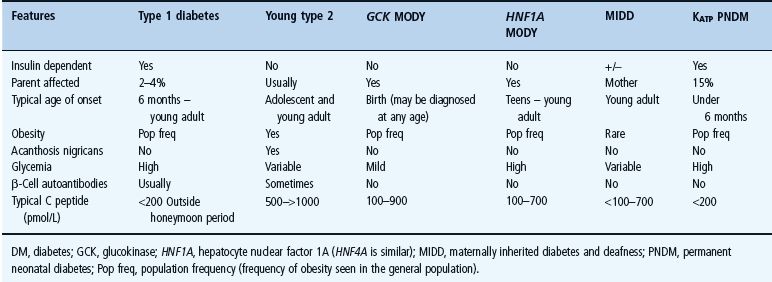
Figure 15.1 Clinical subtypes and management of monogenic β-cell diabetes without extrapancreatic features. To convert plasma glucose measurements to mg/dL multiple by 18. ABCC8, ATB binding cassette subfamily C; GCK, glucokinase gene; HNF, hepatocyte nuclear factor; INS, insulin gene; KCNJ11, potassium inwardly rectifying channel, subfamily J, member 11 gene; OGTT, oral glucose tolerance test.
Maturity-onset diabetes of the young
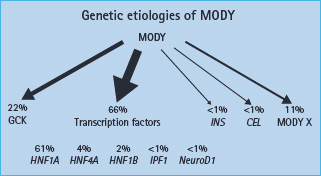
Maturity-onset diabetes of the young (MODY) is autosomal dominantly inherited diabetes that, despite a young age of onset, is not insulin dependent [1,2]. It results from β-cell dysfunction rather than insulin resistance [1]. The underlying genetic etiology has now been defined that allows MODY to be subclassified according to the gene involved [3,4]. Mutations in at least eight genes have been linked to MODY [5,6]. These include mutations in the gene encoding the glucose sensing enzyme glucokinase (GCK) and mutations in several transcription factors that affect β-cell development and function, the frequencies of which are summarized in Figure 15.2. Clinical presentation varies greatly depending on the underlying genetic mutation. Table 15.2 summarizes the clinical features of glucokinase and transcription factor diabetes. The strikingly different subtypes of MODY mean it is important to define the underlying genetic etiology. We recommend the use of clinical categories based on underlying genetic cause-familial mild fasting hyperglycemia resulting from glucokinase gene mutations (GCK MODY), familial young-onset progressive diabetes resulting from HNF1A and HNF4A mutations (transcription factor MODY) and renal cysts and diabetes syndrome (RCAD) resulting from HNF1B mutations.
Table 15.2 Comparison of the clinical characteristics of glucokinase and transcription factor maturity-onset diabetes of the young (MODY).
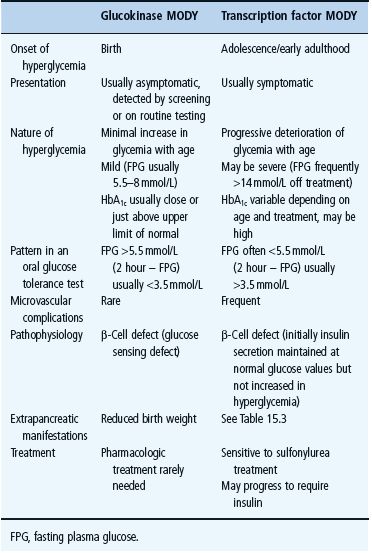
Figure 15.2 The different genetic etiology in a UK maturity-onset diabetes of the young (MODY) series. MODY X denotes dominantly inherited young-onset non- i nsulin dependent diabetes fitting clinical criteria for maturity-onset diabetes of the young where mutations in known MODY genes have not been identified. Adapted from McCarthy & Hattersley [6].
Mutations in the genes associated with MODY should be sought in patients with diabetes diagnosed under 25 years of age, who do not fully fit the phenotypes of T1DM or T2DM and who have a strong family history of diabetes (Table 15.1). Differentiating from apparent T1DM is particularly important as these patients can often be most effectively treated without the use of injected insulin.
