Fig. 3.1
Control of HIF activity. Steady-state levels of HIFα are controlled by its rate of synthesis and degradation. The former is regulated by the TORC1 complex, which contains the mTOR kinase. This is especially true for HIF1α. The rate of degradation is under the control of pVHL. When oxygen is present, HIFα becomes prolyl hydroxylated, which marks it for polyubiquitylation by pVHL and subsequent proteasomal degradation. HIFα can dimerize with its partner protein, HIFβ (also called ARNT), and transcriptionally activate genes such as VEGF and EPO
The interaction between pVHL and HIFα requires oxygen because HIFα must be hydroxylated on one (or both) of two conserved prolyl residues in order to be recognized by pVHL [43–47]. Prolyl hydroxylation of HIFα is catalyzed by members of the EglN family [48–50], which are oxygen-dependent enzymes that serve as cellular oxygen sensors [51]. pVHL contains mutational hotspots called the alpha domain and the beta domain. The alpha domain binds directly to elongin C [30, 52], which recruits the remaining members of the ubitquitin ligase complex, and the beta domain binds directly to hydroxylated HIFα [38, 53, 54].
3.3.1 Role of HIF in Clear Cell Renal Carcinoma
There are three HIFα family members called HIF1α, HIF2α, and HIF3α. Deregulation of HIFα, in particular HIF2α, appears to be a driving force in pVHL-defective kidney cancer. For example, the risk of renal carcinoma linked to different VHL mutations correlates with the degree to which those mutations deregulate HIF [55–57]. VHL−/− renal carcinoma cells frequently silence the expression of FBP1, which is an other endogenous inhibitor of HIF activity [58].
pVHL-defective clear cell renal carcinomas overproduce HIF2α but, in some cases, fail to produce HIF1α [28, 42, 59, 60]. Production of a non-hydroxylatable version of HIF2α, but not HIF1α, can override the tumor suppressor activity of pVHL in preclinical models [61, 62]. Similarly, exogenous overexpression of HIF2α, but not HIF1α, promotes tumor formation by VHL−/− renal cancer cells [63, 64]. Moreover, downregulation of HIF2α, but not HIF1α, is sufficient to suppress tumor formation by pVHL-defective clear cell renal carcinomas [65, 66]. The appearance of HIF2α in premalignant renal lesions in patients with VHL disease heralds malignant transformation [67, 68], and a human single nucleotide polymorphism (SNP) linked to HIF2α on chromosome 2p21 has been associated with the risk of developing clear cell renal carcinomas [69]. Finally, much of the pathology observed after VHL inactivation in genetically engineered mouse models can be linked to the inappropriate accumulation of HIF2α [68, 70–75]. It should be noted that VHL inactivation, but not bona fide hypoxia, is sufficient to induce HIF2α in mouse renal tubular epithelial cells and cause renal cyst formation [68, 72, 76]. Neither VHL inactivation nor increased HIF2α activity, however, is sufficient to cause clear cell renal carcinoma in genetically engineered mouse models [68, 72, 76, 77]. This presumably reflects the need for cooperating genetic events (see below) and perhaps species differences.
As noted above, some clear cell renal carcinoma cell lines and tumors produce low, or undetectable, amounts of HIF1α. Indeed, some VHL−/− clear cell renal carcinoma lines harbor homozygous mutations of the HIF1α locus [60]. Reintroduction of wild-type HIF1α into such lines suppresses their proliferation in cell culture and in nude mice xenograft studies [60, 63, 64]. Conversely, downregulation of HIF1α in HIF1α-proficient VHL−/− clear cell renal carcinoma lines enhances their proliferation in cell culture and in xenograft assays [59, 60]. Interestingly, HIF1α resides on chromosome 14q, which is frequently deleted in clear cell renal carcinomas (together with chromosome 3p loss and chromosome 5q amplification) [60]. Clear cell renal carcinomas with chromosome 14q deletions have gene expression signatures consistent with decreased HIF1α activity [60, 78]. In some VHL−/− clear cell carcinomas that express both HIF1α and HIF2α, the ratio of HIF2α to HIF1α is enhanced by loss of specific microRNAs miR-30c-2-3p and miR-30A-3p that normally serve to repress HIF2α [79]. Finally, loss-of-function intragenic HIF1α mutations have occasionally been identified in VHL−/− clear cell renal carcinomas [60, 80–82]. Collectively, these findings suggest that HIF1α, in contrast to HIF2α, acts as a tumor suppressor in VHL−/− clear cell renal carcinoma.
In apparent disagreement with this contention, expression of a stabilized version of HIF1α, but not a stabilized version of HIF2α, in the proximal renal tubular epithelial cells of mice caused renal cell dysplasia, including evidence of increased proliferation, increased DNA damage, and clear cell histological changes [83, 84]. Similarly, ablation of VHL in primarily mouse collecting ducts caused hyperplastic changes that could be reversed by simultaneous inactivation of HIF1α [85]. Finally, it has also been shown that silencing HIF1α inhibits, rather than augments, tumor growth by human VHL+/+ renal carcinoma growth [86].
