Abstract
Breast cancer, like other cancers, is a genetic disease. The disease is caused by acquired molecular alterations during the lifetime of an individual and by inherited mutant genes. Risk for breast cancer development is also impacted by hormonal factors through control of breast proliferative activity. Although most DNA sequence alterations have no carcinogenic potential, functional changes in critical regulatory genes have the potential to affect important pathways that provide growth advantages for the cells having these alterations. While a wide range of alterations are known, expression array analyses have permitted identification of intrinsic breast cancer subtypes (luminal A, luminal B, HER2-enriched, basal-like), which are associated with differences in patient outcomes as well as responsiveness to therapies. Currently, only a limited number of molecular alterations (expression levels of estrogen receptor-alpha, progesterone receptor, and HER2) are established as critically important for breast cancer patient treatment decisions. These predictive markers permit selection of patients for antihormone, anti-HER2, and CDK4/6 inhibitor therapies. Nevertheless, it is anticipated that better understanding of genome-wide alterations provided by recent genomic, transcriptomic, and epigenomic studies will not only lead to a better understanding of the biological pathways that transform normal cells to malignant growth but will also identify critical alterations that may be susceptible to development of new targeted therapies.
Keywords
acquired alterations, familial breast cancer, genomics, intrinsic subtypes, cancer stem cells, predictive markers, mutations, gene amplification
Cancer is a complex disease that involves progressive accumulation of diverse alterations in genetic and epigenetic regulatory programs of cells in interaction with the host stromal environment. Characterization of cancer-driving alterations that confer specific hallmark traits and thereby transform the normal cell into a malignant cell is essential for linking the molecular pathology of cancers to the molecular fingerprint of cancers. This process is not limited to understanding cancer biology but also involves distinctions between driver and passenger alterations, causal and secondary alterations of clinical significance, and actionable and nonactionable alterations in genes and pathways. Molecular heterogeneity has been an element underlying the complexity of cancer biology and thereby also a challenge for effective clinical management. Molecular heterogeneity is composed of intratumoral (presence of multiple clones representing different molecular profiles within the tumor) and intertumoral (presence of different molecular characteristics between cancers of the same tissue) heterogeneity. Intratumoral heterogeneity can vary across the spatial and temporal dimensions as the clones compete for host resources, and under selective pressure clones with mutations that confer increased advantage for growth and proliferation survive, driving cancer progression. Availability of high throughput technology platforms including next-generation sequencing (NGS) has provided an opportunity to study the cancer genome in high resolution to discover the molecular alterations that are key to disease progression and clinical management.
While focused on clinical utility, understanding the genetic and epigenetic alterations at multiple “omic” levels (genome, transcriptome, and epigenome) provides an understanding of major biological pathways that transform normal cells to acquire malignant characteristics and forms the core of molecular discovery. In this chapter, we summarize the biology of breast cancer development, molecular classification of the disease, and subsequently the molecular oncology of clinical significance.
Hallmarks of Cancer
The processes underlying tumor initiation, progression, and metastasis involves altered activity of multiple signaling pathways, including the pathways that regulate key physiologic functions such as growth and proliferation, differentiation, stress response, DNA repair, metabolism, and cell survival. The recruitment of these pathways in neoplastic transformation and cancer progression is a multistaged process. Precise characterization of these processes is, however, a complex task, especially given the multiple levels of complexity. Intratumoral, intertumoral, and temporal heterogeneity defines cancer as a highly dynamic process. Changes in biological interactions and rewiring of the signaling pathways in response to the environment is another aspect that potentially confounds the understanding of carcinogenesis at the molecular level. The pattern and extent of alterations in genes and pathways are also in part contributed by the interactions of cancerous cells with the stromal microenvironment.
Therefore it is important to describe carcinogenesis as a set of signature traits of cancer cells or hallmarks of cancer ( Fig. 22.1 ). These characteristic traits, described as distinctive and complementary, form a conceptual basis to understand functional organization of cancer cells. Different hallmark traits and known molecular markers having their potential role in acquisition of the cancer hallmark characteristics are illustrated in Fig. 22.1 .
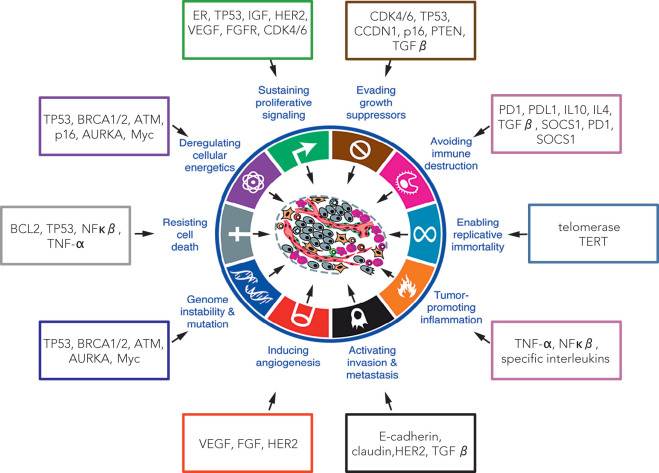
Sustaining Proliferative Signaling
A core cancer-driving trait is acquisition of sustainable growth signaling. Under normal conditions, growth factors bind to specific receptors localized on the cell membrane, leading to release of signal transducers and second messengers in the cell that transmit the signal to the nucleus. As a consequence, nuclear transcription factors are activated and initiate transcription of specific proteins that induce cell division and growth. Regulation of normal cells is related either to their dependency on other cells for growth factors or by an overproduction of a specific growth receptor required to respond to the corresponding growth factor.
Through bypassing the normal regulatory mechanisms, cancer cells acquire self-sufficiency of growth by one of the following common mechanisms:
- 1.
Secreting growth factor ligands themselves (autocrine)
- 2.
Inducing the normal stromal cells to secrete the growth factors (paracrine)
- 3.
Overexpression of growth inducing receptors
- 4.
Altering the structure or type of the receptor, causing ligand-independent activation of proliferative signaling
Abnormal activation of multiple alternative pathways may confer self-sufficiency of growth signaling in breast cancer. Estrogen receptor (ER) signaling, human epidermal growth factor receptor-2 (HER2) membrane receptor signaling, and vascular endothelial growth factor (EGF) receptor family signaling are the most common known proliferation-inducing pathways in breast cancer.
Approximately 70% of breast cancers express ERα, and therefore ER signaling is a key signaling pathway of diagnostic and therapeutic interest. Estrogen could induce proliferation in ERα-mediated and ERα-independent mechanisms. The classical ERα-mediated proliferative response involves estrogen binding to ERα, followed by modulation of target gene regulation through binding to estrogen response elements (ERE) in target gene promoters. In nonclassical activity, ERα can interact with transcription factors, such as SP1 or Fos/Jun, to modulate transcription of genes that do not possess the ERE in their promoter. In addition, ER activates mitogen-activated protein kinase (MAPK) by forming a complex with it in the nucleus to mediate transcription of specific target genes. An increased rate of cell proliferation results in increased mutagenesis. ER-independent proliferative mechanisms, not widely accepted yet, propose genotoxic damage by estradiol metabolites generated by cytochrome P450.
EGF receptor family member HER2, also known as ERBB2 (avian erythroblastosis oncogene B2), is another common proliferative signaling pathway in breast cancer. HER2 gene amplification results in overexpression of the HER2 transmembrane receptor, which is involved in growth signaling. Approximately 15% to 25% of invasive breast cancers have amplification of HER2, and these cancers are associated with aggressive disease behavior and poor prognosis. The overexpressed HER2 protein forms both homodimers and heterodimers with other epidermal growth factor receptor family receptor tyrosine kinases, and these activate downstream signaling. In addition to homodimerization, HER2 can form heterodimers with other members of the EGF receptor family and activate downstream growth signaling pathways, including but not limited to mitogen-activated protein kinase and phosphatidylinositol 3-kinase (PI3K) signaling.