Glucokinase MODY
Glucokinase catalyzes the phosphorylation of glucose to glucose-6-phosphate, the first and rate-limiting step in intracellular glucose metabolism in both β-cells andhepatocytes (Figure 15.3). Owing to the unique catalytic properties of the enzyme, the rate of glucose phosphorylation is proportional to the glucose concentration, thus allowing β-cells and hepatocytes to respond to changes in glycemia. In the β-cell, glucokinase acts as a glucose sensor ensuring insulin release is appropriate to the glucose concentration [7]. Heterozygous loss-of-function mutations in GCK result in a shift of the dose-response curve to the right [8]. Glycemia is therefore regulated at a higher setpoint but remains tightly controlled. Subjects are still able to stimulate their β-cells maximally [8]. Glucokinase is also present in the liver and as a result patients have reduced hepatic glycogen synthesis [9].
Figure 15.3 Glucokinase and its role within the β-cell. Glucokinase is the rate determining step in glucose metabolism, and therefore in the rate of production of ATP, which leads ultimately to insulin secretion
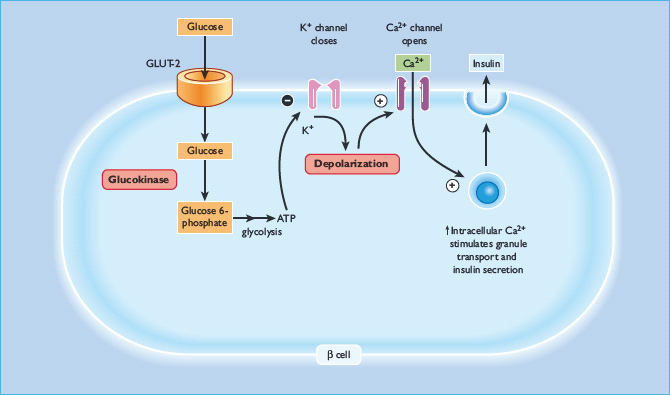
Over 200 loss-of-function mutations in GCK have been identified, all causing a similar clinical picture. Homozygous loss-of-function glucokinase mutations are a rare cause of insulin-requiring diabetes presenting in the neonatal period [10]. Gain-of-function mutations cause congenital hyperinsulinism [11].
Clinical features
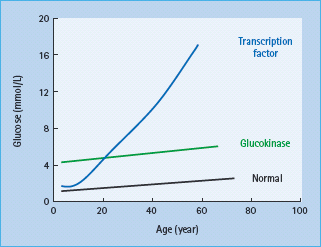
Patients have mild fasting hyperglycemia from birth, usually 5.5–8.0 mmol/L. There is only minor deterioration in fasting glucose with age (Figure 15.4) [12]. Patients do not have symptoms of hyperglycemia. Post meal glucose values are only mildly raised and there is frequently only a small increase (<3 mmol/L in 70% of patients) seen at 2 hours on an oral glucose tolerance test [13] which may explain the near normal HbA1c and rarity of complications [14]. Glycated hemoglobin values above 7.5% would be suggestive of an alternative diagnosis. Marked worsening of the glycemia suggests that the patient has developed T1DM or T2DM in additon to their GCK mutation. Microvascular and macrovascular complications are rare even when no treatment is given [14]. Glucokinase MODY is asymptomatic and, although it is autosomal dominantly inherited, there may be no known family history of diabetes. Testing of apparently unaffected parents can reveal that one parent has mildly raised fasting plasma glucose.
Figure 15.4 Variation of blood glucose concentration with age in patients with glucokinase and transcription factor MODY.
Differentiating from type 1 and 2 diabetes
Diagnosis of glucokinase MODY is most important in young patients who may otherwise be thought to have T1DM and treated with insulin [15]. Unlike T1DM, hyperglycemia remains mild, β-cell antibodies are usually negative and (if tested) one parent is likely to have mild hyperglycemia. Fasting C-peptide will remain detectable and the post meal rise in glucose concentration will be far less than in T1DM. Differentiating glucokinase MODY from T2DM can be more difficult as both conditions can cause mild hyperglycemia with a strong family history. Lack of obesity and features of insulin resistance, a small increment on oral glucose tolerance testing and non-progression all suggest glucokinase MODY.
Management
Outside of pregnancy, hypoglycemic medication is not recommended as hyperglycemia is mild, complications are rare and medication appears to have minimal effect because the regulation of glycemia is preserved [16]. Once diagnosis is confirmed, treatment can usually be discontinued; however, this should be done with caution as it is possible for T1DM or T2DM to coexist with a GCK mutation.
