Molecular endocrinology and endocrine genetics
Isolation and Digestion of DNA and Southern Blotting
Restriction Fragment Length Polymorphism and Other Polymorphic DNA Studies
DETECTION OF MUTATIONS IN HUMAN GENES
POSITIONAL GENETICS IN ENDOCRINOLOGY
EXPRESSION STUDIES (MICROARRAYS, SAGE)
CHROMOSOME ANALYSIS AND MOLECULAR CYTOGENETICS
MOLECULAR BASIS OF PEDIATRIC ENDOCRINOPATHIES
PRINCIPLES OF INTERPRETATION OF GENETIC TESTS IN THE DIAGNOSIS AND MANAGEMENT OF PEDIATRIC ENDOCRINE DISEASES
RECOMBINANT DNA TECHNOLOGY AND THERAPY OF PEDIATRIC ENDOCRINE DISEASES
Copyright © 2014 by Elsevier Inc. All rights reserved. Part of the chapter is in public domain.
Introduction
The study of the endocrine system has undergone a dramatic evolution since the 1990s, from the traditional physiologic studies that dominated the field for many years to the discoveries of molecular endocrinology and endocrine genetics.1,2 At the present time the major impact of molecular medicine on the practice of pediatric endocrinology relates to diagnosis and genetic counseling for a variety of inherited endocrine disorders. In contrast, the direct therapeutic application of this new knowledge is still in its infancy, although recently the results of the first successful human gene therapy trials were reported (but not for endocrine diseases). In addition, the new information has led to a host of molecularly targeted therapies mainly in cancer; endocrine oncology has greatly benefited from the application of new drugs that were designed to battle specific mutations in, for example, thyroid cancer. This chapter is an introduction to the basic principles of molecular biology, common laboratory techniques, and some examples of the recent advances made in clinical pediatric endocrinologic disorders with an emphasis on endocrine genetics. Most new diagnostic testing, pharmacogenetics, and molecular therapies are discussed in the disease-specific chapters of this book, and only examples that highlight the principle/strategy under discussion are listed in this chapter.
Basic molecular tools
Isolation and digestion of DNA and southern blotting
The human chromosome comprises a long double-stranded helical molecule of DNA associated with different nuclear proteins.3,4 As DNA forms the starting point of the synthesis of all the protein molecules in the body, molecular techniques using DNA have proven to be crucial in the development of diagnostic tools to analyze endocrine diseases. DNA can be isolated from any human tissue, including circulating white blood cells. About 200 µg of DNA can be obtained from 10 to 20 mL of whole blood with the efficiency of DNA extraction being dependent on the technique used and the method of anticoagulation employed. The extracted DNA can be stored almost indefinitely at an appropriate temperature. Furthermore, lymphocytes can be transformed with the Epstein-Barr virus (and other means) to propagate indefinitely in cell culture as “immortal” cell lines, thus providing a renewable source of DNA. For performing molecular genetic studies, lymphoid lines are routinely the tissue of choice, because a renewable source of DNA obviates the need to obtain further blood from the family. Fibroblast-derived cultures can also serve as a permanent source of DNA or RNA (once transformed), but they have to be derived from surgical specimens or a biopsy. It should be noted that, because the expression of many genes is tissue specific, immortalized lymphoid or fibroblastoid cell lines cannot be used to anlyze the abundance or composition of messenger RNA (mRNA) for a specific gene. Hence, studies involving mRNA necessitate the analysis of the tissue(s) expressing the gene as outlined in the section on “RNA Analysis” that begins on page 13.
DNA is present in extremely large molecules; the smallest chromosome (chromosome 22) has about 50 million base pairs and the entire haploid human genome is estimated to comprise 3 million to 4 billion base pairs. This extreme size precludes the analysis of DNA in its native form in routine molecular biology techniques. The techniques for identification and analysis of DNA became feasible and readily accessible with the discovery of enzymes termed restriction endonucleases. These enzymes, originally isolated from bacteria, cut DNA into smaller sizes on the basis of specific recognition sites that vary from two to eight base pairs in length.5,6 The term restriction refers to the function of these enzymes in bacteria. A restriction endonuclease destroys foreign DNA (such as bacteriophage DNA) by cleaving the DNA at specific sites, thereby “restricting” the entry of foreign DNA in the bacterium. Several hundred restriction enzymes with different recognition sites are now commercially available. Because the recognition site for a given enzyme is fixed, the number and sizes of fragments generated for a particular DNA molecule remain consistent with the number of recognition sites and provide predictable patterns after separation by electrophoresis.
Analysis of the DNA fragments generated after digestion usually employs the technique of electrophoresis.7 Electrophoresis exploits the property that the phosphate groups in the DNA molecule confer a negative charge to that molecule. Thus, when a mixture of DNA molecules of different sizes is electrophoresed through a sieve (routinely either agarose or acrylamide), the longer DNA molecules migrate more slowly relative to the shorter fragments. Following electrophoresis, the separated DNA molecules can be located by a variety of staining techniques, of which ethidium bromide staining is a commonly used method.