Other upstream growth pathways involved in proliferative signaling in breast cancer are vascular endothelial growth factor (VEGF) signaling, fibroblast growth factor receptor (FGFR) signaling, and autocrine activation of wingless and integration site growth factor (Wnt) signaling pathway signaling. Although somatic mutations are known to activate downstream signaling pathways, a recent study in breast cancer showed amplifications of PIK3CA, KRAS, BRAF, and EGFR, thereby contributing to the abnormal activation of Ras-Raf-MAPK signaling.
Inactivation of negative-feedback mechanisms known to attenuate self-sufficient proliferative signaling is an alternative way by which cancer cells enhance proliferative signaling. PTEN is an example of negative feedback, known to antagonize PI3K signaling. In addition, nuclear PTEN downregulates cyclin D1 and prevents the phosphorylation of MAPK, thus loss of function alterations in PTEN promotes proliferation because of loss of its negative feedback.
Evading Growth Suppressors
Cancer cells are capable of evading a variety of growth suppression mechanisms located in the extracellular matrix or on the surface of nearby cells.
Retinoblastoma gene (RB) is considered a gatekeeper and prototype tumor suppressor protein that has a pivotal role in cell cycle progression because of its role as transducer of extracellular growth inhibitory signals. Alterations in RB signaling can lead to persistent proliferation. Cell proliferation in breast cancer cell lines may be reversed through inhibition of G1 cyclin-dependent kinases (CDKs) with CDK 4/6 inhibitors leading to hypophosphorylation of pRb and a G1/S-phase cell cycle arrest. Studies of breast cancer cell lines show the most sensitive cell lines have elevated expression of Rb and cyclin D1 but decreased expression of CDKN2A (p16 INK4a ). These observations have been confirmed in clinical trials of CDK 4/6 inhibitors (palbociclib, ribociclib, and abemaciclib) in patients with metastatic ER-positive, HER2-negative breast cancer.
Transforming growth factor (TGF)β signaling, another growth suppressor pathway, has been shown to have antiproliferative activity in early breast cancers by downregulation of c-myc oncogene. Altered TGFβ receptors or mutations of components of the signaling pathway might lead to evasion of growth suppression by TGFβ. Mutation or altered expression of SMAD4 protein, which is involved in transducing the TGFβ signals could alter the growth suppression by TGFβ.
Resisting Cell Death
Programmed cell death or apoptosis is an essential pathway for maintaining tissue homeostasis and elimination of cells with irreversible damage. Apoptosis mechanisms are divided into two components: First, an extrinsic pathway, which receives and processes extracellular death signals involving Fas ligand and tumor necrosis factor-α (TNFα); second, an intrinsic pathway that integrates intracellular signals, such as release of mitochondrial cytochrome C. Both pathways result in activation of downstream effector caspases. The intrinsic pathway is regulated by the B-cell lymphoma 2 (BCL2) family proteins. Sensing DNA damage, TP53, a checkpoint regulator, can induce apoptosis by activating BH3-only proteins, activating survival pathways such as PI3K-AKT/PKB pathway, increasing insulin-like growth factor (IGF)1/2 and interleukin 3. Cancer cells evade apoptosis by acquiring loss of function mutations in TP53, observed in approximately 37% of breast cancer samples, downregulating proapoptotic factors (BH3 only proteins) and inducing antiapoptotic proteins, for instance, BCL2.
Enabling Replicative Immortality
In contrast to normal cells, which are capable of passing through a finite number of growth and division cycles before becoming senescent, cancer cells must acquire replicative immortality. It has been proposed that for cancer cells to acquire this hallmark trait, they upregulate telomerase, a specialized DNA polymerase enzyme that is involved in maintaining telomere length or can add nucleotide repeats—TTAGGG oriented 5′-to-3′ toward and at the end of the chromosomes. More recently a protein subunit of telomerase, known as telomerase reverse transcriptase or TERT, has been shown to possess other functions, even after elimination of the canonical telomerase activity.
Telomerase expression observed in approximately 53% of breast cancers was associated with poor outcomes in patients treated with chemotherapy, while the opposite effect was observed with endocrine therapy. ERα can increase the telomerase activity, whereas the anti-ER tamoxifen is known to suppress telomerase activity.
Inducing Angiogenesis
In contrast to transient activity of angiogenesis in normal tissues, activation of an “angiogenic switch” in cancer cells underlies sustained angiogenesis, thereby stimulating abnormal and sustained angiogenesis in cancer that transports oxygen and nutrients to cancer cells and evacuates the waste and carbon dioxide. An imbalance between the factors that promote and oppose angiogenesis results in activation of persistent angiogenesis. In response to hypoxia or oncogenic stimuli, prototype angiogenesis initiator VEGF is expressed and binds to VEGF receptors (VEGFR1, VEGFR2, and VEGFR3) on cell membrane of endothelial cells. In addition, fibroblast growth factor 1/2 are potent proangiogenic factors, whereas TSP-1 (thrombospondin-1) counteracts the proangiogenic factors by binding the transmembrane receptors on endothelial cells. Although initial clinical trial results using humanized anti-VEGF-A monoclonal antibodies (bevacizumab) in breast cancer patients appeared promising, subsequent clinical trials failed to demonstrate a significant improvement in either progression-free (PFS) or overall (OS) survival.
Additionally, hematopoietic progenitors such as bone marrow–derived progenitor cells may potentially play a role in cancer angiogenesis. However, more comprehensive evidence is necessary to understand migration and invasion of these cells into developing breast tumors in vivo.
Activation of Invasion and Metastasis
Transformation of well-differentiated normal epithelial cells into often poorly differentiated cancer cells with migratory and invasive properties is a consequence of specific molecular alterations. The multistep process of invasion and metastasis is described as the invasion-metastasis cascade, which results in local invasion, intravasation of cancer cells, transit, extravasation at distant tissue sites, followed by colonization with a small fraction of the cancer cells, resulting in formation of disseminated cancer nodules (micrometastasis) and, lastly, some proportion of micrometastatic nodules adapt to the local microenvironment and proliferate to become mature metastases. The invasion-metastasis cascade is regulated by a multifaceted program, referred as epithelial-mesenchymal transition (EMT), which permits transformed epithelial cells to acquire mesenchymal features including motility, invasiveness, and resistance to apoptosis. The entry of cancer cells into EMT may be transient, stable, or partial.
E-cadherin is a key protein involved in formation of cell-to-cell adherens junctions that assembles adjacent epithelial cells and maintains quiescence of the cells. Loss of functional E-cadherin by transcriptional repression, mutation, chromosomal deletion, or protein degradation may confer metastatic potential and ultimately provoke EMT, in addition to loss of cell-to-cell adherens junctions.
Among other factors, specific members of the integrin family, matrix metalloproteinases, and crosstalk between tumor and stromal cells contribute to invasion and metastasis.
Genome Instability and Mutation
Unlike normal cells, which only infrequently contain mutations, cancer cells have significantly high rates of acquired mutations. Initial occurrence of mutations in gatekeeper genes result in genomic instability, and multiple genomic alterations are a key feature of cancer cells because of their role in developing other cancer hallmark capabilities. Whereas most cancers originate from a single ancestor cell due to a specific mutation(s), their progression is attributed to clonal evolution of the cells with acquisition of additional genomic alterations that confer selective growth advantages and lead to more aggressive behavior.
Mutations in DNA repair genes can lead to a mutator phenotype that subsequently gives rise to more mutations and eventually genomic instability. Genomic instability is a primary event in hereditary cancers with germline mutations in genes, such as BRCA1 and BRCA2, that are linked to DNA repair. In sporadic cancers, oncogene-induced collapse of DNA replication forks may lead to DNA double strand breaks and genomic instability.