Glucokinase MODY and pregnancy
Clinical features
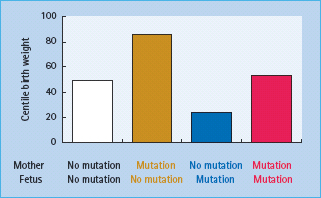
Patients with GCK mutations are frequently found to have hyper-glycemia during screening in pregnancy and represent approximately 3% of Caucasian patients with gestational diabetes [17]. Their identification is important because they have a different clinical course, both within and outside pregnancy, than other subjects with gestational diabetes. The birth weight of the newborn infant will depend on the mutation status of both the mother and the fetus (Figure 15.5). Where only the mother carries the mutation, maternal hyperglycemia may result in increased fetal insulin secretion and growth causing the fetus to be large for gestational age [18]. If the fetus inherits the mutation from the father, however, birth weight is reduced by approximately 500 g as a result of reduced fetal insulin secretion and insulin-mediated fetal growth [18]. If both mother and fetus have the GCK mutation the two opposing effects are cancelled out and the newborn infant is of normal weight.
Figure 15.5 The centile birth weight of children in families with glucokinase mutations. The weight is increased by the presence of a maternal mutation and decreased by the presence of a fetal mutation. Data from Hattersley et al. [18].
Genetic testing for GCK mutations in pregnancy
We recommend testing for GCK mutations when a pregnant patient is found to have persistently raised fasting plasma glucose 5.5–8 mmol/L and an increment of <4.6 mmol/L on at least one oral glucose tolerance test (either during or outside pregnancy). An absence of family history should not exclude the diagnosis as asymptomatic hyperglycemia in a parent may not have been detected.
Management
Patients with hyperglycemia resulting from glucokinase mutations are often treated with insulin during pregnancy in an attempt to correct the fasting hyperglycemia. Fetal genotype, however, is a far greater determinant of fetal birth weight than treatment of the mother and insulin treatment appears to have little effect on fetal growth [19]. This probably reflects the difficulty in lowering the blood glucose in glucokinase patients because of increased counter-regulation [20]. Patients stop producing their own insulin and produce counter-regulatory hormones if blood glucose is reduced to normal levels, making successful control of blood glucose with insulin difficult. This results in frequent hypoglycemic symptoms at non-hypoglycemic blood sugar levels and means that large doses of insulin may be required to reduce fasting hyperglycemia to normal levels [20,21]. In some cases where the fetus has inherited the mutation, intensive insulin treatment has resulted in a low birth weight child [21]. This is to be expected as a small baby is seen when the fetus inherits a mutation from the father and is born to a normoglyc-emic mother [18,22]. Testing fetal genotype in utero is not without risk. Treatment decisions in glucokinase gestational diabetes should therefore be related to fetal growth as shown by scans rather than being made solely on maternal glycemia [21]. If the abdominal circumference is greater than the 75th centile insulin may be used but early delivery is the most successful strategy.
HNF1A and HNF4A (transcription factor MODY)
Transcription factors are proteins that bind to DNA and form part of a complex regulatory network controlling gene expression. The majority of patients with MODY have a heterozygous mutation in a transcription factor gene, by far the most common being mutations in the hepatic nuclear factors 1A and 4A (HNF1A and HNF4A). Diabetes resulting from mutations in other transcription factor encoding genes including HNF1B. insulin promoter factor 1 (IPF-1) and NEUROD1 are discussed elsewhere in this chapter.
Transcription factor mutations alter insulin secretion in the mature β-cell as well as altering β-cell development, proliferation and cell death. Mutations in the hepatic nuclear factors appear to alter levels of proteins critical in metabolism including the GLUT-2 glucose transporter and key enzymes in the mitochondrial metabolism of glucose [23–25]. Reduced β-cell proliferation and preserved or increased apoptosis could explain the progressive deterioration in β-cell function seen in these patients [25–28].
Mutations in HNF1A account for up to 70% of cases of MODY with nearly 200 different mutations reported. HNF4A. the next most common, accounts for approximately 3% of cases [29].
Clinical features
Heterozygous transcription factor mutations cause autosomal dominant diabetes presenting in adolescence or early adulthood resulting from progressive failure of insulin secretion. While diabetes is similar in HNF1A and HNF4A mutation carriers as a result of a common pattern of β-cell dysfunction, a number of differences in extrapancreatic features occur (Table 15.3).