Although staining with ethidium bromide is a versatile technique, analysis of a few hundred base pairs of DNA in the region of interest is difficult when the DNA from all the human chromosomes are cut and separated on the same gel. These limitations are circumvented by the technique of Southern blotting (named after its originator, Edward Southern) and the use of labeled radioactive or more commonly nonradioactive probes. Southern blotting involves digestion of DNA and separation by electrophoresis through agarose.8 After electrophoresis, the DNA is transferred to a solid support (such as nitrocellulose or nylon membranes), enabling the pattern of separated DNA fragments to be replicated onto the membrane (Figure 2-1). The DNA is then denatured (i.e., the two strands are physically separated), fixed to the membrane, and the dried membrane is mixed with a solution containing the DNA probe. A DNA probe is a fragment of DNA that contains a nucleotide sequence specific for the gene or chromosomal region of interest. For purposes of detection, the DNA probe is labeled with an identifiable tag, such as radioactive phosphorus (e.g.,32P) or a chemiluminescent moiety; the latter has almost exclusively replaced radioactivity. The process of mixing the DNA probe with the denatured DNA fixed to the membrane is called hybridization, the principle being that there are only four nucleic acid bases in DNA—adenine (A), thymidine (T), guanine (G), and cytosine (C)—that always remain complementary on the two strands of DNA, A pairing with T, and G pairing with C. Following hybridization, the membrane is washed to remove the unbound probe and exposed to an x-ray film either in a process called autoradiography to detect radioactive phosphorus or in a process used to detect the chemiluminescent tag. Only those fragments that are complementary and have bound to the probe containing the DNA of interest will be evident on the x-ray film, enabling the analysis of the size and pattern of these fragments. As routinely performed, the technique of Southern analysis can detect a single copy gene in as little as 5 µg of DNA, the DNA content of about 106 cells.
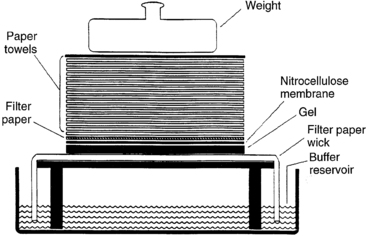
FIGURE 2-1  Southern blot. Fragments of double-stranded DNA are separated by size by agarose gel electrophoresis. To render the DNA single stranded (denatured), the agarose gel is soaked in an acidic solution. After neutralization of the acid, the gel is placed onto filter paper, the ends of which rest in a reservoir of concentrated salt buffer solution. A sheet of nitrocellulose membrane is placed on top of the gel and absorbent paper is stacked on top of the nitrocellulose membrane. The salt solution is drawn up through the gel by the capillary action of the filter paper wick and the absorbent paper towels. As the salt solution moves through the gel, it carries along with it the DNA fragments. Because nitrocellulose binds single-stranded DNA, the DNA fragments are deposited onto the nitrocellulose in the same pattern that they were placed in the agarose gel. The DNA fragments bound to the nitrocellulose are fixed to the membrane by heat or UV irradiation. The nitrocellulose membrane with the bound DNA can then be used for procedures such as hybridization to a labeled DNA probe. Techniques to transfer DNA to other bonding matrices, such as nylon, are similar. (Adapted from Turco E, Fritsch R, Trucco M [1990]. Use of immunologic techniques in gene analysis. In Herberman RB, Mercer DW [eds.], Immunodiagnosis of cancer. New York: Marcel Dekker, 205.)
Southern blot. Fragments of double-stranded DNA are separated by size by agarose gel electrophoresis. To render the DNA single stranded (denatured), the agarose gel is soaked in an acidic solution. After neutralization of the acid, the gel is placed onto filter paper, the ends of which rest in a reservoir of concentrated salt buffer solution. A sheet of nitrocellulose membrane is placed on top of the gel and absorbent paper is stacked on top of the nitrocellulose membrane. The salt solution is drawn up through the gel by the capillary action of the filter paper wick and the absorbent paper towels. As the salt solution moves through the gel, it carries along with it the DNA fragments. Because nitrocellulose binds single-stranded DNA, the DNA fragments are deposited onto the nitrocellulose in the same pattern that they were placed in the agarose gel. The DNA fragments bound to the nitrocellulose are fixed to the membrane by heat or UV irradiation. The nitrocellulose membrane with the bound DNA can then be used for procedures such as hybridization to a labeled DNA probe. Techniques to transfer DNA to other bonding matrices, such as nylon, are similar. (Adapted from Turco E, Fritsch R, Trucco M [1990]. Use of immunologic techniques in gene analysis. In Herberman RB, Mercer DW [eds.], Immunodiagnosis of cancer. New York: Marcel Dekker, 205.)
Restriction fragment length polymorphism and other polymorphic DNA studies
The number and size of DNA fragments resulting from the digestion of any particular region of DNA form a recognizable pattern. Small variations in a sequence among unrelated individuals may cause a restriction enzyme recognition site to be present or absent; this results in a variation in the number and size pattern of the DNA fragments produced by digestion with that particular enzyme. Thus this region is said to be polymorphic for the particular enzyme tested—that is, a restriction fragment length polymorphism (RFLP) (Figure 2-2). The value of RFLP is that it can be used as a molecular tag for tracing the inheritance of the maternal and paternal alleles. Furthermore, the polymorphic region analyzed does not need to encode the genetic variation that is the cause of the disease being studied, but only to be located near the gene of interest. When a particular RFLP pattern can be shown to be associated with a disease, the likelihood of an offspring inheriting the disease can be determined by comparing the offspring’s RFLP pattern with the RFLP pattern of the affected or carrier parents. The major limitation of the RFLP technique is that its applicability for the analysis of any particular gene is dependent on the prior knowledge of the presence of convenient (“informative”) polymorphic restriction sites that flank the gene of interest by at most a few kilobases. Because these criteria may not be fulfilled in any given case, the applicability of RFLP cannot be guaranteed for the analysis of a given gene.