There are a number of caveats to these studies, however. For example, the cell of origin for VHL−/− clear cell renal carcinoma is still debated but likely involves a distal tubular epithelial cell that is permissive for HIF2α accumulation and the expression of specific HIF2α target genes (e.g. cyclin D1) following pVHL loss [67, 68, 87]. In this regard, forced expression of a stabilized version of HIF2α in the murine proximal renal tubule did not recapitulate the induction of HIF targets seen in VHL−/− clear cell renal carcinoma [83], perhaps because the wrong cell type was targeted. The genetically engineered mouse studies might also be confounded by biological differences between mice and men, as has been observed with many other cancer genes. Finally, the apparent dependence of human VHL+/+ renal carcinomas on HIF1α for tumor growth does not preclude a tumor suppressor role for HIF1α in VHL−/− renal carcinomas, especially bearing in mind potential differences in cell of origin and cooperating genetic events.
There are a number of quantitative and qualitative differences between HIF1α and HIF2α that could account for their seemingly antagonistic effects in VHL−/− clear cell renal carcinoma. These differences likely reflect the fact that some HIF target genes are preferentially activated by specific HIFα family members as well as by the existence of non-canonical HIF functions that are unique to specific HIFα proteins. HIF2α cooperates with c-Myc to promote the proliferation of VHL−/− clear cell renal carcinoma cells, while HIF1α is capable of inhibiting c-Myc [88–91]. Both HIF1α and HIF2α can induce REDD1 and thereby suppress the activity of the TORC1 complex, which contains mTOR, and Cap-dependent translation [92–95]. HIF2α, however, and not HIF1α, can also stimulate translation. HIF2α transcriptionally induces the amino acid transporter SLC7A5 and thereby increases intracellular amino acid availability, which activates TORC1 [96]. In addition, HIF2α forms a complex with RBM4 and eIF4E that promotes Cap-dependent translation in cells with depressed TORC1 activity [97]. HIF1α and HIF2α also appear to differentially regulate p53 and the DNA damage response [59, 63, 98, 99].
pVHL has a number of other functions that, although incompletely understood biochemically, appear to be a least partly HIF-independent. These include a role in the maintenance of a specialized structure called the primary cilium on the cell surface that serves as a mechanosensor [76, 100–103], possibly by virtue of pVHL’s role in stabilization of microtubules [104–106]. Interestingly, a number of diseases characterized by visceral cyst formation, including VHL disease, are caused by mutations that disrupt the primary cilium [107, 108]. pVHL also suppresses autophagy via both HIF-independent and HIF-dependent pathways, perhaps contributing to the increased autophagy seen in clear cell renal carcinomas [109, 110]. In addition, pVHL plays roles in extracellular matrix formation by fibronectin [111–114], epithelial-epithelial contacts [115, 116], NFκB signaling [117–120], control of atypical PKC activity [121–125], Rpb1 expression and activity [126–128], receptor internalization [129–131], and mRNA turnover [20, 26, 132–135]. It is possible that these other functions also contribute to tumor suppression by pVHL.
3.3.2 Cooperating Events
It is clear that pVHL loss is an important, but not sufficient, step in renal carcinogenesis. This is most clearly demonstrated by studies of the natural history of von Hippel-Lindau disease. Patients with von Hippel-Lindau disease can develop hundreds of premalignant renal cysts, very few of which will go on to become clear cell renal carcinomas [67, 136] (Fig. 3.2). This bottleneck presumably reflects the requirement for additional genetic events, occurring stochastically, to fully transform renal epithelial cells. Indeed, a number of nonrandom genomic abnormalities have been described in clear cell renal carcinoma including, most notably, 5q amplification and 14q loss [6, 137–143] (Fig. 3.2). The triad of 3p loss, 14q loss, and 5q gain is a signature of clear cell renal carcinoma, and some clear cell renal carcinomas have unbalanced translocations involving 3p and 5q that result in loss of 3p and gain of 5q sequences [60, 144–150].
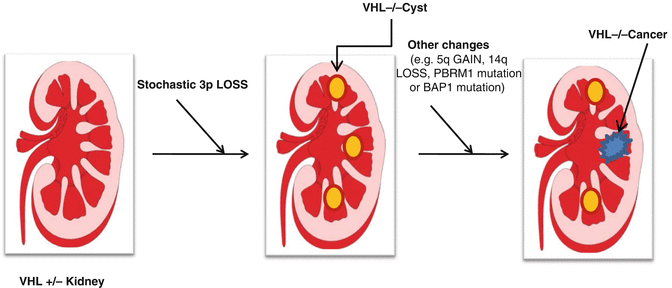
Fig 3.2
Development of renal cell carcinoma in VHL patients. VHL patients are VHL heterozygotes, having one normal VHL allele and one defective allele. Loss of the remaining normal allele in kidney cells, occurring stochastically, leads to the development of preneoplastic renal cysts. A minority of such cysts will ultimately accumulate additional genetic changes and become clear cell renal carcinomas. Such genetic changes include gain of 5q, loss of 14q, as well as intragenic mutations of specific genes such as PBRM1 or BAP1
Loss of chromosome 3p, which harbors the VHL tumor suppressor gene, is the most common genetic event in kidney cancer. Chromosome 3p has been suspected for many years, however, to contain at least one additional kidney cancer suppressor gene. Indeed, it is now clear that 3p harbors several renal cancer suppressor genes other than VHL including PBRM1, which encodes the BAF180 chromatin-associated protein; SETD2, which encodes a histone H3 lysine 36 methyltransferase; and BAP1, which encodes a ubiquitin hydrolase [82, 151–156] (Fig. 3.3). PBRM1 is, after VHL, the most frequently mutated gene in clear cell renal carcinoma. PBRM1 and BAP1 mutations are largely mutually exclusive and appear to define clinically distinct subgroups of renal cancers [152, 157, 158].