Occurrence of mutations may have diverse, often overlapping or linked functional contributions in conferring hallmark traits including genomic instability, resulting in cancer progression ( Fig. 22.2 ). Tumor suppressor genes are of prominent interest in development of cancer hallmarks. The gatekeeper mutations occur in early cancers and contribute to loss of growth regulation and poor differentiation of cancer cells. TP53 is a well-studied gene involved in direct regulation of cell cycle, apoptosis, and DNA repair, thereby maintaining genomic integrity. Germline or early occurring somatic mutations of TP53 drive cancer by loss of their gatekeeper function. Mutation in E-cadherin is another such example. Caretaker genes are involved in DNA repair and checkpoint control; however, they cannot initiate carcinogenesis as a direct consequence of mutations. Mutations in BRCA1 and BRCA2 in breast cancer represent good examples of caretaker mutations. The landscaper mutations modulate the microenvironment and can promote cell proliferation. No comprehensive evidence is available that identifies any mutations in the role of a landscape mutation. However, among hereditary BRCA1/2 mutant cases, alterations in stromal cells have been proposed to contribute as landscape mutations. In addition, mutations may be broadly categorized as driver and passenger mutations. The driver mutations are capable of conferring selective advantage to cancer cells promoting carcinogenesis and survival of the cancerous cells. Mutations in TP53, PIK3CA, MAP3K1, GATA3, CDH1, BRCA1, PTEN, PIK3R1, AKT1, and RB1 are among the prominent driver mutations in breast cancer. Passenger mutations are other nonsynonymous mutations that do not play a direct role in carcinogenesis or progression. Mutations could also cause partial or complete loss of function that applies to tumor suppressor functions. Specific mutations of TP53 cause gain of novel functions. For instance, interference with the p53 function preventing DNA damage and promoting genetic instability, is the opposite of its wild-type role.
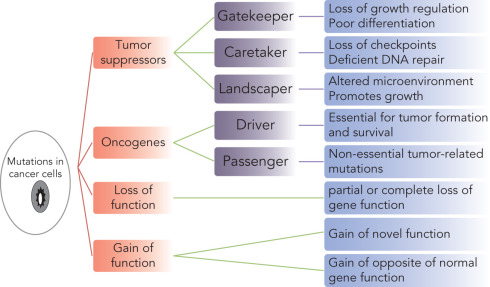
In addition to mutations, loss of telomeric DNA and specific copy number alterations are other factors that may contribute to genomic instability.
Tumor Promoting Inflammation
Cancers induce immune response, resulting in immune infiltration and inflammation. Inflammation could contribute to cancer progression by expression of growth factors, survival factors, and proangiogenic factors to the local microenvironment to promote tumor formation.
Reprogramming Energetics
Normal cells favor glycolysis under anaerobic stress. In contrast, cancer cells, even under aerobic conditions, favor metabolism of glucose to lactate, a state referred as anaerobic glycolysis. This means that the usual preference for oxidative phosphorylation (OXPHOS) in normal cells is shifted to metabolically inefficient glycolysis despite the presence of oxygen, mainly attributed to mitochondrial malfunction and a hypoxic microenvironment. Cancer cells face two challenges: first, the need to continue support for proliferation and growth by reprogramming metabolism and, second, achieving metabolic adaptation to survive metabolic stress. Regulation of adenosine triphosphate (ATP) synthesis by regulating substrate uptake and glycolytic enzymes is a possible mechanism that enables cells to adapt to the microenvironment.
Mutations in cancer cells also play a role in reprogramming of cancer cell energetics. Mutations in PIK3CA are common in breast cancer. Mutant PIK3CA or loss of its regulator PTEN can promote PI3K/Akt signaling and ultimately changes in metabolism, by increased expression of nutrient transporters, stimulation of hexo-/phosphor-fructokinase to drive glycolysis, enhanced transcription of genes involved in glycolysis, and/or by an enhanced rate of protein synthesis through Akt-dependent mTOR activation.
Considering reprogramming of metabolism as a consequence of oncogenic activity brings up the question of whether it is a core hallmark of cancer. Regardless, it remains a potential target for novel therapeutic strategies.
Evading Immune Destruction
Cancer cells evade the immunosurveillance and thereby continue to grow locally, metastasize to distant tissues and ultimately result in a macroscopic tumor. There are two potential mechanisms underlying dissemination of cancers: First, mutations accumulated either in a subpopulation of primary tumor or in disseminated cells to confer metastatic potential to a fraction of cells and second, expression of immune mediators (i.e., TGFβ, TP53, HIF, VEGF) by the tumor cells could modulate immune response. T regulatory cells (Tregs) that normally suppress T-cell response and natural killer (NK)-cell activity are observed to be significantly associated with larger tumor size and trend toward an association with estrogen receptor negativity. In addition, the same study observed an association with ER/progesterone receptors (PR)/HER2-negative or triple-negative breast cancer (TNBC) and Nottingham grade III cases that were significantly associated with high number of FOXP3 (fork-head/winged-helix transcription factor, which is involved in the development in Treg cells).
The presence of immune cells in breast cancers may also reflect the potential for harnessing the patients’ own immune system to eliminate tumor cells. The presence of tumor-infiltrating lymphocytes (TILs), especially in lymphocyte-predominant breast cancers, has been associated with significant improvements in pathologic complete response to chemotherapy. The potential protective role of lymphocytes has been appreciated in cancers with microsatellite instability. The inability of these cancers to efficiently repair errors in exons leads to expression of proteins containing neo-antigens that may be identified by a patient’s own immune system ostensibly leading to a high density of TILs. These neo-antigens expressed in proteins on the cell surface are speculated to be most likely to have this effect. This concept has led to the development of novel immunomodulatory therapies, such as programmed cell death 1 inhibitors for the treatment of metastatic cancers, including breast cancers.
Normal Mammary Development and Carcinogenesis
Endogenous hormones have been shown to be key nongenetic factors underlying breast carcinogenesis. Endogenous hormones, on one hand, have essential roles in normal development of the breast, whereas on the other hand, they also increase the risk of breast cancer. Epidemiology studies have shown that early menarche, late menopause, nulliparity, lack of lactation and breastfeeding, and delayed childbearing are associated with increased risk of developing breast cancer, underscoring the profound role endogenous hormones play in cancer development.
Endogenous Hormones and Growth Factors
Understanding breast carcinogenesis requires an understanding of the normal development of the breast. Breast tissue is composed of epithelial and mesenchymal components. Opening onto the skin surface and lined by squamous cells, breast ducts are lined by a double-layered epithelium composed of luminal and myoepithelial cells for the vast majority of the ductal system throughout the breast. The luminal epithelial cells are most prone to malignant transformation. During development steroid hormone receptors, especially ER and PR, facilitate cellular responses to hormones that, among other activities, lead to proliferation of the luminal cells.
Although all mammals are born with a rudimentary mammary gland, after birth, through a process of cyclical growth and involution coupled to body hormone status ( Fig. 22.3 ), the primitive breast undergoes extensive morphogenesis through invasion of the surrounding fat pad and progressive branching to the outer limits of the mesenchymal fat pad to create an extensive treelike structure, the terminal duct lobule unit (TDLU), specialized for milk production during pregnancy.