Table 15.3 Extrapancreatic features assisting in the differential diagnosis of transcription factor maturity-onset diabetes of the young ( MODY ).
| Transcription factor | Extrapancreatic clinical features |
| HNF1A | Low renal glucose threshold (glycosuria) |
| Raised HDL | |
| Raised cardiovascular risk (in excess of type 2 diabetes) | |
| HNF4A | Increased birth weight/macrosomia |
| Low HDL, low lipoprotein A1 and A2, raised LDL | |
| HNF1B | Renal cysts and renal development disorders and multiple others. See Table 15.5 |
| NeuroD1 | None described |
HDL, high density lipoprotein cholesterol; LDL, low density lipoprotein cholesterol.
Diabetes
Patients are usually born with normal glucose tolerance and then show progressive β-cell dysfunction until they develop diabetes, usually between 10 and 30 years of age (Figure 15.4). Sixty-three percent of HNF1A carriers are diagnosed with diabetes by the age of 25 years and 79% by the age of 35 years; the age of diagnosis is partly related to the location of the underlying mutation within the gene [30–32]. Patients show deteriorating glycemia with age and require pharmacologic treatment. In the oral glucose tolerance test, in contrast to patients with glucokinase mutations, the fasting glucose is often normal initially but there is marked elevation of glycemia at 2 hours and consequently a large 2-hour increment (>5.0 mmol/L) [13]. This occurs because insulin secretion rates in early HNF1A MODY remain appropriate, with blood glucose values less than 8.0 mmol/L but are reduced significantly in comparison to non-diabetic non-mutation carriers above this level [33]. Microvascular complications are frequent particularly when hyperglycemia is inadequately treated [34]. Patients tend to be lean and insulin-sensitive. Obesity occurs at similar levels to the normal population.
Extrapancreatic clinical features
These are summarized in Table 15.3 and discussed in more detail below.
HNF1A
Patients with HNF1A mutations have elevated levels of high density lipoprotein cholesterol (HDL) which contrasts with the reduced HDL levels seen in T2DM [35]. Despite this they appear to have a greater risk of coronary heart disease than patients with T1DM [34]. Frequency of microvascular complications is similar to that seen in T1DM and T2DM and relates to degree of glycemic control [34]. Patients have a reduced renal threshold for glucose. Mutation carriers without diabetes may develop glycosuria after a glucose challenge even if glycemia remains within normal limits [36].
HNF4A
HNF4A mutations are associated with an 800 g increase in birth weight compared with non-mutation carrying siblings [37]. This means the offspring of HNF4A mutation carrying fathers, as well as the offspring of HNF4A mothers, are at risk of marked mac-rosomia. There is also an increased risk of hypoglycemia in affected neonates. These features appear to relate to increased insulin secretion in utero and in early infancy which evolves into reduced insulin secretion and diabetes in later life [37]. HNF4A mutation carriers have reduced levels of HDL (and lipoprotein A1 and A2) and frequently have raised LDL, while triglyceride levels are similar to population norms [38].
Differentiating from type 1 diabetes
These patients are usually diagnosed as having T1DM as they have symptomatic diabetes occurring in adolescence or young adulthood. We recommend genetic testing for HNF1A mutations in any young adult with apparent T1DM, a parent with diabetes, and who is antibody-negative at diagnosis. Evidence of non-insulin dependence increases the likelihood of a positive result; this would include no ketosis in the absence of insulin treatment, good glycemic control on low doses of insulin, or detectable C peptide with plasma glucose >8 mmol/L 3–5 years after diagnosis (outside the honeymoon period) [39]. While glutamine acid decarboxylase (GAD) antibodies are usually negative, positive GAD antibodies may be expected in up to 1–2% of the non-diabetic normal population, and therefore positive antibodies (particularly at low titer) may not exclude monogenic diabetes; testing should be considered where clinical suspicion is high [40,41]. HNF4A testing should be performed in any patient with a high suspicion of having HNF1A who tests negative for HNF1A mutations, particularly where there is evidence of increased birth weight and/or neonatal hypoglycemia.