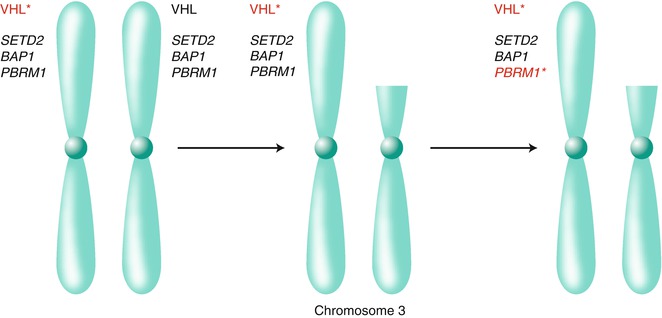
Fig 3.3
Chromosome 3p harbors multiple renal cancer suppressors. Biallelic inactivation of the VHL tumor suppressor gene on chromosome 3p, usually as the result of intragenic mutation (indicated by the asterisk) followed by loss of the remaining wild-type allele because of a gross 3p deletion, is a critical early event in most clear cell renal carcinomas. The 3p deletions in clear cell renal carcinoma typically span VHL, on 3p25, as well as the additional renal cancer suppressors SETD2, BAP1, and PBRM1 on 3p21. As a result, subsequent intragenic mutations of these genes deprive renal cells of their wild-type protein products (for illustrative purposes PBRM1 is shown to be mutated)
As described above, HIF1α is a likely target of the 14q deletions in VHL−/− clear cell renal carcinomas. These deletions are very large, however, suggesting there are additional renal cancer suppressor genes located at 14q. It should also be noted that most 14q deleted VHL−/− clear cell renal tumors (in contrast to cell lines) appear to retain a wild-type HIF1α allele [60]. This suggests that HIF1α is a haploinsufficient clear cell renal carcinoma suppressor and that loss of the remaining allele is associated with tumor progression in vivo or establishment of cell lines ex vivo.
SQSTM1, encoding p62, appears to be one of the renal carcinoma 5q oncogenes [159]. Increased expression of p62 promotes the growth of VHL−/− renal carcinoma cells in cell culture and tumor xenograft assays and increases their resistance to redox stress [159]. p62 plays important roles in autophagy and also signals to renal carcinoma relevant proteins including NRF2, NFκB, and mTOR [160–162].
Sequencing of kidney cancer genomes has identified additional genes that, when mutated, contribute to renal carcinogenesis including several more genes linked to chromatin regulation such as JARID1C (also known as KDM5C), which encodes a histone H3 lysine 4 demethylase; UTX (KMD6A), which encodes a histone H3 lysine 27 demethylase; and ARID1A, a component of a chromatin remodeling complex [82, 151–156, 163]. Notably, many histone demethylase genes are themselves transcriptionally induced by HIF [164–169]. It is possible that their inappropriate expression pursuant to VHL loss alters chromatin structure and creates the selection pressure to mutate specific chromatin regulators.
Genes linked to the mTOR pathway including PIK3CA, PTEN, TSC1, TSC2, and MTOR itself are occasionally mutated in clear cell renal carcinomas [7, 82, 152, 155, 156]. Preliminary data suggest that such mutations identify a subset of renal cell carcinoma patients more likely to derive significant benefit from TORC1 inhibitors [170].
The NFE2L gene, encoding NRF2, and the NRF2-negative regulator KEAP1 are occasionally mutated in clear cell renal carcinoma [82, 159]. Such mutations appear to be mutually exclusive with higher level SQSTM1 amplification [159]. Genes involved in the response to DNA damage, including p53, MDM4, and ATM, are also occasionally mutated in clear cell renal carcinoma [7, 82, 155, 156]. p53 loss cooperates with Vhl loss in mouse models to promote renal carcinogenesis [171].
3.3.3 Treatment of Renal Cell Carcinoma: HIF Antagonists
The preclinical data outlined above suggest that drugs that inhibit HIF, and particularly HIF2α, would have antitumor activity in kidney cancer. Unfortunately, DNA-binding transcription factors, with the exception of the steroid hormone receptors, have historically been difficult to target with drug-like small molecules. Nonetheless, a number of approaches to targeting HIF are being explored in the laboratory, including the use of DNA-binding polyamides [172–174] and short interfering RNAs [175]. Moreover, HIF2α, but not HIF1α, has a potentially druggable pocket, and lead compounds have been identified that can inhibit HIF2α in biochemical, cell-based, and animal models [176–178].