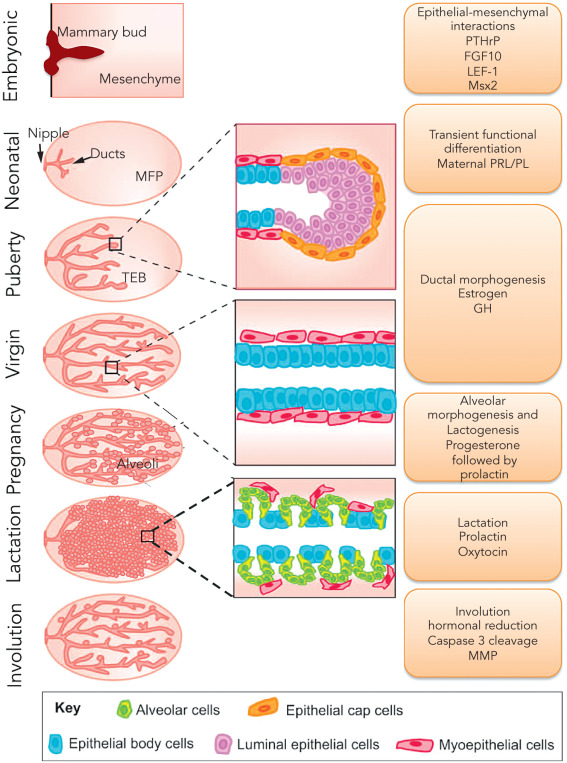
The branching of large ducts eventually leads to the TDLUs that are composed of terminal ducts and blind-ended ductules. TDLUs in adult women branch into small acini to form the lobules. During adulthood, TDLUs demonstrate proliferative activity, predominantly during the second half of each menstrual cycle, and subsequent epithelial apoptosis at the conclusion of each cycle. Presumably as a result of this repetitive proliferative activity with each menstrual cycle, it is the TDLUs that are responsible for the development of the vast majority of breast cancers.
Estradiol and other estrogens interact with two specific intracellular (nuclear) receptors, which are both inducible transcription factors, ERα and ERβ. Some normal luminal epithelial cells express the ERα and are considered relatively mature cells that infrequently cycle in the adult breast. ERα is expressed in infant, pubertal, and cycling adult breast epithelial cells. ERβ is expressed in most luminal, myoepithelial cells, fibroblasts, and lymphocytes. PR is expressed in the breast as two isoforms, PR-A and PR-B, based on alternative initial transcription start sites from a single PGR gene. PR is expressed in a minority of cells in the luminal epithelium concentrated in the terminal bud cells. Expression of PR is induced by estrogen action at the transcriptional level and is decreased by progestins at both transcriptional and translational levels. In addition to ER/PR-mediated proliferation, a paracrine or juxtacrine mode of regulation of proliferation has been proposed.
Multiple stages of mammary development are depicted in Fig. 22.3 . Here we briefly present an overview of the process, the factors involved in it, and the role of stem cells in breast morphogenesis, the details of which are described in other chapters.
Prenatal or Fetal
Mammary glands originate from mammary buds, which are ectodermal ingrowths along the mammary lines and are present in 4-week-old embryos. Between 14 to 18 weeks of gestation, the mammary gland appears as an epithelial anlage resulting from focal ingrowth of the epidermis into the underlying mesenchyme. It subsequently undergoes branching and ductal morphogenesis into the mesenchyme. BCL2 expression is observed at 18 weeks and is known to prevent apoptosis and permit expansion of the epithelial cell population. Differentiation of nipple and areola and development of a rudimentary ductal system occurs during the second half of fetal life. Prolactin is involved in ductal budding by sensitizing the cells to insulin, resulting in a mitogenic effect. By week 28, the luminal cells are separated from the basement membrane by an outer layer of myoepithelial cells. The basal cells express EGFR and TGFα. BCL2 is expressed in stromal cells at this stage and protects them from apoptosis, permitting their expansion as future fibroblasts. Parathyroid hormone-related protein (PTHrP), together with bone morphogenic proteins (BMP), have been shown to have a role in the development of mammary buds and induction of Msh Homeobox 2 (MSX2).
Postnatal
At birth, the ductal system opens onto the skin surface, through the breast pit. With birth, estrogen, progesterone, and prolactin levels decline, resulting in involution of the breast. From 2 years of age until puberty, the mammary gland is composed of small ductal structures in fibroblastic stroma in both sexes.
Puberty
As a response to a surge in hypothalamic-pituitary-ovarian activity, also referred to as gonadarche, increased secretion of gonadotrophins and estrogen initiate the development of the breast glandular architecture, referred as thelarche. Apart from estrogen, other factors, including insulin, cortisol, thyroxin, prolactin, and growth hormone, are involved in the gradual process of gland differentiation. An increase in growth hormone secretion occurring in adolescence sensitizes the mammary cells to the mitogenic effects of insulin. The mammary gland at puberty contains multiple types of stem and progenitor cells, including bipotent mammary associated stem cells (MaSCs), which then differentiate into luminal and myoepithelial cells of the duct. The terminal end buds undergo branching, forming blind-ended ductules, known as acini. Collection of acini arising from a terminal duct form a functional unit, terminal ductal-lobular unit (TDLU), a site where most cancers arise. After menarche, the mammary gland continues cyclic epithelial proliferation with an associated increase in the number of alveoli in the TDLU, especially during the luteal phase, when levels of estrogen and progesterone are high, followed by apoptosis with menses during each successive cycle. This association of hormonal levels and proliferation supports the role of a hormonal stimulation in mitotic cell division, thus increasing the chance of errors in DNA replication and repair, leading to carcinogenesis. Luminal cell type is a primary target for carcinogenesis because luminal cells account for more than 90% of epithelial proliferation, as well as the observations that more than 90% of breast carcinomas synthesize cytokeratins and 70% are positive for hormone receptors.
Pregnancy and Lactation
Under the influence of estrogens and progesterone, mammary stem cells induce controlled ductal and alveolar proliferation during pregnancy. The number of TDLUs and alveoli increase as well. Increased rate of proliferation and potential errors in DNA synthesis and accumulation of mutations contributes to transient elevation of breast cancer risk. Human placental lactogen results in increased secretion of fluid and protein causing alveolar dilation. During the late stages of pregnancy, increased DNA synthesis and mitosis occur, related to extensive lobuloalveolar growth. Estrogen and progesterone in the presence of prolactin induce differentiation of alveolar epithelial cells into milk-secreting lobular alveolar epithelial cells, after prior exposure to cortisol, growth hormone, thyroxine, and insulin. After delivery, estrogen, progesterone, and human placental lactogen decrease, resulting in elimination of their inhibitory effect on prolactin, initiation of lactation, and inhibition of proliferation. Weaning is a gradual process, involves apoptosis of alveolar cells, alveolar collapse, and ensuing involution of the breast tubule-alveolar glands. The ability of mammary epithelium to undergo proliferation and differentiation with each pregnancy provides an indication of the important role of stem cells in this process.
Postmenopausal Involution
During postmenopausal involution, both ducts and lobules are reduced in number, in contrast to the postlactational involution that is limited to lobules. The intralobular stroma is replaced by collagen. The glandular epithelium and intralobular and interlobular connective tissue is replaced by fat, leading to reduced mammographic density. Lobular involution and a decreased mitotic rate reduces the rate and number of mutations, thus a relative protective effect of menopause as expressed in the reduced slope of breast cancer induction with age, although the majority of breast cancer occurs in postmenopausal women.