Differentiating from type 2 diabetes
HNF1A should be suspected and mutation screening performed in patients otherwise suspected to be have T2DM where the following features are present ([39] and www.diabetesgenes.org):
In addition, a large increment in the glucose tolerance test (>5 mmol/L), presence of glycosuria with blood glucose less than 10 mmol/L, marked sensitivity to sulfonylureas and a lipid profile showing normal or raised HDL and normal or low triglycerides (atypical for T2DM) would all be supportive of a diagnosis of HNF1A instead of T2DM [35]. HNF4A should be suspected and tested in those patients who are suspected to have HNF1A mutations but test negative on HNF1A screening. These patients have a normal renal glucose threshold and frequently have a personal and/or family history of high birth weights and/or neonatal hypoglycemia.
Management
Patients with both HNF1A and HNF4A mutations are sensitive to sulfonylurea therapy which we recommend as first line treatment [38,42]. Glycemic control with sulfonylureas is often better than with insulin and the fasting glucose lowering effect is 4 times greater than that seen in T2DM [42,43]. Transfer to sulfonylurea treatment is successful in the majority of patients although insulin therapy may be required as diabetes progresses [44]. Even very low sulfonylurea doses may cause hypoglycemia. The starting dose should therefore be low-we use a starting dose of 40 mg/ day gliclazide or 2.5 mg/day glibenclamide in adults. If there is hypoglycemia with low doses of standard agents, a short-acting agent such as nateglinide may be appropriate [45]. Because of the apparent increased risk of cardiovascular disease in HNF1A, statin therapy should be considered and we suggest it for all patients aged over 40 years.
Management in pregnancy
Evidence to support management strategies for HNF1A and HNF4A in pregnancy is very limited. Our current practice is to continue sulfonylureas if glycemic control is good before preg-nancybut otherwise institute treatment with insulin.Consideration should be given to switching to glibenclamide in the prepreg-nancy period as this sulfonylurea has the most evidence for safety in pregnancy [46,47]. If a fetus carries HNF4A, the risk of mac-rosomia and neonatal hypoglycemia is high whether the mutation comes from the mother or father [37]. If either parent is known to carry a HNF4A mutation, we recommend:
Other transcription factor MODY
Other transcription factor mutations causing autosomal dominant β-cell diabetes have been identified in the genes IPF1, NEUROD1, KLF11 and PAX 4 but all are very rare [48–53].
Neonatal diabetes and diabetes diagnosed within 6 months of life
Children diagnosed with diabetes within the first 6 months of life (referred to as neonatal diabetes) are likely to have monogenic diabetes and not T1DM [54–57]. These patients commonly present with ketoacidosis and absent C-peptide. Neonatal diabetes is rare, affecting 1 in 100 000–200 000 live births [58]. Approximately half of cases remit spontaneously and are therefore termed transient neonatal diabetes mellitus (TNDM) as opposed to permanent neonatal diabetes mellitus (PNDM) where diabetes persists. TNDM often recurs in later life [59]. Neonatal diabetes results from mutations of key genes involved in β-cell development or function. Table 15.4 summarizes the known genetic causes of neonatal diabetes.
Table 15.4 Causes of neonatal diabetes.
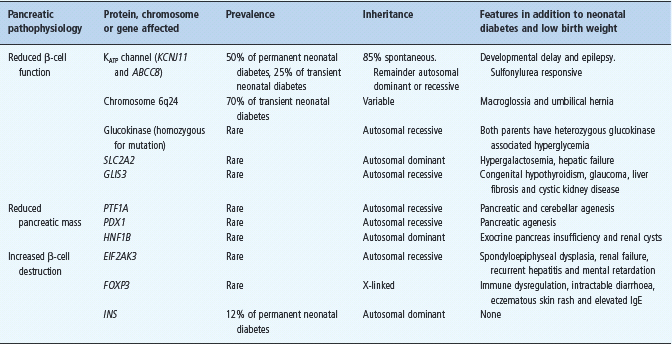
Permanent neonatal diabetes
Approximately half of PNDM is caused by mutations in the genes KCNJ11 and ABCC8 which encode the Kir6.2 and SUR1 subunits, respectively, of the β-cell ATP-sensitive potassium channel (KATP channel) [60–63]. This channel is constitutively open and regulates insulin secretion by closing in response to the raised intracel-lular ATP levels that occur as a consequence of hyperglycemia. Channel closure triggers depolarization of the β-cell membrane which leads to insulin secretion. Activating mutations in KCNJ11 and ABCC8 prevent closure of the potassium channel in response to increased ATP so the β-cell remains hyperpolarized and unable to secrete insulin [64]. Sulfonylureas close the β-cell KATP channel by an ATP independent route and they have been used successfully in the management of the majority of patients with neonatal diabetes resulting from KCNJ11 and ABCC8 mutations [65]. The KATP channel is also present in the brain, nerves and muscles. Reflecting this distribution of channels, 20% of patients with KCNJ11 mutations (and occasional patients with ABCC8 mutations) have associated neurologic features [57,60,61,64].