3.3.4 Treatment of Renal Cell Carcinoma: mTOR Inhibitors
mTOR participates in two complexes, called TORC1 and TORC2 [179]. The former can be inhibited with rapamycin-like drugs. Two such drugs, temsirolimus and everolimus, have been FDA approved for the treatment of renal cell carcinoma based on positive randomized clinical trial data [180, 181]. In theory the activity of these agents reflects direct effects on tumor cells, including modulation of HIF [182], and effects downstream of VEGF signaling in endothelial cells (see below). In preclinical models, VHL−/− renal carcinoma lines are more sensitive to rapamycin than are their pVHL-proficient counterparts [183]. As noted above, preliminary data suggest that concurrent mutations of the PI3K-MTOR pathway are enriched among renal carcinoma patients who exhibit the greatest clinical benefit from rapamycin-like drugs [170].
Two factors might, however, limit the effectiveness of rapamycin-like drugs in the treatment of renal cell carcinoma. First, the TORC1 complex feedback inhibits signaling by certain receptor tyrosine kinases [184, 185, 185a, 186–188]. As a result, treatment of tumor cells with rapamycin-like drugs can cause a paradoxical increase in receptor kinase activity leading to activation of TORC2, which is relatively rapamycin resistant, PI3K, and AKT, all of which might promote tumor growth [184, 185, 185a, 186–188]. Second, inhibition of TORC1 appears to preferentially inhibit HIF1α, which as argued above appears to act a renal cell carcinoma suppressor, rather than HIF2α [189]. In contrast, inhibition of TORC2 preferentially affects HIF2α [189]. Second-generation, ATP-like, mTOR inhibitors can inhibit both TORC1 and TORC2 and hence might be more active than rapamycin-like drugs in the treatment of clear cell renal carcinoma [190, 191]. Emerging preclinical data support such a view [192].
3.3.5 Treatment of Renal Cell Carcinoma: Angiogenesis Inhibitors
3.3.5.1 VEGF
Renal cell carcinoma is one of the most angiogenic solid tumors. Indeed, renal angiography was once an important tool to diagnose this neoplasm. Renal cell carcinoma hypervascularity reflects the overproduction of HIF-dependent angiogenic factors such as VEGF. Notably, the remarkable upregulation of VEGF observed upon pVHL loss, and consequent increase in new blood vessel production, probably diminishes the selection pressure to upregulate additional angiogenic factors in this setting. In contrast, a host of angiogenic factors in addition to, or instead of, VEGF likely contributes to neoangiogenesis associated with other solid tumor types.
In keeping with this view, a variety of drugs that inhibit VEGF, such as bevacizumab, or its receptor KDR, such as sorafenib, sunitinib, axitinib, and pazopanib, have now demonstrated significant activity in the treatment of renal cell carcinoma and were approved by the FDA [193–197]. These agents induce significant disease stabilization and, in some cases, frank regressions. Newer VEGF inhibitors that are more potent, more specific, or both are in various stages of development. It is anticipated that greater potency will translate into greater clinical efficacy although there might be limits regarding the degree to which VEGF signaling can be safely interrupted in man. Microangiopathic hemolytic anemia was observed in patients in which two VEGF inhibitors were combined [198, 199], and both preclinical and clinical data suggest that chronic VEGF inhibition could lead to cardiomyopathic changes [200, 201]. Developing VEGF inhibitors that exhibit greater specificity is important because some of the existing agents are difficult to combine with other agents, presumably because of their off-target effects. The history of curative cancer therapy suggests that the eventual cure of renal cell carcinoma will require a combination of agents that have novel mechanisms of action and that are non-cross resistant. A VEGF inhibitor will probably be cornerstone of such a combination.
In the simplest view, pVHL status would serve as a predictive biomarker, with VEGF inhibitors being more active in pVHL-defective renal cell carcinomas than in pVHL-proficient tumors. Although some studies support this contention, others do not [202–205]. This lack of consistency might be due, at least partly, to technical differences related to how pVHL status was determined and how therapeutic response was measured. It appears that the vast majority of clear cell renal carcinomas (especially those that do not exhibit mixed histological patterns with areas of non-clear cell features) have transcriptional signatures indicative of pVHL inactivation and HIF activation, including some without demonstrable VHL mutations or hypermethylation [6]. Studies with newer sequencing platforms suggest that some of these tumors do, indeed, have VHL mutations that would be missed using conventional DNA sequencing approaches [4, 7]. Suffice it to say that VHL status is not currently a sufficient robust predictive biomarker to be used in clinical decision-making.
