cellular energetics.5 In the field of leukemia research, attention has been focused on deregulation of proliferation (Class I mutations), and a block in differentiation (Class II mutations).4 The block in differentiation is visually striking under the microscope, as blasts have morphologic characteristics of hematopoietic stem cells (HSCs). However, what is relevant to leukemogenesis is that HSCs have self-renewal. Many of the well-characterized recurrent translocations in acute leukemia produce fusion genes that encode mutant transcription factors that can no longer activate the genes required for differentiation. However, some translocations (notably BCR-ABL1), and some of the genes most commonly affected by point mutations (FLT3) encode kinases that cause deregulated proliferation.4 Through new advances in technology outlined in the next section scientists have discovered a host of additional gene mutations that occur in leukemia, suggesting many more than two hits in the path toward leukemia. In addition, a further layer of regulation at the epigenetic level has been hypothesized due to the number of genes mutated in leukemia that are active in modification of histones or in regulation of DNA methylation.
TABLE 72.1 RISK GROUPS IN ADULT ACUTE MYELOID LEUKEMIA BASED ON CYTOGENETIC AND MOLECULAR ANALYSIS | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
TABLE 72.2 RISK GROUPS IN PEDIATRIC B-CELL ACUTE LYMPHOBLASTIC LEUKEMIA BASED ON CYTOGENETIC AND MOLECULAR ANALYSIS | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||
deletions and amplifications in acute leukemias. Similar microarrays were designed to cover common single nucleotide polymorphisms (SNPs) for genotyping purposes, but they too have been used to identify CNVs, particularly copy number neutral loss of heterozygosity (also known as uniparental disomy) in leukemia.12 Array CGH and SNP arrays display the human genome as small probes and will not detect balanced translocations. Most excitingly, major advances in whole genome sequencing now provide single base pair resolution so that point mutations may be identified as well as chromosomal rearrangements.13 These powerful technologies are readily deployed in the diagnosis of acute leukemia and provide a complete genomic perspective of the disease; however, there are important caveats. Human genomes are full of rare and common structural variants and SNPs.14, 15, 16, 17 and 18 Therefore, mutations can only be interpreted as being acquired in leukemogenesis if they are not present in normal somatic tissue. Thus, matched normal somatic tissue is frequently subjected to the same genomic analyses as leukemic blasts.
TABLE 72.3 RESOLUTION OF TECHNOLOGIES AVAILABLE TO IDENTIFY GENETIC ALTERATIONS IN ACUTE LEUKEMIA | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
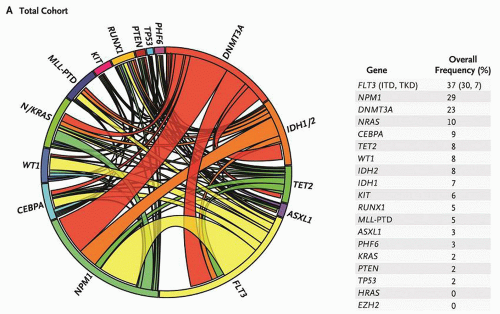 FIGURE 72.1. Mutational complexity of acute myeloid leukemia (AML). The Circos diagram depicts the relative frequency and pairwise co-occurrence of mutations in patients with newly diagnosed AML who were enrolled in the Eastern Cooperative Oncology Group E1900 clinical trial. The length of the arc along the outer circle corresponds to the frequency of mutations in the first gene, and the width of the ribbon corresponds to the percentage of patients who also had a mutation in the second gene listed on the opposite end of the ribbon. Pairwise co-occurrence of mutations is denoted only once, going in the clockwise direction. The frequency of occurrence in the test cohort of the 18 genes in the test panel is listed to the right of the Circos diagram. From Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia NEJM 2012;366:1079-1089, Copyright 2012 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society. |