Heterozygous mutations in the insulin gene (INS) have been identified in 12% of cases of isolated PNDM and insulin treatment is required [66,67]. A number of other genetic causes have been found which all appear to be relatively rare [58] as outlined in Table 15.4.
The majority (85%) of PNDM resulting from KATP channel mutations arise spontaneously from de novo heterozygous mutations, with the remainder being familial and inherited mainly in an autosomal dominant pattern. About 40% of neonatal diabetes resulting from ABCC8 mutations, however, are inherited in an autosomal recessive fashion [63].
Clinical features
Diabetes caused by KCNJ11 mutations typically present in the first 26 weeks of life (median 4–6 weeks) with marked hyperglycemia often accompanied by ketosis. C-peptide is usually unde-tectable and islet cell antibodies negative [60]. As with all neonatal diabetes subtypes, infants are often small for gestational age as a result of reduced fetal insulin secretion with consequent decreased insulin mediated growth. About 20% of patients with PNDM and KCNJ11 mutations have neurologic features, the most common being developmental delay, sometimes with muscle weakness and/or epilepsy. The most severe form where neonatal diabetes is accompanied by developmental delay and epilepsy has been named developmental delay, epilepsy and neonatal diabetes (DEND). “Intermediate DEND” refers to neonatal diabetes with less severe developmental delay and no epilepsy. The severity of the clinical condition relates closely to the underlying mutation and its effect on KATP channel ATP sensitivity [64,68].
Neonatal diabetes caused by ABCC8 mutation has a similar phenotype but leads to transient neonatal diabetes more commonly than PNDM and has associated neurologic features only rarely [61–63]. Patients with neonatal diabetes and INS mutations present at a median age of 9 weeks and are also often small for gestational age but do not have extrapancreatic features [66].
Management
Although insulin therapy is commonly used in the initial period after diagnosis, the majority of patients with KCNJ11 and ABCC8 mutations can successfully transfer from insulin to sulfonylurea therapy, usually with significant improvements in glycemic control [62,65]. Ninety percent of those with KCNJ11 mutations are able to discontinue insulin, while HbA1c appears to improve in all patients with a mean drop from 65 to 46 mmol/mol (8.1 to 6.4%) after 12 weeks [65]. Glibenclamide was initially selected as it is non-selective and widely available; it has been used in the majority of cases and may be more effective than other sulfony-lurea agents [69]. The doses needed are often higher than those needed for the treatment of T2DM: a median dose of 0.45 mg/kg/ day is required with doses up to 1.5 mg/kg/day needed in some cases [65,70]. Diarrhoea is a possible side effect but this usually only lasts 1–3 days [71]. Sulfonylurea therapy may result in some improvement in neurologic features even where they are commenced in adulthood [69,72,73]. Further information on transferring patients from insulin to sulfonylureas can be found at www.diabetesgenes.org.
Neonatal diabetes resulting from INS mutations requires insulin treatment [66]. Affected individuals with a heterozygous KCNJ11 mutation contemplating parenthood should be counseled that they have a 50% chance of passing on the mutation to their offspring. Where unaffected parents whose child is affected by a heterozygous mutation are planning further pregnancies, the risk of further affected children is low because of the possibility of a germline mutation is approximately 5–10% [74]. Where parents have a child with neonatal diabetes caused by a recessive ABCC8 mutation, there is a 25% chance of each further offspring being affected but the risk is low for subsequent generations.