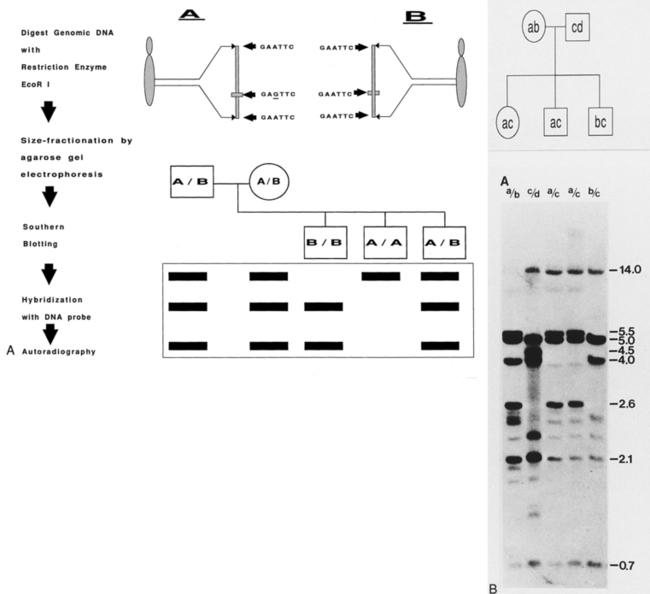
FIGURE 2-2  Restriction fragment length polymorphism. A, Schematic illustration. A and B represent two alleles that display a polymorphic site for the restriction enzyme EcoR I. EcoR I will cut DNA with the sequence “GAATTC”; hence, allele B will be cut by EcoR I at three sites to generate two fragments of DNA, whereas allele A will be cut by EcoR I only twice and not at the site (indicated by horizontal bar) where nucleotide G (underlined) replaces the nucleotide A present in allele B. Following digestion, the DNA is size-fractionated by agarose gel electrophoresis and transferred to a membrane by Southern blot technique (see Figure 2-1 for details). The membrane is then hybridized with a labeled DNA probe, which contains the entire sequence spanned by the three EcoR I sites. Autoradiography of the membrane will detect the size of the DNA fragments generated by the restriction enzyme digestion. In this particular illustration, both parents are heterozygous and possess both A and B alleles. Matching the pattern of the DNA bands of the offspring with that of the parents will establish the inheritance pattern of the alleles. For example, if allele A represents the abnormal allele for an autosomal recessive disease, then examination of the Southern blot will establish that (from left to right) the first offspring (B/B) is homozygous for the normal allele, the second offspring (A/A) is homozygous for the abnormal allele, and the third offspring (A/B) is a carrier. B, RFLP analysis of the DQ-beta gene of the HLA locus. Genomic DNA from the members of the indicated pedigree was digested with restriction enzyme Pst I, size-fractionated by agarose gel electrophoresis, and transferred to nitrocellulose membrane by Southern blot technique. The membrane was then hybridized with a cDNA probe specific for the DQ-beta gene; the excess probe was removed by washing at appropriate stringency and was analyzed by autoradiography. The sizes of the DNA fragments (in kilobases, kb) are indicated on the right. The pedigree chart indicates the polymorphic alleles (a, b, c, d) and the bands on the Southern blot corresponding to these alleles (a [5.5 kb], b [5.0 kb], c [14.0 kb], d [4.5 kb]) indicate the inheritance pattern of these alleles. (Adapted from Turco E, Fritsch R, Trucco M [1998]. First domain encoding sequence mediates human class II beta-chain gene cross-hybridization. Immunogenetics 28:193.)
Restriction fragment length polymorphism. A, Schematic illustration. A and B represent two alleles that display a polymorphic site for the restriction enzyme EcoR I. EcoR I will cut DNA with the sequence “GAATTC”; hence, allele B will be cut by EcoR I at three sites to generate two fragments of DNA, whereas allele A will be cut by EcoR I only twice and not at the site (indicated by horizontal bar) where nucleotide G (underlined) replaces the nucleotide A present in allele B. Following digestion, the DNA is size-fractionated by agarose gel electrophoresis and transferred to a membrane by Southern blot technique (see Figure 2-1 for details). The membrane is then hybridized with a labeled DNA probe, which contains the entire sequence spanned by the three EcoR I sites. Autoradiography of the membrane will detect the size of the DNA fragments generated by the restriction enzyme digestion. In this particular illustration, both parents are heterozygous and possess both A and B alleles. Matching the pattern of the DNA bands of the offspring with that of the parents will establish the inheritance pattern of the alleles. For example, if allele A represents the abnormal allele for an autosomal recessive disease, then examination of the Southern blot will establish that (from left to right) the first offspring (B/B) is homozygous for the normal allele, the second offspring (A/A) is homozygous for the abnormal allele, and the third offspring (A/B) is a carrier. B, RFLP analysis of the DQ-beta gene of the HLA locus. Genomic DNA from the members of the indicated pedigree was digested with restriction enzyme Pst I, size-fractionated by agarose gel electrophoresis, and transferred to nitrocellulose membrane by Southern blot technique. The membrane was then hybridized with a cDNA probe specific for the DQ-beta gene; the excess probe was removed by washing at appropriate stringency and was analyzed by autoradiography. The sizes of the DNA fragments (in kilobases, kb) are indicated on the right. The pedigree chart indicates the polymorphic alleles (a, b, c, d) and the bands on the Southern blot corresponding to these alleles (a [5.5 kb], b [5.0 kb], c [14.0 kb], d [4.5 kb]) indicate the inheritance pattern of these alleles. (Adapted from Turco E, Fritsch R, Trucco M [1998]. First domain encoding sequence mediates human class II beta-chain gene cross-hybridization. Immunogenetics 28:193.)