Stem Cells
Normal mammary stem cells (MaSC), a type of somatic stem cell, are multipotent cells with a unique capacity for self-renewal as well as the potential to differentiate into all specialized mammary epithelial cell types. MaSCs are key to pubertal elongation of ducts and gestational lobuloalveolar differentiation. Just as normal stem cells are essential for maintaining tissue homeostasis and functional integrity given their properties of self-renewal and differentiation in the normal gland, cancer stem cells (CSCs) are, similarly, a subpopulation of cancer cells that are proposed to have a tumorigenic role in addition to characteristics of “stemness” displayed by normal stem cells. A small number of cancer stem cells may act as potential reserve cells for cancer recurrence, relapse, and metastasis. Cancer stem cells may (but do not necessarily) originate from normal stem cells and can be one of the origins of cancers. MaSCs with suitable genomic alterations may initiate cancer. The potential role of MaSCs is explained by a parity-associated protective effect against breast cancer, attributable to a decrease in MaSCs during breast epithelial involution after pregnancy and lactation. The MaSCs do not express steroid hormone receptors (ERα and PR) or HER2, the key clinical predictive molecular markers for contemporary breast cancer treatment, favoring their association with specific subsets of breast cancer. Mutations altering key regulatory pathways and epigenetic aberrations could potentially transform stem cells into CSC. Stem cell niches, or stroma associated with cancer stem cells, represent distinct regions of the tumor microenvironment. These niches may play a key role in maintaining stemlike characteristics, which could promote cancer progression and metastasis. Both MaSCs and CSCs express aldehyde dehydrogenase 1 (ALDH1), and ALDH1 expression correlates with proliferation index and HER2 / ERBB2 overexpression in breast carcinomas. In addition, CSCs are known to express CD44 with low/nonexpressed CD24 and Lin proteins. The potential tumorigenic and metastatic roles of CSCs have inspired substantial clinical interest and investigation.
Stemness and Clonal Evolution
Genomic instability and the occurrence of mutations as cancer hallmarks, described in the previous section of this chapter, comply with the classic clonal evolution model. The cancer stem cell model is different from the clonal evolution model, which proposes that any cell accumulating specific genomic alterations has the capability to initiate cancer development and stemness. In contrast, the CSC hypothesis proposes that only the CSCs that have the property of self-renewal can differentiate into cancer cells that are both tumorigenic and nontumorigenic when transplanted to animals. The tumorigenic CSCs are responsible for tumor relapse, recurrence, and metastasis. The two models are not considered mutually exclusive, given available evidence. CSCs have not been detected in all cancers. Cancers that do not show evidence of hierarchical organization (undifferentiated precursor—differentiated descendant relationship), or heterogeneity resulting from a therapeutic response may be explained by clonal evolution, rather than by epigenetic differences between CSCs and their progeny.
Clinical Perspective on Carcinogenesis and Progression
Initiated as premalignant or preinvasive lesions, such as atypical ductal or lobular hyperplasia, ductal carcinoma in situ (DCIS), and lobular carcinoma in situ (LCIS), breast carcinomas acquire multiple hallmarks of cancer in multiple stages and evolve into invasive carcinomas, as demonstrated by the modified Wellings-Jensen-Marcum model. This model provides a context for identification of the underlying molecular alterations in the cancer hallmarks theory and, thereby, the study of cancer progression is viewed as a consequence of acquisition of driver and passenger mutations, copy number aberrations, and other genomic and epigenomic changes in the terminal duct lobular unit (TDLU). In addition, the intertumor, intratumoral spatial and temporal heterogeneity, as explained by the clonal evolution model ( Fig. 22.4 ), provides a broad overview of clinical cancer progression as a phenotypic outcome of interactions within multiple competing clones within the stromal microenvironment under selective pressure, including the dominant and outcome-determinant subclones in combination with minor subclones, in the context of treatment. From a clinical perspective, this implies the need for multimodal diagnostic and therapeutic approaches to achieve effective disease management.
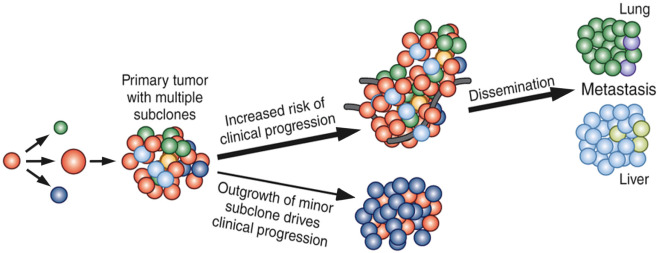
The following are considered priorities for multimodal clinical management:
- 1.
Large studies to identify specific genomic alterations that underlie altered activity of specific biological pathways that can drive tumor progression and clinical outcome.
- 2.
Development of high-resolution, robust, and reproducible tools to identify molecular drivers of breast cancers.
- 3.
Availability of decision-making tools to expand the use of effective targeted therapeutics for particular cancer-driver pathways.
Molecular Profiling
Technological advances in high-throughput methods, as well as parallel improvements in computation over the past 2 decades, have enabled the generation, analysis, and integration of data from multiple biological levels, including the genome, transcriptome, epigenome, and proteome. Studies based on NGS and microarrays have shown breast cancer to be a molecularly heterogeneous disease, with broad diversity in biological characteristics that correspond to differences in clinical outcome. The number of novel biomarker genes and pathways deciphered from NGS data focusing on cancer driver mechanisms, including clinically significant disease variants, copy number changes, pharmacogenomics, and corresponding clinical trials investigating their therapeutic utility, are anticipated to expand in the near future. However, the current 2016 American Society of Clinical Oncology (ASCO) guidelines for breast cancer management recommend ER, PR, and HER2 as markers for clinical decision-making. Besides their use as predictive markers for antiestrogen or anti-HER2 therapy, they are also used as predictive markers for novel mTOR inhibitors and CDK4/6 inhibitors. Despite the continuing progress toward gaining insight into potential driver mechanisms in the recent years, translation related to predictive markers for these targeted therapeutic alternatives have been a challenge, probably due to the requirement for large prospective confirmatory studies with a focus on clinical efficacy, safety, and reproducibility of the potential biomarkers.
Estrogen Receptor
ERα, a member of the steroid hormone receptor superfamily and a ligand-dependent transcription factor, is a regulator of various essential developmental and physiologic processes. Estradiol, the predominant form of biologically active estrogen, interacts with ER for physiologic development and regulation of the reproductive system, bone metabolism, and effects on the cardiovascular and central nervous systems. Given its core involvement in growth and proliferation of breast tissue, a dysregulation of the estrogen hormonal axis in developmental aberrations associated with breast carcinogenesis is not surprising. The link between estrogen and breast cancer was observed as early as 1896 by George Beatson, who reported regression of advanced breast cancers in patients after oophorectomy. Since then a number of clinical studies have shown that both endogenous estrogen levels and the duration of hormone replacement therapy, especially estrogen plus progestin, are correlated with an increased risk of breast cancer and disease-related mortality, which has evolved into the current view of ER and PR signaling as a core determinant of breast cancer biology and patient outcome.
The effects of estrogen are mediated through two receptor proteins, ERα and ERβ, encoded by corresponding genes on chromosome 6q24 and 14q22, respectively. Both receptors share about 97% homology in the DNA binding domain, with relatively low homology in other domains ( Fig. 22.5 ), indicating that they may share specificity for similar estrogen-responsive elements (EREs) on target gene promoters or enhancers. Although the role of ERβ in normal tissue is not known, loss of ERβ expression in breast cancer is associated with increased proliferation. Nevertheless, ERα, but not ERβ, is currently considered to be relevant for patient management and treatment decisions, and therefore only ERα is discussed further.