Transient neonatal diabetes
The genetic etiology of more than 90% of transient neonatal diabetes has been established. The majority (70%) of cases result from abnormalities the q24 region of chromosome 6 (6q24) affecting imprinted genes [58,75]. Genetic imprinting occurs when only the maternal or paternally inherited allele of a gene is expressed. In TNDM paternal uniparental disomy, paternal duplication of 6q24 or abnormal methylation of the maternal copy of the chromosome causes overexpression of the paternal copies of the genes PLAGL1 (also known as ZAC) and HYMAI [75,76]. Paternal duplication of 6q24 can be inherited, therefore this abnormality causes the majority of inherited TNDM cases. Uniparental disomy causes sporadic TNDM; cases resulting from abnormal methylation of the maternal copy of chromosome 6 may be sporadic or inherited [75,76]. The majority (90%) of TNDM not associated with 6q24 abnormalities are caused by mutations in KCNJ11 and ABCC8 [57,59,62,77–81].
Clinical features
6q24 diabetes usually presents in the first week of life often with severe hyperglycemia and dehydration but usually without ketosis [75]. Islet cell antibodies are usually negative and C-peptide is low or negligible [75]. Low birth weight is common (mean birth weight 2.1 kg), and there may be associated macroglossia and/or umbilical hernia. Insulin treatment is required for a median of 12 weeks before the patient goes into remission. Diabetes recurs later in life in 50–60% of patients as a result of β-cell dysfunction. The average age of recurrence is 14 years. In some cases hyperg-lycemia may be intermittent and seen only at times of stress [75,82]. Where TNDM is caused by KCNJ11 and ABCC8 mutations, diabetes tends to present later (median 4 weeks), takes longer to remit and is associated with less intrauterine growth restriction (median birth weight 2.6 kg) [59].
Management
Insulin is required in the neonatal period whereas treatment requirements following relapse vary from diet to oral hypoglyc-emics or insulin [82]. In TNDM cases resulting from KCNJ11 and ABCC8 mutations, diabetes may be successfully managed with sulfonylureas [59,62].
Genetic counseling depends on the underlying genetic etiology. Cases caused by uniparental disomy are sporadic and therefore have low risk of occurrence in either siblings or offspring of the affected child. Methylation defects often result from homozygous mutations in the transcription factor gene ZFP57 and therefore may be inherited in an autosomal recessive manner [76]. Offspring of males with 6q24 duplication have a 50% chance of developing TNDM whereas if the abnormality is inherited from the mother they will not be affected but the TNDM may occur in the following generation [82].
Genetic testing in n eonatal diabetes
At the time of diagnosis of neonatal diabetes it is not known whether the diabetes will be transient or permanent. We recommend testing for 6q24 abnormalities, KCNJ11, ABCC8 and INS mutations at diagnosis in all diabetes diagnosed before 6 months. Identifying mutations in these genes is important as it will influence treatment. An early diagnosis and very low birth weight make 6q24 most likely. A genetic cause (KCNJ11 or INS) can be established in approximately 7% of diabetes diagnosed between 6 months and 1 year of age so consideration should be given to testing this age group, especially where autoantibody tests are negative [56].
Diabetes with extrapancreatic features
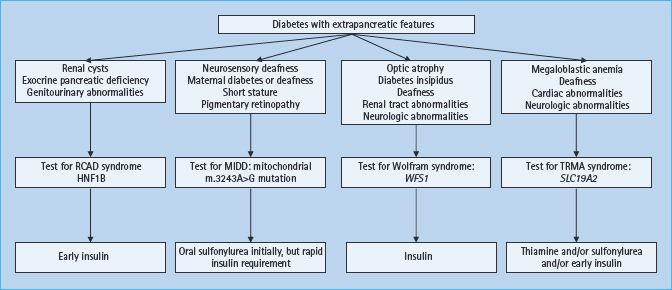
A number of monogenic causes of diabetes are associated with distinct features occurring outside the pancreas. In many cases extrapancreatic disease may be the presenting feature, for example in cystic fibrosis and hemochromatosis (see Chapter 18). Clinical subtypes and management of monogenic β-cell diabetes that have extrapancreatic features are summarized in Figure 15.6.
Figure 15.6 Clinical subtypes and management of monogenic β-cell diabetes that has extrapancreatic features. HNF1B, hepatocyte nuclear factor 1B; MIDD, maternally inherited diabetes and deafness; RCAD, renal cysts and diabetes; SLC19A2, solute carrier family 19, member 2 gene; TRMA, thiamine responsive megaloblastic anemia; WFS1, Wolfram syndrome 1 gene.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