Polymerase chain reaction
The polymerase chain reaction (PCR) is a technique that was developed in the late 1980s and has indeed revolutionized molecular biology (Figure 2-3). PCR allows the selective logarithmic amplification of a desired fragment of DNA from a complex mixture of DNA that theoretically contains at least a single copy of the target fragment. In the typical application of this technique, some knowledge of the DNA sequences in the region to be amplified is necessary, so that a pair of short (approximately 18 to 25 bases in length) specific oligonucleotides (“primers”) can be synthesized. The primers are synthesized in such a manner that they define the limits of the region to be amplified. The DNA template containing the segment that is to be amplified is heat denatured such that the strands are separated and then cooled to allow the primers to anneal to the respective complementary regions. The enzyme Taq polymerase, a heat stable enzyme originally isolated from the bacterium Thermophilus aquaticus, is then used to initiate synthesis (extension) of DNA. The DNA is repeatedly denatured, annealed, and extended in successive cycles in a machine called the “thermocycler” that permits this process to be automated. In the usual assay, these repeated cycles of denaturing, annealing, and extension result in the synthesis of approximately 1 million copies of the target region in about 2 hours. To establish the veracity of the amplification process, the identity of the amplified DNA can be analyzed by electrophoresis, hybridization to RNA or DNA probes, digestion with informative restriction enzyme(s), or subjected to direct DNA sequencing. The relative simplicity combined with the power of this technique has resulted in widespread use of this procedure and has spawned a wide variety of variations and modifications that have been developed for specific applications.9,10 From a practical point of view, the major drawback of PCR is the propensity to get cross-contamination of the target DNA. This drawback is the direct result of the extreme sensitivity of the method that permits amplification from one molecule of the starting DNA template. Thus, unintended transfer of amplified sequences to items used in the procedure will amplify DNA in samples that do not contain the target DNA sequence (i.e., a false positive result). Cross-contamination should be suspected when amplification occurs in negative controls that did not contain the target template. One of the most common modes of cross-contamination is via aerosolization of the amplified DNA during routine laboratory procedures such as vortexing, pipetting, and manipulation of microcentrifuge tubes. Meticulous care to experimental technique, proper organization of the PCR workplace, and inclusion of appropriate controls are essential for the successful prevention of cross-contamination during PCR experiments.
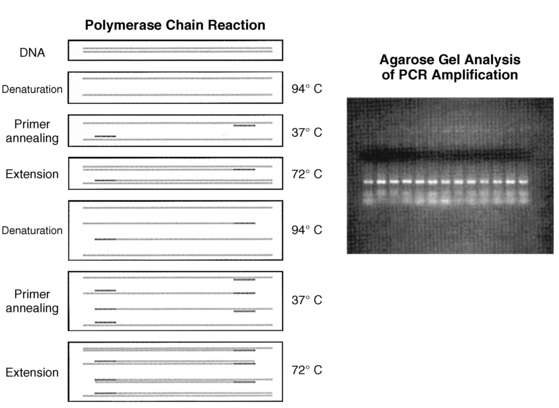
FIGURE 2-3  Polymerase chain reaction (PCR). A pair of oligonucleotide primers (solid bars), complementary to sequences flanking a particular region of interest (shaded, stippled bars), are used to guide DNA synthesis in opposite and overlapping directions. Repeated cycles of DNA denaturation, primer annealing, and DNA synthesis (primer extension) by DNA polymerase enzyme result in an exponential increase in the target DNA (i.e., the DNA sequence located between the two primers) such that this DNA segment can be amplified 1 × 106-7 times after 30 such cycles. The use of a thermostable DNA polymerase (i.e., Taq polymerase) allows for this procedure to be automated. Inset: The amplified DNA can be used for subsequent analysis (i.e., size-fractionation by agarose gel electrophoresis). (Adapted from Trucco M [1992]. To be or not to be ASP 57, that is the question. Diabetes Care 15:705.)
Polymerase chain reaction (PCR). A pair of oligonucleotide primers (solid bars), complementary to sequences flanking a particular region of interest (shaded, stippled bars), are used to guide DNA synthesis in opposite and overlapping directions. Repeated cycles of DNA denaturation, primer annealing, and DNA synthesis (primer extension) by DNA polymerase enzyme result in an exponential increase in the target DNA (i.e., the DNA sequence located between the two primers) such that this DNA segment can be amplified 1 × 106-7 times after 30 such cycles. The use of a thermostable DNA polymerase (i.e., Taq polymerase) allows for this procedure to be automated. Inset: The amplified DNA can be used for subsequent analysis (i.e., size-fractionation by agarose gel electrophoresis). (Adapted from Trucco M [1992]. To be or not to be ASP 57, that is the question. Diabetes Care 15:705.)