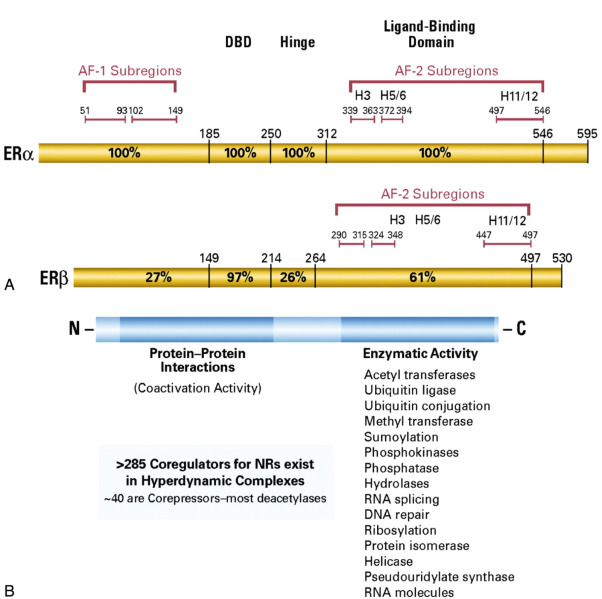
Upon binding of 17β estradiol, nuclear ER monomers dimerize, bind to EREs, and, in cooperation with coregulators (coactivators and corepressors), facilitate transcription of target genes, referred as classical genomic activity of ER (see Fig. 22.5 ). The nuclear coregulators modulate transcription through multiple mechanisms, particularly chromosome remodeling and posttranslational modification. The indirect genomic activity involves ER binding to other transcription factors, such as SP1, AP1, and NFκB, and thereby regulation of transcription through their binding to the target gene promoters. Cyclin D1 is a direct ER target that can activate CDK4 and CDK6, which phosphorylate RB with release of E2F, leading to induction of the cell cycle proliferative activity.
In recent years, cytoplasmic/membrane-bound ER cross-talk with HER2, EGFR, IGF1R, and FGFR has been proposed as a trigger contributing to the activation of AKT and downstream signaling components of the PI3K pathway. However, the mechanism for this cross-talk is not clear, and comprehensive evidence is needed to establish the presence of significant quantities of cytoplasmic ER, as well as its functional impact in patients.
Breast cancers that express ER are potentially sensitive to endocrine therapy in both primary and metastatic disease settings. ER-positive breast cancer patients have relatively better 5-year survival compared with ER-negative breast cancer patients, although substantially reduced difference in 10-year survival because of the delayed recurrence observed in ER-positive disease. ER expression status is a core predictor of disease aggressiveness, therapeutic response, and patient outcome. The objective of endocrine therapy is to target the key growth-driving pathway—ER signaling—by one of three alternative approaches:
- 1.
Reduction of circulating estrogen levels through surgical ovarian ablation or, more recently, through inhibition of peripheral conversion of steroids to estrogens using drug-mediated inhibition of aromatase with aromatase inhibitor (AI) therapies.
- 2.
Competitive inhibition of estrogen binding to ER using selective estrogen receptor modulators (SERM) such as tamoxifen to inhibit recruitment of activated coregulators through conformational displacement of ER helix 12.
- 3.
Downregulation of ER through irreversible binding of small molecules, such as fulvestrant, to ER with 100 times higher affinity to ER compared with tamoxifen, leading to selective degradation of the ER in breast cancer cells and thereby preventing estrogen-mediated transcriptional activation.
Although AIs are superior to tamoxifen for prevention of contralateral breast cancers as well as prevention of recurrence and metastases in postmenopausal women, tamoxifen is more suitable for adjuvant therapy in premenopausal women. Tamoxifen is also used for prolonged antihormonal therapy beyond the initial 5 years of treatment. A recent meta-analysis demonstrated a 30% reduction in recurrences associated with 5-year therapy with AIs compared with tamoxifen in postmenopausal women with early breast cancers as well as a 15% reduction in mortality.
Approximately 30% of women treated by antihormone therapy may have a recurrence within 15 years. Among the mechanisms of acquired resistance, deregulation of estrogen receptor signaling, either by loss of ERα expression or mutation in the ESR1 gene encoding ERα, are the most common associations. Loss of ER positivity is a common mode of resistance to endocrine therapy. Mutations clustering in the ligand-binding domain (LBD) are observed in advanced metastatic ER-positive samples ( Fig. 22.6A ) but are seldom observed in the primary carcinomas, suggesting a role for acquisition of ESR1 point mutations in advanced-stage disease, leading to resistance to endocrine therapy due to constitutive ligand-independent activation of ER. Recently a plasma circulating DNA-based prospective-retrospective study focused on patients with ER-positive metastatic cancers who developed disease progression after aromatase inhibitor therapy demonstrated that ESR1 mutations were predictive of resistance to Exemestane and sensitivity to fulvestrant. Notably, this study observed that a combination of palbociclib (CDK4/6 inhibitor) and fulvestrant was equally effective in patients with or without ESR1 mutations. Larger studies are required to further establish the predictive role of ER mutations and their relationship to response to various endocrine treatment options.
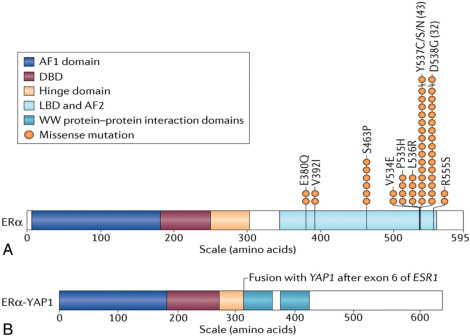
In addition, ESR1 chromosomal translocation, leading to fusion genes with ESR1, for instance, ESR1 (exons 1–6)- YAP1 (as illustrated in Fig. 22.6B ), has been proposed to be another potential mechanism underlying resistance to endocrine therapy.
Progesterone Receptor
The PR is a nuclear receptor closely associated to the ER, being a direct target of ERα as well as its involvement in ERα chromatin binding. Expressed as two highly studied isoforms, PRA (94 kDa) and PRB (116 kDa), and coded from a single gene, PR forms upon hormone binding to form homo/hetero-dimers, binding to the PR response elements (PREs) on target gene promoter regions via the DNA binding domain and thereby modulate transcriptional activation of target genes. PRB has been shown to be the predominant regulator for several of the target genes and uniquely possesses an activation function 3 (AF3) domain in addition to common AF1 and AF2 domains on both isoforms, suggesting both common as well as unique targets. PRA may suppress PRB activation, although genes regulated uniquely by PRA are known and PRA expression is higher. Both isoforms are equally expressed in normal breast tissues but altered in breast cancer, with higher PRA in many cases.
PR positivity is slightly less common than ER positivity in invasive cancers, although PR positivity indicates active ER signaling. The independent role of PR status in breast cancer phenotypic outcome has been a matter of debate. ER-positive tumors without PR expression are shown to have increased occurrence of copy number changes compared with ER-positive/PR-positive tumors, was well as enrichment of growth factor signaling via PI3K/Akt/mTOR. Loss of PR expression during tamoxifen therapy tends to be dramatic compared with loss of ER and marks potential resistance to therapy. Loss of PR expression is explained by transcriptional inhibition of PR by other growth pathways that can also modify ER functions, thus supporting a potentially beneficial role of SERDs that can effectively block ER signaling or of simultaneous inhibition of other growth pathways. However, a predictive role of PR has not been observed in early breast cancers.
HER2 Membrane Receptor
HER2 or epidermal growth factor receptor 2, a member of the human EGFR family of tyrosine kinases (RTKs), through homodimerization or heterodimerization with other family members, induces tyrosine kinase activity and consequential activation of pathways involved in cell growth, differentiation, and survival. The expression of HER2 is normally found at low levels in all epithelial cells of both fetal and adult tissues, but not in cells of hematopoietic origin. Amplification and/or overexpression of HER2 is found in a number of solid tumors including breast cancer. HER2 protein is a key driver of growth, an aggressive phenotype in cancers and, therefore, a therapeutic target in the subset of breast and gastric cancers that have alterations in the gene encoding this receptor. Amplification of the HER2 gene (also known as ERBB2 ) is observed in preinvasive breast carcinomas (DCIS) as well as 20% to 30% of invasive breast carcinomas. A role for HER2 gene amplification as a driver alteration that determines biological behavior including progression, resistance to apoptosis, invasion, and metastasis is well established for invasive carcinomas but not DCIS.