TABLE 72.4 WORLD HEALTH ORGANIZATION CLASSIFICATION OF ACUTE MYELOID LEUKEMIA | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
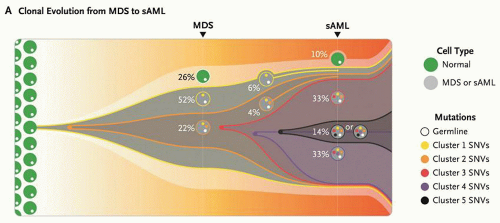 FIGURE 72.2. Clonal progression of MDS to secondary acute myeloid leukemia (AML). This model summarizes the clonal evolution from MDS to secondary AML (sAML) in one patient. Cells in clone 1 (yellow) contain cluster 1 mutations, 323 somatic single nucleotide variants (SNVs) present in approximately 74% of the bone marrow cells. Cells in clone 2 (orange) originated from a single cell in clone 1 and therefore contain all cluster 1 and 2 mutations. This clone became dominant in the sAML sample, in which three subsequent subclones (red, purple, and black) evolved through serial acquisition of SNVs (clusters 3, 4, and 5). From Walter MJ, Shen D, Ding L, et al. Clonal architecture of secondary acute myeloid leukemia. NEJM 2012;366:1090-1098. Copyright 2012 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society. |
as promyelocytic zinc finger), a transcriptional repressor. The N-terminal portion of the fusion protein encoded by ZBTB16 includes the N-terminal POZ/BTB protein interaction domain, transcriptional activation and repression domains, and a variable number of zinc fingers important for protein and DNA interactions (reviewed in Refs. 43,45). ZBTB16 interacts with N-CoR, SMRT, mSin3A, and HDAC1 via the POZ/BTB domain,51,52 and therefore contributes a second binding site for corepressor proteins. Therefore, although pharmacologic doses of ATRA induce release of corepressors from the RARA portion of the fusion protein, the corepressors binding to ZBTB16 are unaffected (Fig. 72.3C).43,53 Significantly, concomitant treatment of cells with HDAC inhibitors such as trichostatin A (TSA) restores ATRA sensitivity, since TSA inhibits the deacetylase activity of the corepressors on the ZBTB16 moiety.49,52
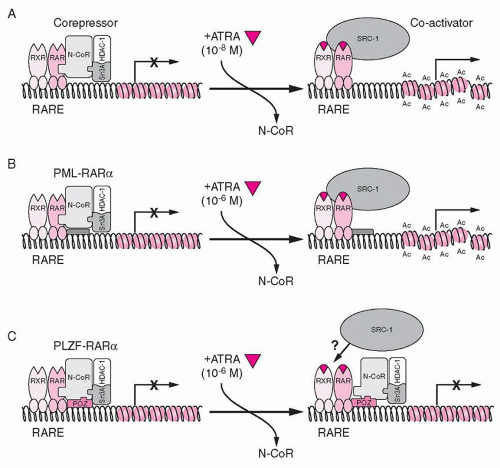 FIGURE 72.3. Model for the role of nuclear corepressors and retinoid acid receptor α (RARA) fusion proteins in the pathogenesis and treatment of acute promyelocytic leukemia. A: In the absence of all-trans retinoic acid (ATRA), RARA, promyelocytic leukemia (PML)-RARA, and promyelocytic leukemia zinc finger (PLZF; now known as ZBTB16)-RARA associate with N-CoR/sin3A/HDAC1 corepressor complex, which deacetylates histone tails, resulting in a compressed chromatin and transcriptional repression. Binding of ATRA at a physiologic concentration induces a conformational change in RARA, causing release of the corepressor complex and binding of coactivator (SRC-1) with histone acetyltransferase activity. Acetylation (Ac) of histone tails opens up the chromatin, facilitating transcriptional activation. B: In the case of PML-RARA protein, pharmacologic doses of ATRA are required to achieve dissociation of the N-CoR repressor complex. C: Because of additional interactions of the PLZF (ZBTB16) moiety of PLZF-RARA fusion protein with corepressors, they do not dissociate even in the presence of pharmacologic doses of ATRA. Therefore, the chromatin still remains in the repressed state. From Guidez F, Ivins S, Jhu J, et al. Reduced retinoic acid-sensitivities of nuclear receptor corepressor binding to PML- and PLZF-RARα underlie molecular pathogenesis and treatment of acute promyelocytic leukemia. Blood 1998;91:2634-2642, with permission. |
nervy gene.78 The four homology domains shared with the Drosophila protein include a region of similarity to TAF110, a hydrophobic heptad repeat (HHR), an ND domain of undetermined function, and two zinc finger motifs that may be a protein-protein interaction domain (Fig. 72.4A).68 RUNX1T1 does not appear to bind DNA specifically on its own. However, it may act as a corepressor protein.79 It associates with N-CoR and mSin3A, and directly binds to the Class I HDACs, HDAC-1, HDAC-2, and HDAC-3 (Fig. 72.4A).80