In general, PCR applications are either directed toward the identification of a specific DNA sequence in a tissue or body fluid sample or used for the production of relatively large amounts of DNA of a specific sequence, which then are used in further studies. Examples of the first type of application are common in many fields of medicine, such as in microbiology, wherein the PCR technique is used to detect the presence DNA sequences specific for viruses or bacteria in a biological sample. Prototypic examples of such an application in pediatric endocrinology include the use of PCR of the SRY gene for detecting Y chromosome material in patients with karyotypically defined Turner syndrome and the rapid identification of chromosomal gender in cases of fetal or neonatal sexual ambiguity11 (Figure 2-4).
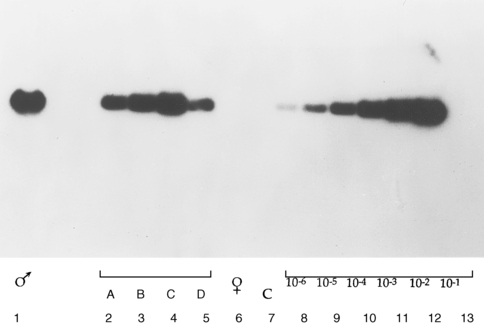
FIGURE 2-4  Detection of SRY gene–specific sequence in Turner syndrome by polymerase chain reaction (PCR) amplification and Southern blot. SRY-specific primers were used in PCR to amplify DNA from patients with 45X karyotype. The amplified DNA was size-fractionated by agarose gel-electrophoresis and transferred to membrane by Southern blotting. The membrane was then hybridized to labeled SRY-specific DNA and autoradiographed. From left to right: amplified male DNA (lane 1); amplified DNA from patients with 45X karyotype (lanes 2-5); amplified female DNA (lane 6); negative control with no DNA (lane 7); serial dilution of male DNA (lanes 8-13). (Adapted from Kocova M, Siegel SF, Wenger SL, et al. [1993]. Detection of Y chromosome sequence in Turner’s syndrome by Southern blot analysis of amplified DNA. Lancet 342:140. © Copyright by the Lancet Ltd.)
Detection of SRY gene–specific sequence in Turner syndrome by polymerase chain reaction (PCR) amplification and Southern blot. SRY-specific primers were used in PCR to amplify DNA from patients with 45X karyotype. The amplified DNA was size-fractionated by agarose gel-electrophoresis and transferred to membrane by Southern blotting. The membrane was then hybridized to labeled SRY-specific DNA and autoradiographed. From left to right: amplified male DNA (lane 1); amplified DNA from patients with 45X karyotype (lanes 2-5); amplified female DNA (lane 6); negative control with no DNA (lane 7); serial dilution of male DNA (lanes 8-13). (Adapted from Kocova M, Siegel SF, Wenger SL, et al. [1993]. Detection of Y chromosome sequence in Turner’s syndrome by Southern blot analysis of amplified DNA. Lancet 342:140. © Copyright by the Lancet Ltd.)
RNA analysis
There are many techniques for analyzing mRNA. Northern blotting (so named because it is based on the same principle as the Southern blot) is one of the original methods used for mRNA analysis. In Northern blotting, RNA is denatured by treating it with an agent such as formaldehyde to ensure that the RNA remains unfolded and in the linear form.12,13 The denatured RNA is then electrophoresed and transferred onto a solid support (such as nitrocellulose membrane) in a manner similar to that described for the Southern blot.8 The membrane with the RNA molecules separated by size is probed with the gene-specific DNA probe labeled with an identifiable tag that, as in the case of Southern blotting, is either a radioactive label (e.g., 32P) or more commonly a chemiluminescent moiety. The nucleotide sequence of the DNA probe is complementary to the mRNA sequence of the gene and is hence called complementary DNA (cDNA). It is customary to use labeled cDNA (and not labeled mRNA) to probe Northern blots because DNA molecules are much more stable and easier to manipulate and propagate (usually in bacterial plasmids) than mRNA molecules. The Northern blot provides information regarding the amount (estimated by the intensity of the signal on autoradiography) and the size (estimated by the position of the signal on the gel in comparison to concurrently electrophoresed standards) of the specific mRNA. Although the Northern blot technique represents a versatile and straightforward method to analyze mRNA, it has major drawbacks. Northern analysis is a relatively insensitive technique, both in terms of the concentration of mRNA that can be detected and in terms of the fine structure. This technique cannot detect small changes in size, nucleotide composition, or the abundance of the mRNA being analyzed. At present, reverse transcriptase-PCR (RT-PCR) has become the technique of choice for the routine analysis of mRNA.