3.3.5.2 PDGF
Platelet-derived growth factor B (hereafter called PDGF) is another well-studied HIF target [206, 207]. PDGF supports the expansion of pericytes that surround new blood vessels and provide survival signals to the associated endothelial cells. In preclinical models, newly sprouting blood vessels that lack pericyte coverage are more sensitive to VEGF blockade than are more mature vessels that are associated with pericytes [208–210]. This might explain why the objective tumor response (regression) rate in renal cell carcinoma is higher with small-molecule KDR inhibitors, many of which inhibit PDGFR, than with bevacizumab, which solely inhibits VEGF. On the other hand, it should be borne in mind that PDGFR inhibitors such as imatinib mesylate have not yet demonstrated utility as single agents in renal cell carcinoma and have not been shown to enhance the activity of bevacizumab [211–213]. Moreover, many of the existing KDR inhibitors might have off-target effects other than PDGFR inhibition that fortuitously contribute to their antitumor activity.
3.3.5.3 IL-8
VEGF inhibitors, although highly active in renal cell carcinoma, are not curative as single agents, and renal cell carcinoma patients treated with these agents will eventually experience disease progression. The mechanisms underlying de novo or acquired resistance to VEGF inhibitors are poorly understood at the molecular level. One study suggested that upregulation of the angiogenic cytokine IL-8, which cooperates with VEGF in some settings [214], contributes to resistance to VEGF inhibitors [215] and IL-8 polymorphisms and circulating IL-8 levels have been linked to clinical outcomes in patients treated with VEGF inhibitors [216, 217]. Interestingly, IL-8 is regulated by HIF and NFκB, both of which are controlled by pVHL [214, 218–222] (Fig. 3.3). These considerations warrant exploration of inhibitors of IL-8, or its receptors CXCR1 and CXCR2, in renal cell carcinoma.
3.3.5.4 TIE2
The receptor tyrosine kinase TIE2 plays an important role in angiogenesis [223]. Activation of TIE2 by ligands such as angiopoietin 1 stabilizes blood vessels, while antagonists such as angiopoietin 2 destabilize blood vessels, rendering them permissive for sprouting and new blood vessel formation but also hyperdependent on VEGF as a survival factor. Although there have been conflicting reports on the regulation of angiopoietins by pVHL [224, 225], knowledge of TIE2 biology suggests that dual inhibition of VEGF and TIE2 might block angiogenesis more effectively than would VEGF blockade alone. Circulating levels of a soluble form of TIE2 have also been touted as a means of monitoring antiangiogenic therapy in this patient population [226]. Unfortunately, the TIE2 antagonist AMG386 in combination with the VEGFR inhibitor sorafenib was not more active than sorafenib alone [227].
3.3.5.5 CXCR4 and SDF
Both CXCR4 and its ligand, CXCL12/SDF, are HIF targets and upregulated in pVHL-defective tumors [228, 229]. In some mouse models, blocking CXCR4 inhibits the recruitment of circulating bone marrow-derived cells that can contribute to new blood vessel formation and can enhance the antiangiogenic activity of VEGF inhibitors [230]. CXCR4 might also play cell autonomous roles in renal cell carcinoma invasion and metastasis. In this regard, neutralizing antibodies to CXCL12 were shown to decrease metastasis, without affecting angiogenesis, in an orthotopic renal tumor model in mice [231]. Conversely, upregulation of CXCR4 on an epigenetic basis was associated with increased renal cell carcinoma metastasis [232].
3.3.6 Treatment of Renal Cell Carcinoma: Tumor Cell Receptor Tyrosine Kinases
3.3.6.1 EGFR
Renal cell carcinomas frequently overexpress EGFR and its ligand TGFα [233–236]. TGFα is a transcriptional HIF target, while HIF has been reported to increase the rate of EGFR translation [97, 237, 238]. In addition, pVHL loss might decrease the rate of EGFR internalization and recycling [129]. In preclinical models, inhibiting EGFR decreases tumor growth in vivo [239, 240].
Despite these observations, EGFR inhibitors have been very disappointing in the treatment of renal cell carcinoma, both alone and in combination with VEGF inhibitors [241, 242]. Why have EGFR inhibitors failed thus far in the clinic? One possibility, in addition to a possible failure to achieve adequate EGFR inhibition in vivo, stems from recent work showing that c-MET, which is frequently active in renal cell carcinoma (see below), can confer resistance to EGFR blockade [243–245]. Preclinical xenograft studies performed in mice frequently underestimate the importance of c-MET because mouse HGF, the ligand for c-MET, does not activate human c-MET (present on implanted human tumor cells) [246, 247].
3.3.6.2 c-MET
pVHL-defective tumor cells exhibit increased c-MET activity and are hypersensitive to HGF [248–250]. Precisely how pVHL regulates c-MET is somewhat controversial, with some report suggesting c-MET is a HIF target [250–252] and others focusing on the effects of pVHL on signaling downstream of c-MET [248, 249]. Interestingly, activating germline MET mutations are linked to the development of papillary renal cell carcinoma [253]. HGF and c-MET play an important role in both tumorigenesis and angiogenesis. pVHL-defective tumor cells are hypersensitive to c-MET loss [254], and inhibition of c-MET might, for the reasons outlined above, augment the activity of EGFR inhibitors. Cabozantinib (XL184), which inhibits both VEGFR and c-MET, demonstrated clinical activity in heavily pretreated renal cell carcinoma patients who had failed prior VEGF inhibitor therapy in a phase 1 study [255]. To what extent these responses were due specifically to c-Met inhibition remains to be determined.