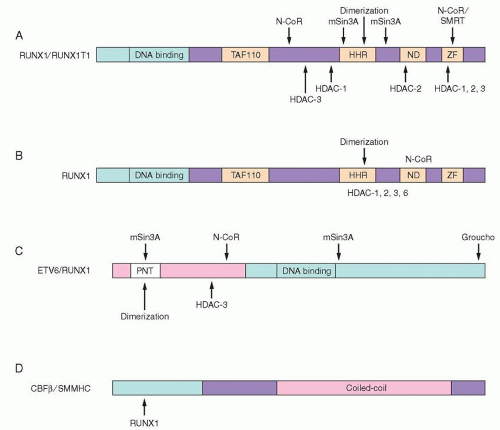 FIGURE 72.4. Schematic diagram of the t(8;21), t(16;21), t(12;21), and inv(16) fusion proteins with known corepressor contacts. A: t(8;21) RUNX1/RUNX1T1. The RUNX1 portion is shown in light pink, with the DNA-binding domain indicated. The RUNX1T1 portion is the dark pink box with domains conserved between RUNX1T1 and its Drosophila homolog in light gray boxes. Known contacts with corepressors and histone deacetylases are shown. B: t(16;21) RUNX1-MTG16 (now known as CBFA2T3). RUNX1 is shown as a light pink box, and MTG16 is shown in a similar manner to RUNX1T1 in A. C: t(12;21) ETV6-RUNX1. ETV6 is the dark pink box, with the conserved pointed (PNT) domain indicated. The RUNX1 portion is the light pink box. Interactions with corepressors and HDACs are shown. D: Inv(16) CBFB-SMMHC (now known as MYH11). The CBFB portion, which interacts with RUNX1, is light pink, and the SMMHC is dark pink, with the coiled-coil domain indicated as well as the C-terminal portion, which is necessary for interaction with mSin3A and HDAC8.77 HHR, hydrophobic heptad repeat; ND, nervy domain; TAF110, a domain with homology to the TAF110 coactivator; ZF, zinc finger domain. From Hiebert SW, Lutterbach B, Amann J. Role of corepressors in transcriptional repression mediated by the t(8;21), t(16;21), t(12;21), and inv(16) fusion proteins. Curr Opin Hematol 2001;8:197-200, with permission. |
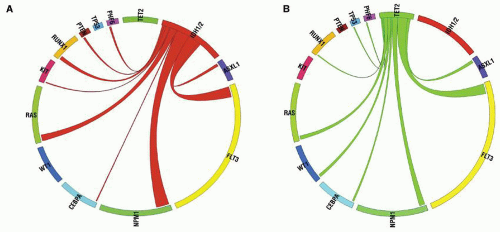 FIGURE 72.5. IDH1/2 mutations are mutually exclusive with mutations in TET2 in de novo AML. A: Circos diagram revealing relative frequency and pairwise co-occurrences of mutations in IDH1 and IDH2 in a cohort of 385 patients with de novo AML. B: Circos diagram revealing relative frequency and pairwise co-occurrences of mutations in TET2 in a cohort of 385 patients with de novo AML. Reprinted from Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010;18:553-567, with permission from Elsevier. |
that fewer cytosines in a CpG island are methylated.120 The effect on tetramerization explains why the DNMT3A mutations are dominant negative, usually occurring in just one allele. Global methylation does not seem to be affected in DNA from AML with mutated DNMT3A, but analysis of DNA methylation by MeDIP-Chip (methylated DNA immunoprecipitation-Chip) analysis in a matched set of DNAs from 5 AML patients with mutated DNMT3A and 5 AML patients with wild-type DNMT3A demonstrated 182 genomic sites where the DNA from patients with mutated DNMT3A was hypomethylated.31 Studies of DNA methylation in Dnmt3a-null murine HSCs demonstrated a complex story with poor correlation between changes in methylation sites and changes in gene expression comparing wild-type to Dnmt3a-null HSCs. However, changes in gene expression patterns may be due to changes in methylation of regulatory regions of directly affected genes, whose expression then alters regulation of many other genes by mechanisms other than methylation. Transcriptional profiling of Dnmt3a-null HSCs did reveal that genes involved in the multipotency of normal HSCs were upregulated, whereas genes necessary for differentiation of the HSCs were downregulated. This suggests that the DNMT3A mutations may contribute to the block in differentiation that occurs in leukemic blasts. Interestingly the Dnmt3a-null mice have not yet developed leukemia, suggesting that DNMT3A mutation alone is not sufficient for leukemogenesis.121
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