One of the most sensitive methods for the detection and quantitation of mRNA currently available is the technique of quantitative RT-PCR (qRT-PCR).9 This technique combines the unique function of the enzyme reverse transcriptase with the power of PCR. qRT-PCR is exquisitely sensitive, permitting analysis of gene expression from very small amounts of RNA. Furthermore, this technique can be applied to a large number of samples or many genes (multiplex) in the same experiment. These two critical features endow this technique with a measure of flexibility unavailable in more traditional methods such as Northern blot or solution hybridization analysis. Whereas the detection of a specific mRNA by this technique is relatively straightforward, the precise quantitation of the mRNA in a given sample is more complicated. The first step in qRT-PCR analysis is the production of DNA complementary (cDNA) to the mRNA of interest. This is done by using the enzymes with RNA-dependent DNA polymerase activity that belong to the reverse transcriptase (RT) group of enzymes (e.g., Moloney murine leukemia virus [MMLV], avian myeloblastosis virus [AMV] reverse transcriptase, an RNA dependent DNA polymerase). The RT enzyme, in the presence of an appropriate primer, will synthesize DNA complementary to RNA. The second step in the qRT-PCR analysis is the amplification of the target DNA, in this case the cDNA synthesized by the reverse transcriptase enzyme. The specificity of the amplification is determined by the specificity of the primer pair used for the PCR amplification. To establish the veracity of the amplification process, the identity of the amplified DNA can be analyzed by electrophoresis, hybridization to RNA or DNA probes, digestion with informative restriction enzyme(s), or subjected to direct DNA sequencing
Whereas the detection of a specific mRNA by this technique is relatively straightforward, the precise quantitation of the mRNA in a given sample is more complicated. Because the production of DNA by PCR involves an exponential increase in the amount of DNA synthesized, relatively minor differences in any of the variables controlling the rate of amplification will cause a marked difference in the yield of the amplified DNA. In addition to the amount of template DNA, the variables that can affect the yield of the PCR include the concentration of the polymerase enzyme, magnesium, nucleotides (dNTPs), and primers. The specifics of the amplification procedure including cycle length, cycle number, annealing, extension and denaturing temperatures also affect the yield of DNA. Because of the multitude of variables involved, routine RT-PCR is unsuitable for performing a quantitative analysis of mRNA. To circumvent these pitfalls alternate strategies have been developed. One technique for determining the concentration of a particular mRNA in a biological sample is a modification of the basic PCR technique called competitive RT-PCR.9,14,15 This method is based on the co-amplification of a mutant DNA that can be amplified with the same pair of primers being used for the target DNA. The mutant DNA is engineered in such a way that it can be distinguished from the DNA of interest by either size or the inclusion of a restriction enzyme site unique to the mutant DNA. The addition of equivalent amounts of this mutant DNA to all the PCR reaction tubes serves as an internal control for the efficiency of the PCR process, and the yield of the mutant DNA in the various tubes can be used for the equalization of the yield of the DNA by PCR. It is important to ensure for accurate quantitation of the DNA of interest that the concentrations of the mutant and target template should be nearly equivalent. Because the use of mutated DNA for normalization does not account for the variability in the efficiency of the reverse transcriptase enzyme, a variation of the original method has been developed. In this modification, competitive mutated RNA transcribed from a suitably engineered RNA expression vector is substituted for the mutant DNA in the reaction prior to initiating the synthesis of the cDNA. Competitive RT-PCR can be used to detect changes of the order of two- to threefold of even very rare mRNA species. The major drawback of this method is the propensity to get inaccurate results because of the contamination of samples with the mRNA of interest. In theory, as the technique is based on PCR, contamination by even one molecule of mRNA of interest can invalidate the results. Hence, scrupulous attention to laboratory technique and set up is essential for the successful application of this technique.
In general, two types of methods are used for the detection and quantitation of PCR products: the traditional “end-point” measurements of products and the newer “real-time” techniques. End-point determinations (e.g., the competitive RT-PCR technique described earlier) analyze the reaction after it is completed, whereas real-time determinations are made during the progression of the amplification process. Overall the real-time approach is more accurate and is currently the preferred method. Advances in fluorescence detection technologies have made the use of real-time measurement possible for routine use in the laboratory. One of the popular techniques that takes advantage of real-time measurements is the TaqMan (fluorescent 5′ nuclease) assay (Figure 2-5).16,17 The unique design of TaqMan probes, combined with the 5′ nuclease activity of the PCR enzyme (Taq polymerase), allows direct detection of PCR product by the release of fluorescent reporter during the PCR amplification by using specially designed machines (ABI Prism 5700/7700). The TaqMan probe consists of an oligonucleotide synthesized with a 5′-reporter dye (e.g., FAM; 6-carboxy-fluorescein) and a downstream, 3′-quencher dye (e.g., TAMRA; 6-carboxy-tetramethyl-rhodamine). When the probe is intact, the proximity of the reporter dye to the quencher dye results in suppression of the reporter fluorescence, primarily by Forster-type energy transfer. During PCR, forward and reverse primers hybridize to a specific sequence of the target DNA. The TaqMan probe hybridizes to a target sequence within the PCR product. The Taq polymerase enzyme, because of its 5′-3′ nuclease activity, subsequently cleaves the TaqMan probe. The reporter dye and the quencher dye are separated by cleavage, resulting in increased laser-stimulated fluorescence of the reporter dye as a direct consequence of target amplification during PCR. This process occurs in every cycle and does not interfere with the exponential accumulation of product. Both primer and probe must hybridize to the target for amplification and cleavage to occur. The fluorescence signal is generated only if the target sequence for the probe is amplified during PCR. Because of these stringent requirements, nonspecific amplification is not detected. Fluorescent detection takes place through fiber optic lines positioned above optically nondistorting tube caps. Quantitative data are derived from a determination of the cycle at which the amplification product signal crosses a preset detection threshold. This cycle number is proportional to the amount of starting material, thus allowing for a measurement of the level of specific mRNA in the sample. An alternate machine (Light Cycler) also uses fluorogenic hydrolysis or fluorogenic hybridization probes for quantification in a manner similar to the ABI system.