3.3.6.3 IGFR
HIF upregulates IGF-1 and IGF-2 as well as IGFB-2 and IGFB-3 [256, 257]. pVHL, in a HIF-independent manner, downregulates IGFR levels by inhibiting SP1 and the RNA-binding protein HuR [134] and IGFR-dependent signaling through PKCδ [123, 124]. Inhibition of IGFR sensitizes renal cell carcinoma cells to cytotoxic drugs as well as to rapamycin-like drugs [258]. This latter observation might relate to the role of rapamycin in feedback inhibition of receptor tyrosine kinase signaling, as described above. In addition, downregulation of IGFR-1 with shRNA technology decreases VHL−/− renal carcinoma growth in nude mouse xenograft assays [259].
3.3.6.4 ROR2
ROR2 (RTK-like orphan receptor 2) was identified in an unbiased screen for receptor tyrosine kinases that are upregulated and activated by pVHL loss in renal carcinoma cells [260, 261]. The biological functions of ROR2 are incompletely understood although it has been linked to tumor cell invasiveness through the upregulation of matrix metalloproteinases and may act as a receptor for Wnt ligands. Inhibition of ROR2 in renal carcinoma cells with short hairpin RNAs suppresses tumor growth in orthotopic tumor models [261].
3.3.7 Other Targets
3.3.7.1 Cdk4/6
Deregulation of HIF2α in renal cell carcinoma cells drives the overproduction of the cyclin D1 oncoprotein that, together with the cdk4 or cdk6 kinase, promotes cell-cycle progression [64, 87, 262, 263]. In contrast, hypoxia and HIF activation lowers cyclin D1 levels in most other cell types [87]. Some renal cell carcinomas have also sustained deletions of the INK4A tumor suppressor protein [6, 138, 140], which acts as an inhibitor of cdk4 and cdk6, and pVHL-defective tumor cells appear to be hypersensitive to loss of cdk6 in vitro [254]. Moreover, cdk6 is located on a large region of chromosome 7 that is amplified in a subset of renal cell carcinomas [6]. Downregulation of cyclin D1 with shRNA technology is sufficient to inhibit tumor formation by VHL−/− renal carcinoma cells in mouse models [259]. Although a relatively promiscuous cdk inhibitor was relatively ineffective in the treatment of kidney cancer at maximally tolerated doses, newer, more selective cdk inhibitors targeted against cdk4 and cdk6 might now be explored for this indication [264, 265].
3.3.7.2 NFκB
pVHL suppresses NFκB via HIF-dependent and HIF-independent pathways [117–120, 266]. With respect to the latter, pVHL, bound to casein kinase 2, promotes the inhibitory phosphorylation of the NFκB agonist Card9 [120]. NFκB activity is increased in human renal cell carcinoma and might contribute to both tumor development and therapeutic resistance [267, 268]. HIF and NFκB coregulate targets such as cyclin D1 and VEGF, and preclinical studies suggest that inhibiting NFκB activity, such as might be achieved with inhibitors of IKK, would have salutary effects in the treatment of kidney cancer [269].
3.3.7.3 IL6
Renal cell carcinomas frequently overexpress interleukin 6, which is suspected of acting as an autocrine growth factor in this disease [270–272]. Binding of IL-6 to its receptor activates the JAK-STAT pathway that, in turn, can stimulate renal carcinoma cell proliferation [273]. IL-6 was shown to be pVHL responsive in one study [262] and has been implicated as both a prognostic biomarker in clear cell renal carcinoma and as a predictive biomarker for clear cell renal carcinoma patients being treated with the VEGFR inhibitor pazopanib [217]. A neutralizing antibody against IL-6 stabilized disease in approximately 50 % of patients with metastatic renal cancer in a phase 2 study [274].
3.3.8 Carbonic Anhydrase and Lactate Dehydrogenase
HIF1α upregulates a number of genes that promote glycolysis and lactate acid production. This potentially places a burden on pVHL-defective tumor cells to maintain pH homeostasis. Preclinical studies suggest that inhibition of lactate dehydrogenase A or carbonic anhydrase IX, both of which are HIF targets, would be a viable therapeutic strategy for treating pVHL-defective renal cell carcinomas [275–278].
3.3.9 Histone Methylases and Demethylases
Resequencing of renal cell carcinoma genomes has identified mutations affecting enzymes that regulate histone methylation, as described above. In addition, HIF transcriptionally activates a number of histone demethylases including JMJD1A, JMJD2B, and JARID1B [164–169]. In one study, inhibition of JMJD1A with a short hairpin RNA inhibited renal carcinoma growth [168]. Histone methylases and demethylases can, in principle, be inhibited with drug-like small molecules, and the identification of these enzymes as mutational targets in renal cell carcinoma and other neoplasms is motivating a deeper understanding of their biological functions as well as nascent drug discovery efforts.