Amplification of HER2 is associated with high histologic grade, high proliferative rate, reduced expression of steroid hormone receptors, and lymph node metastases and is a key predictor of shorter patient survival and time to relapse. Besides being a prognostic marker, HER2 amplification, and the associated directly related protein overexpression, predicts responsiveness to trastuzumab, a humanized anti-HER2 monoclonal antibody, with either anthracycline-based chemotherapy or non–anthracycline-based chemotherapy. Another humanized anti-HER2 monoclonal antibody, pertuzumab, as well as an antibody-drug cognate, ado-trastuzumab emtansine (T-DM1), have also been approved for treatment of HER2-positive breast cancer patients. In addition, small molecule inhibitors of the HER2 tyrosine kinase have shown promise in the treatment of HER2-positive breast cancer patients with lapatinib, a dual targeting agent that inhibits both HER2 and HER1 receptors and is Food and Drugs Administration (FDA)-approved for treatment of women with metastatic disease. Other small molecule inhibitors of HER2 are currently in clinical trials.
The downstream consequences of HER2 amplification and its messenger RNA (mRNA) overexpression include formation of active hetero-/homo-dimers of HER2 protein with other RTKs, activation of intracellular tyrosine kinase domain and phosphorylation of the intracellular domain of HER2 and recruitment of adaptor proteins that forms a complex, leading to activation of intracellular signaling pathways, including PI3-kinase/AKT, and RAS/MAPK signaling. HER2 and other downstream signaling pathways increase cell proliferation, cell survival, and loss of apoptosis. Production of VEGF and induction of angiogenesis is one of the downstream consequences of HER2 activation ( Fig. 22.7 ).
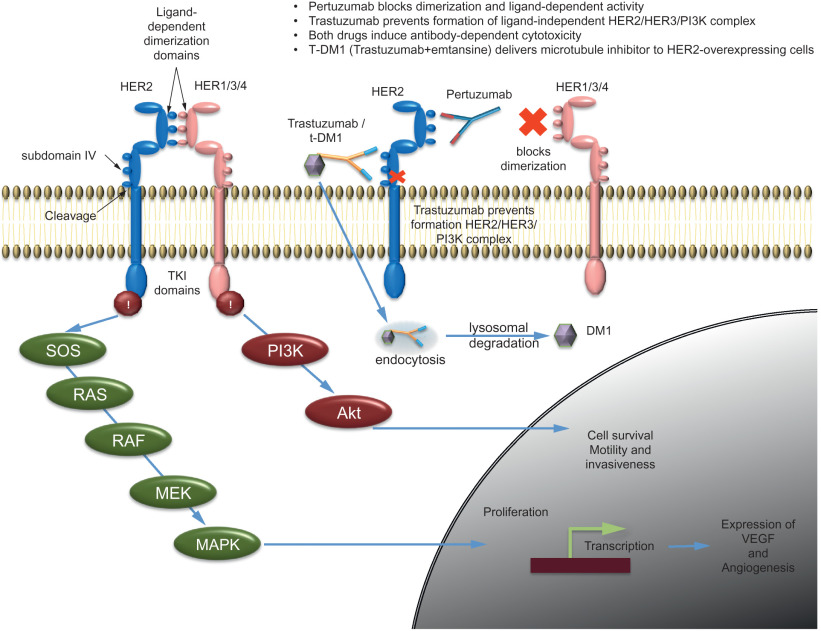
Additional therapeutic strategies target this signaling pathway by simultaneously targeting HER2 and HER3 with trastuzumab and pertuzumab, a second humanized anti-HER2 antibody that prevents HER2 dimerization, with concurrent cytotoxic chemotherapy. An antibody-drug conjugate, T-DM1, directed at HER2 has also been developed for treatment of HER2-positive breast cancer patients.
Trastuzumab, a humanized monoclonal antibody was FDA-approved for the treatment of HER2-amplified metastatic breast cancers in 1998 and subsequently for early-stage HER2-positive, node-positive breast cancer in 2006. The antigen-specific site of trastuzumab binds the juxtamembrane portion of the extracellular subdomain 4 of the HER2 receptor (see Fig. 22.7 ). The mechanism of action of trastuzumab is not completely understood but multiple possible mechanisms include inhibition of the intracellular activation of the HER2 tyrosine kinase domain, antibody-dependent cellular cytotoxicity (ADCC), through binding of trastuzumab with induction of natural killer cell responses against the HER2-overexpressing cells, inhibition of receptor dimerization, receptor degradation through endocytosis, inhibition of shedding of the extracellular domain, and inhibition of ligand-independent, HER2-mediated mitogenic signaling. Trastuzumab activity has been shown in early as well as metastatic breast cancers with HER2 amplification. HER2 activity is ligand independent. Among the possible heterodimers, the HER2-HER3 pair has been shown to be the most potent in activation of cell proliferation. HER3, upon binding to its ligand, dimerizes with HER2 and activates the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3-K) signaling pathway. The clinical benefit of trastuzumab therapy can be improved by simultaneously blocking both ligand-independent and ligand-dependent activity. Accordingly, therapy with trastuzumab and pertuzumab could achieve more comprehensive control of HER2 signaling. The CLEOPATRA trial showed significantly reduced risk of disease progression (improved PFS) or death with trastuzumab, pertuzumab, and a docetaxel regimen compared with placebo plus trastuzumab with the same docetaxel therapy in metastatic breast cancers. Resistance to trastuzumab remains an important issue. One or more mechanisms could activate alternate growth signaling pathways and, thereby, overcome the HER2 or HER2-HER3 blockade. Factors such as activation of other members of HER family, activation of PI3K/AKT/mTOR by alternative pathway, IGF1 activation, increased activity of VEGF or src, c-MET overexpression or loss of PTEN are some of the potential mechanisms considered possible underlying mechanisms of resistance. Conjugation of trastuzumab with maytansine (DM1), a tubulin inhibitor, also referred as T-DM1, has demonstrated activity against the HER2-overexpressing cells that were resistant to trastuzumab and lapatinib. The combination of T-DM1 with pertuzumab provided superior pathologic complete response rate compared with the paclitaxel with trastuzumab in the I-SPY 2 (Investigation of Serial Studies to Predict Your Therapeutic Response With Imaging And moLecular Analysis 2) trial.
Molecular Subtypes of Breast Cancer
In addition to ER, PR, and HER2, which are the key predictive biomarkers, advances in high-throughput technologies have shown breast cancer to be a molecularly, highly heterogeneous disease. The initial aim of classifying breast cancers had been for prognostication and prediction of response to chemotherapy, which with technological advances, has evolved into a broader aim of defining molecular classes by causal or driver alterations that determine patient outcome, disease aggressiveness, metastatic potential, or drug responsiveness.