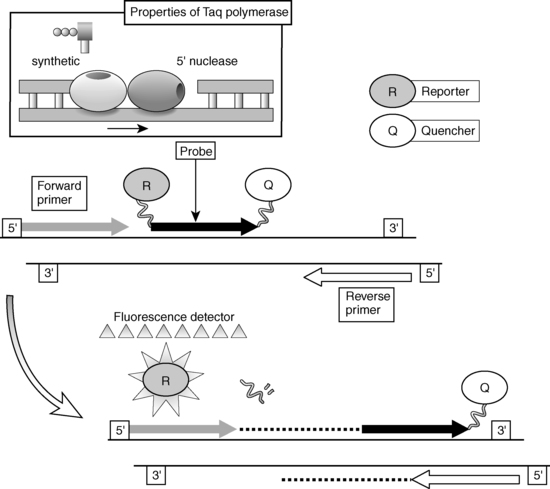
FIGURE 2-5  Fluorescent 5′ nuclease (TaqMan) assay.
Fluorescent 5′ nuclease (TaqMan) assay.
Three synthetic oligonucleotides are utilized in a fluorescent 5’ nuclease assay. Two oligonucleotides function as “forward” and “reverse” primers in a conventional polymerase chain reaction (PCR) amplification protocol. The third oligonucleotide, termed the TaqMan probe, consists of an oligonucleotide synthesized with a 5’-reporter dye (e.g., FAM; 6-carboxy-fluorescein) and a downstream, 3’-quencher dye (e.g., TAMRA; 6-carboxy-tetramethyl-rhodamine). When the probe is intact, the proximity of the reporter dye to the quencher dye results in suppression of the reporter fluorescence, primarily by Forster-type energy transfer. During PCR, forward and reverse primers hybridize to a specific sequence of the target DNA. The TaqMan probe hybridizes to a target sequence within the PCR product. The Taq polymerase enzyme, because of its 5’-3’ exonuclease activity, subsequently cleaves the TaqMan probe. The reporter dye and the quencher dye are separated by cleavage, resulting in increased fluorescence of the reporter dye as a direct consequence of target amplification during PCR. Both primers and probe must hybridize to the target for amplification and cleavage to occur. Hence, the fluorescence signal is generated only if the target sequence for the probe is amplified during PCR. Fluorescent detection takes place through fiberoptic lines positioned above the caps of the reaction wells. Inset: The two distinct functions of the enzyme Taq polymerase: the 5’-3’ synthetic polymerase activity and the 5’-3’ polymerase-dependent exonuclease activity.
Detection of mutations in genes
Changes in the structural organization of a gene that impact its function involve deletions, insertions, or transpositions of relatively large stretches of DNA, or more frequently single-base substitutions in functionally critical regions. Southern blotting and RFLP analysis can usually detect the deletion or insertion of large stretches of DNA. However, these analytic methods can be used for detecting point mutations only if the mutation involves the recognition site for a particular restriction enzyme, such that the absence of a normally present restriction site or the appearance of a novel site unmasks the presence of the point mutation. More commonly these techniques cannot be used for such an analysis, necessitating alternate procedures. High throughput or next-generation sequencing has revolutionized the identification of mutations in genes.48
Direct methods
DNA sequencing is the current gold standard for obtaining unequivocal proof of a point mutation. However, DNA sequencing has its limitations and drawbacks. A clinically relevant problem is that current DNA sequencing methods do not reliably and consistently detect all mutations. For example, in many cases where the mutation affects only one allele (heterozygous), the heights of the peaks of the bases on the fluorescent readout corresponding to the wild-type and mutant allele are not always present in the predicted (1:1) ratio. This limits the discerning power of “base calling” computer protocols and results in inconsistent or erroneous assignment of DNA sequence to individual alleles.18 Because of this limitation, clinical laboratories routinely determine the DNA sequence of both the alleles to provide independent confirmation of the absence/presence of a putative mutation. DNA sequencing can be labor intensive and expensive, although advances in pyrosequencing (discussed later), for example, have made it technically easier and cheaper.
Although the first DNA sequences were determined with a method that chemically cleaved the DNA at each of the four nucleotides,19 the enzymatic or dideoxy method developed by Sanger and colleagues in 1977 is the one most commonly used for routine purposes20 (Figure 2-6). This method uses the enzyme DNA polymerase to synthesize a complementary copy of the single-stranded DNA (“template”) whose sequence is being determined. Single-stranded DNA can be obtained directly from viral or plasmid vectors that support the generation of single-stranded DNA or by partial denaturing of double-stranded DNA by treatment with alkali or heat.21 The enzyme DNA polymerase cannot initiate synthesis of a DNA chain de novo but can only extend a fragment of DNA. Hence, the second requirement for the dideoxy method of sequencing is the presence of a “primer.” A primer is a synthetic oligonucleotide, 15 to 30 bases long, whose sequence is complementary to the sequence of the short corresponding segment of the single-stranded DNA template. The dideoxy method exploits the observation that DNA polymerase can use both 2′-deoxynucleoside triphosphate (dNTP) and 2′,3′-dideoxynucleoside triphosphates (ddNTPs) as substrates during elongation of the primer. Whereas DNA polymerase can use dNTP for continued synthesis of the complementary strand of DNA, the chain cannot elongate any further after addition of the first ddNTP because ddNTPs lack the crucial 3′-hydroxyl group. To identify the nucleotide at the end of the chain, four reactions are carried out for each sequence analysis, with only one of the four possible ddNTPs included in any one reaction. The ratio of the ddNTP and dNTP in each reaction is adjusted so that these chain terminations occur at each of the positions in the template where the nucleotide occurs. To enable detection by autoradiography, the newly synthesized DNA is labeled, usually by including in the reaction mixture radioactively labeled dATP (for the older manual methods) or, most commonly, currently fluorescent dye terminators in the reaction mixture (now in use in automated techniques). The separation of the newly synthesized DNA strands manually is done via high-resolution denaturing polyacrylamide electrophoresis or with capillary electrophoresis in automatic sequencers. Fluorescent detection methods have enabled automation and enhanced throughput. In capillary electrophoresis, DNA molecules are driven to migrate through a viscous polymer by a high electric field to be separated on the basis of charge and size. Though this technique is based on the same principle as that used in slab gel electrophoresis, the separation is done in individual glass capillaries rather than gel slabs, facilitating loading of samples and other aspects of automation. Whereas manual methods allow the detection of about 300 nucleotide of sequence information with one set of sequencing reactions, automated methods using fluorescent dyes and laser technology can analyze 7500 or more bases per reaction. To sequence larger stretches of DNA it is necessary to divide the large piece of DNA into smaller fragments that can be individually sequenced. Alternatively, additional sequencing primers can be chosen near the end of the previous sequencing results, allowing the initiation point of new sequence data to be moved progressively along the larger DNA fragment.
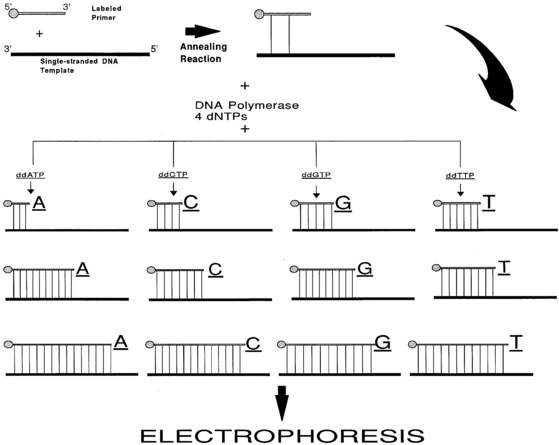
FIGURE 2-6  DNA sequencing by the dideoxy (Sanger) method. A 5’-end-labeled oligonucleotide primer with sequence complementarity to the DNA that is to be sequenced (DNA template) is annealed to a single-strand of the template DNA. This primer is elongated by DNA synthesis initiated by the addition of the enzyme DNA polymerase in the presence of the four dNTPs (2’-deoxynucleoside triphosphates) and one of the ddNTPs (2’,3’-dideoxynucleoside triphosphates); four such reaction tubes are assembled to use all the four ddNTPs. The DNA polymerase enzyme will elongate the primer using the dNTPs and the individual ddNTP present in that particular tube. Because ddNTPs are devoid of 3’ hydroxyl group, no elongation of the chain is possible when such a residue is added to the chain. Thus, each reaction tube will contain prematurely terminated chains ending at the occurrence of the particular ddNTP present in the reaction tube. The concentrations of the dNTPs and the individual ddNTP present in the reaction tubes are adjusted so that the chain termination takes place at every occurrence of the ddNTP. Following the chain elongation-termination reaction, the DNA strands synthesized are size-separated by acrylamide gel electrophoresis and the bands visualized by autoradiography.
DNA sequencing by the dideoxy (Sanger) method. A 5’-end-labeled oligonucleotide primer with sequence complementarity to the DNA that is to be sequenced (DNA template) is annealed to a single-strand of the template DNA. This primer is elongated by DNA synthesis initiated by the addition of the enzyme DNA polymerase in the presence of the four dNTPs (2’-deoxynucleoside triphosphates) and one of the ddNTPs (2’,3’-dideoxynucleoside triphosphates); four such reaction tubes are assembled to use all the four ddNTPs. The DNA polymerase enzyme will elongate the primer using the dNTPs and the individual ddNTP present in that particular tube. Because ddNTPs are devoid of 3’ hydroxyl group, no elongation of the chain is possible when such a residue is added to the chain. Thus, each reaction tube will contain prematurely terminated chains ending at the occurrence of the particular ddNTP present in the reaction tube. The concentrations of the dNTPs and the individual ddNTP present in the reaction tubes are adjusted so that the chain termination takes place at every occurrence of the ddNTP. Following the chain elongation-termination reaction, the DNA strands synthesized are size-separated by acrylamide gel electrophoresis and the bands visualized by autoradiography.
The most exciting new technique in mutation identification is pyrosequencing, which is based on an enzymatic real-time monitoring of DNA synthesis by bioluminescence; this read-as-you-go method uses nucleotide incorporation that leads to a detectable light signal from the pyrophosphate released when a nucleotide is introduced in the DNA strand.22 The rapidity and reliability of this method far exceed other contemporary DNA sequencing techniques. However, the major limitation of this technique is that it can only be used to analyze short stretches of DNA sequence.
Pyrosequencing, introduced in the early 2000s, provided the background for the explosion of new techniques collectively known as high-throughput or next-generation sequencing (NGS). NGS provides longer read length and cheaper price per base of sequencing compared to Sanger sequencing. NGS is based on the uncoupling of the traditional nucleotide-identifying enzymatic reaction and the image capture and doing so in an ever-speedier way allows for essentially unlimited capacity. Currently, two systems are being used for NGS: the SOLiD (by Applied Biosystems, Inc.) and the Illumina (Solexa) systems. The first discoveries of gene mutations for endocrine diseases exploiting NGS were published in 2011.21