Molecular expression subtypes were initially based on microarray-based differential mRNA expression patterns across the spectrum of breast cancer samples. A set of 496 genes, defining the subtypes, was composed of genes that showed significantly greater variation in expression between different cancers than between paired samples from the same tumor. These genes also represented the biological characteristics of the cancers and were referred as an intrinsic gene set. The expression clusters were separated into five subtypes: luminal A, luminal B, basal, ERBB2-enriched (HER2-positive) and normal-like. Subsequently, using a 50-gene predictor gene set (PAM50) in a study of 710 node-negative cancers, the molecular subtypes were shown to be associated with differential relapse-free survival and were associated with response to chemotherapy. A randomized trial (MA.12 study) of 672 premenopausal, stage I–III breast cancer patients receiving adjuvant tamoxifen showed the PAM50 subtype to be prognostic for both disease-free survival (DFS; p = .0003) and overall survival (OS; p = .0002). This study also showed the luminal subtype to be predictive of response to tamoxifen therapy compared with the nonluminal cases. Using a large sample set of 10,000 pooled samples from multiple studies, immunohistochemistry-based expression of ER, PR, HER2, and basal markers (cytokeratins CK5/6, and EGFR) was applied to identify six subtypes: luminal 1 (HER2-negative) with or without expression of basal markers, luminal 2 (HER2-positive) with or without expression of the basal markers, nonluminal HER2-positive, and TNBC. This classification distinguished breast cancers by their short and long-term survival. Overlapping largely with TNBC with high frequency of metaplastic and medullary differentiation, a novel class of tumors, the claudin-low subtype, was identified that are characterized by low or absent expression of luminal differentiation markers, high expression of EMT markers, immune response genes, and CSC-like features.
Although expression subtypes have improved our understanding of molecular heterogeneity, the known expression subtypes do not represent the complete spectrum of the biological variation. Subtype characterization has been based on mRNA expression using microarrays in small sample sets, but its clinical utility is limited, given the lack of reproducibility and robustness, technological difficulties, and insufficient consideration to tumor heterogeneity. These limitations in clinical utility are discussed in greater detail elsewhere.
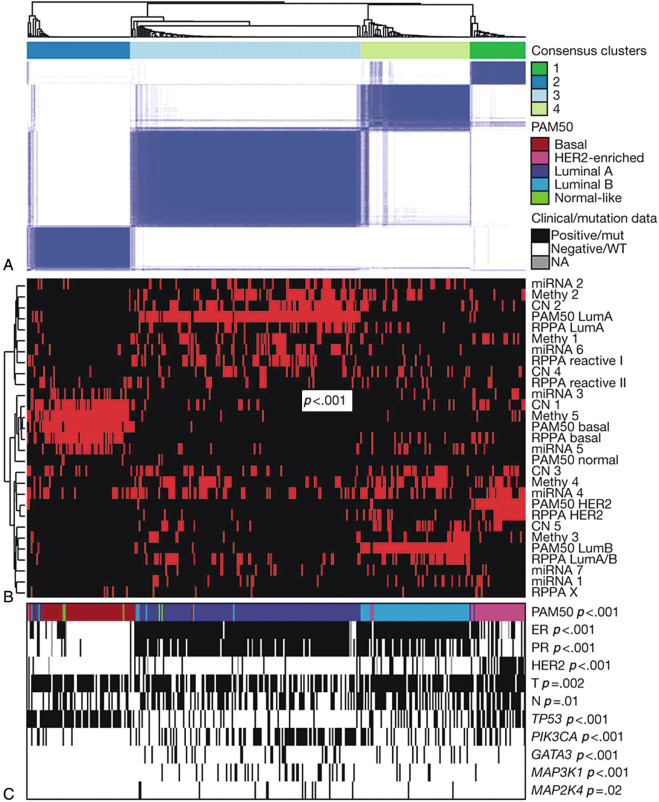
The concept of molecular subtyping has continued to evolve with advances in identifying the underlying somatic and germline mutations, copy number alterations and other alterations. Five types of data, including mRNA expression, DNA methylation, single nucleotide polymorphism (SNP) arrays, microRNA (miRNA) sequencing, whole-exome sequencing, and protein expression (reverse phase protein array or RPPA) from tumor and germline DNA showed consensus patterns between samples by unsupervised consensus clustering analysis. The four major clusters obtained from these analyses have been correlated with the mRNA expression-subtypes as well as clinical characteristics. Frequent mutation of PIK3CA (45%), MAP3K1, GATA3, TP53, CDH1, and MAP2K4 has been observed in luminal A subtype breast cancers, whereas the luminal B subtype most frequently showed mutations in TP53 and PIK3CA (29% each). The highest occurrence (80% samples) of TP53 mutations was observed in the basal-like cancers. The HER2-positive subtype showed frequent HER2 amplification (80%) and a high rate of mutations of TP53 (72%) and PIK3CA (39%) ( Fig. 22.8 ).
Molecular Subtypes of Triple-Negative Breast Cancers
TNBCs are a heterogeneous group. Using the k-means clustering and consensus clustering on gene expression data of 587 TNBC of which 386 samples were used as a discovery data set compiled from 14 breast cancer data sets and 201 samples were used as a validation data set compiled from 7 data sets, six TNBC subtypes were identified: basal-like 1 (BL1), basal-like 2 (BL2), immunomodulatory (IM), mesenchymal (M), mesenchymal stem–like (MSL), luminal androgen receptor (LAR), and unstable (UNS). The BL1 and BL2 subtypes showed high ki67 proliferation scores and higher response rates after taxane chemotherapy. The BL1 subtype showed enrichment of cell cycle genes, whereas the BL2 subtype showed enrichment of growth factor signaling (IGF1R, MET, EGF, NGF pathways) and showed features suggestive of basal/myoepithelial origin as demonstrated by higher expression levels of TP63 and MME (CD10). The IM subtype showed overlap with the medullary signature and was enriched with genes from immune cell signaling pathways, antigen processing and presentation, and signaling through core immune signal transduction pathways (NFKB, TNF, and JAK/STAT signaling). The M subtype displayed enrichment of cell motility, extracellular matrix receptor interaction, and cell differentiation pathways. The MSL subtype shared enrichment of similar biological processes as with the M subtype and in addition was enriched by inositol phosphate metabolism, EGFR, platelet-derived growth factor, calcium signaling, G-protein coupled receptor, and ERK1/2 signaling as well as ATP-binding cassette transporters and adipocytokine signaling. The LAR subtype expressed luminal markers CK18 and was enriched with androgen receptor signaling and therefore corresponded to the apocrine variety of tumors.
Although precision in discovery of cancer drivers and propagating events is the key for clinical utility, discovery of more molecular subtypes based on less common driver events will be the result of the ongoing effort.
Hereditary Breast Cancers
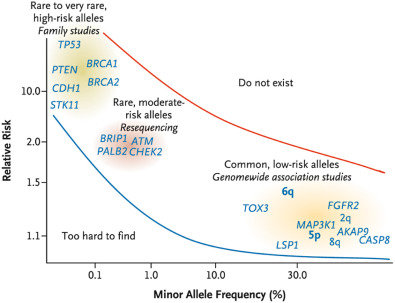
Although approximately 20% of breast cancers may be associated with a familial inheritance pattern, only 10% to 12% are considered hereditary with inheritance of alterations in known susceptibility genes or loci. Hereditary breast cancers have been frequently associated with an autosomal dominant pattern of inheritance.
Fig. 22.9 depicts known susceptibility genes and loci. Mutations in high-penetrance genes confer fivefold or greater breast cancer risk, whereas the mutations in moderate-penetrance genes may confer two- to fourfold risk, compared with the reference population. Mutations in low-penetrance loci confer low risk ranging between one- and twofold increased risk ( Table 22.1 ).
Related posts:
 Anatomy of the Breast, Axilla, Chest Wall, and Related Metastatic Sites
Anatomy of the Breast, Axilla, Chest Wall, and Related Metastatic Sites
 Mesenchymal Neoplasms and Primary Lymphomas of the Breast
Mesenchymal Neoplasms and Primary Lymphomas of the Breast
 Breast Conservation Therapy for Invasive Breast Cancer
Breast Conservation Therapy for Invasive Breast Cancer
 Radiation Complications and Their Management
Radiation Complications and Their Management
 Immunologic Approaches to Breast Cancer Therapy
Immunologic Approaches to Breast Cancer Therapy
 Delayed Diagnosis of Symptomatic Breast Cancer
Delayed Diagnosis of Symptomatic Breast Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