3.3.9.1 CTLA4 and PD1
It has been appreciated for decades that renal cell carcinoma has a highly variable natural history and that some patients can experience spontaneous regressions. Although the mechanisms underlying such spontaneous regressions are unknown, a role for the immune system has been suspected. Moreover, immune modulators have been used in the treatment of this disease for many years, including high-dose interleukin 2 [279]. High-dose interleukin 2 can induce durable remissions in patients with metastatic kidney cancer. Unfortunately, this therapy is sufficiently toxic that it should only be given at specialized care centers, and it is impossible to predict the small subset of patients who will achieve such lasting remissions.
A growing appreciation of the signals that are used by tumor cells to evade immune recognition has led to new cancer immunotherapeutic agents, including antibodies directed against CTLA4 and PD1, which are proteins that serve to dampen the immune response. Interestingly, a particular CTLA4 polymorphism was found in one study to be associated with the risk of developing renal cell carcinoma [280].
Anti-CTLA4 has demonstrated activity in the treatment of renal cell carcinoma and is now being explored in combinations [281, 282]. A cautionary note is that acute renal failure was observed when anti-CTLA4 was combined with sunitinib [282]. Early data with anti-PD1 antibodies for the treatment of renal cell carcinoma also appear promising [283–285].
It is not yet known whether pVHL loss influences the recognition of tumor cells by the immune system although VEGF has, itself, been implicated as an immune suppressant [286–288]. Moreover, HIF stimulates the production of adenosine, which can suppress the immune response via the A2A adenosine receptor [289, 290]. Future therapies against renal cell carcinoma will likely involve combinations of agents that directly kill tumor cells with agents that enhance the host immune response.
3.4 Conclusions
Renal cell carcinoma is a common cancer that, historically, has been refractory to therapy with standard chemotherapeutic agents and radiation. High-dose interleukin-2 can induce durable remissions in a small subset of patients, but it is impossible to predict which patients will benefit from this toxic and expensive form of therapy. The von Hippel-Lindau tumor suppressor protein, pVHL, is frequently inactivated in clear cell renal carcinoma, which is the most common form of kidney cancer. The knowledge that pVHL inhibits the HIF transcription factor provided a conceptual framework for drugs that inhibit the HIF-responsive gene product VEGF. The clinical activity of mTOR inhibitors might also relate to HIF biology because mTOR regulates HIF synthesis and might also act downstream of VEGF. A number of other HIF-responsive gene products are also known or suspected of playing roles in tumorigenesis and are worthy of exploration as kidney cancer drug targets. Elucidation of the genetic events that cooperate with pVHL loss in clear cell carcinoma will hopefully yield additional targets. In this regard, it is anticipated that the frequent occurrence of mutations affecting chromatin regulatory proteins in clear cell renal carcinoma will create exploitable therapeutic vulnerabilities. Finally, there is a growing appreciation of how, mechanistically, clear cell renal carcinoma subverts immune recognition. The studies, in total, should provide a platform for the design and testing of effective therapeutic combinations for this disease.
References
2.
Latif F, Tory K, Gnarra J, Yao M, Duh F-M, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L, Schmidt L, Zhou F, Li H, Wei MH, Chen F, Glenn G, Choyke P, Walther MM, Weng Y, Duan D-SR, Dean M, Glavac D, Richards FM, Crossey PA, Ferguson-Smith MA, Pasiler DL, Chumakov I, Cohen D, Chinault AC, Maher ER, Linehan WM, Zbar B, Lerman MI (1993) Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 260:1317–1320PubMed
3.
Kim WY, Kaelin WG (2004) The role of VHL gene mutation in human cancer. J Clin Oncol 22(24):4991–5004PubMed
4.
Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko V, Navratilova M, Szeszenia-Dabrowska N, Mates D, Mukeria A, Holcatova I, Schmidt LS, Toro JR, Karami S, Hung R, Gerard GF, Linehan WM, Merino M, Zbar B, Boffetta P, Brennan P, Rothman N, Chow WH, Waldman FM, Moore LE (2008) Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res 14(15):4726–4734PubMedCentralPubMed
5.
Young AC, Craven RA, Cohen D, Taylor C, Booth C, Harnden P, Cairns DA, Astuti D, Gregory W, Maher ER, Knowles MA, Joyce A, Selby PJ, Banks RE (2009) Analysis of VHL gene alterations and their relationship to clinical parameters in sporadic conventional renal cell carcinoma. Clin Cancer Res 15(24):7582–7592PubMedCentralPubMed
6.
Beroukhim R, Brunet JP, Di Napoli A, Mertz KD, Seeley A, Pires MM, Linhart D, Worrell RA, Moch H, Rubin MA, Sellers WR, Meyerson M, Linehan WM, Kaelin WG Jr, Signoretti S (2009) Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res 69(11):4674–4681PubMedCentralPubMed
7.
Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, Martinez P, Phillimore B, Begum S, Rabinowitz A, Spencer-Dene B, Gulati S, Bates PA, Stamp G, Pickering L, Gore M, Nicol DL, Hazell S, Futreal PA, Stewart A, Swanton C (2014) Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 46(3):225–233PubMed
8.
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366(10):883–892PubMed
9.
Fisher R, Horswell S, Rowan A, Salm MP, de Bruin EC, Gulati S, McGranahan N, Stares M, Gerlinger M, Varela I, Crockford A, Favero F, Quidville V, Andre F, Navas C, Gronroos E, Nicol D, Hazell S, Hrouda D, OB T, Matthews N, Phillimore B, Begum S, Rabinowitz A, Biggs J, Bates PA, McDonald NQ, Stamp G, Spencer-Dene B, Hsieh JJ, Xu J, Pickering L, Gore M, Larkin J, Swanton C (2014) Development of synchronous VHL syndrome tumors reveals contingencies and constraints to tumor evolution. Genome Biol 15(8):433PubMedCentralPubMed
10.
Sankin A, Hakimi AA, Mikkilineni N, Ostrovnaya I, Silk MT, Liang Y, Mano R, Chevinsky M, Motzer RJ, Solomon SB, Cheng EH, Durack JC, Coleman JA, Russo P, Hsieh JJ (2014) The impact of genetic heterogeneity on biomarker development in kidney cancer assessed by multiregional sampling. Cancer Med 3(6):1485–1492PubMedCentralPubMed
11.
Schoenfeld A, Davidowitz E, Burk R (1998) A second major native von Hippel-Lindau gene product, initiated from an internal translation start site, functions as a tumor suppressor. Proc Natl Acad Sci U S A 1(95):8817–8822
12.
Iliopoulos O, Ohh M, Kaelin W (1998) pVHL19 is a biologically active product of the von Hippel-Lindau gene arising from internal translation initiation. Proc Natl Acad Sci U S A 95:11661–11666PubMedCentralPubMed
13.
Blankenship C, Naglich J, Whaley J, Seizinger B, Kley N (1999) Alternate choice of initiation codon produces a biologically active product of the von Hippel Lindau gene with tumor suppressor activity. Oncogene 18:1529–1535PubMed
14.
Corless CL, Kibel A, Iliopoulos O, Kaelin WGJ (1997) Immunostaining of the von Hippel-Lindau gene product (pVHL) in normal and neoplastic human tissues. Hum Pathol 28:459–464PubMed
15.
Iliopoulos O, Kibel A, Gray S, Kaelin WG (1995) Tumor suppression by the human von Hippel-Lindau gene product. Nat Med 1(8):822–826PubMed
16.
Lee S, Chen DYT, Humphrey JS, Gnarra JR, Linehan WM, Klausner RD (1996) Nuclear/cytoplasmic localization of the von Hippel-Lindau tumor suppressor gene product is determined by cell density. Proc Natl Acad Sci 93:1770–1775PubMedCentralPubMed
17.
Lee S, Neumann M, Stearman R, Stauber R, Pause A, Pavlakis G, Klausner R (1999) Transcription-dependent nuclear-cytoplasmic trafficking is required for the function of the von Hippel-Lindau tumor suppressor protein. Mol Cell Biol 19(2):1486–1497PubMedCentralPubMed
18.
Shiao YH, Resau JH, Nagashima K, Anderson LM, Ramakrishna G (2000) The von Hippel-Lindau tumor suppressor targets to mitochondria. Cancer Res 60(11):2816–2819PubMed
19.
Schoenfeld A, Davidowitz E, Burk R (2001) Endoplasmic reticulum/cytosolic localization of von Hippel-Lindau gene products is mediated by a 64-amino acid region. Int J Cancer 91:457–467PubMed
20.
Gnarra JR, Zhou S, Merrill MJ, Wagner J, Krumm A, Papavassiliou E, Oldfield EH, Klausner RD, Linehan WM (1996) Post-transcriptional regulation of vascular endothelial growth factor mRNA by the VHL tumor suppressor gene product. Proc Natl Acad Sci 93:10589–10594PubMedCentralPubMed
21.
Baba M, Hirai S, Kawakami S, Kishida T, Sakai N, Kaneko S, Yao M, Shuin T, Kubota Y, Hosaka M, Ohno S (2001) Tumor suppressor protein VHL is induced at high cell density and mediates contact inhibition of cell growth. Oncogene 20(22):2727–2736PubMed
22.
Davidowitz E, Schoenfeld A, Burk R (2001) VHL induces renal cell differentiation and growth arrest through integration of cell-cell and cell-extracellular matrix signaling. Mol Cell Biol 21:865–874PubMedCentralPubMed
23.
Mohan S, Burk RD (2003) von Hippel-Lindau protein complex is regulated by cell density. Oncogene 22(34):5270–5280PubMed
24.
Lieubeau-Teillet B, Rak J, Jothy S, Iliopoulos O, Kaelin W, Kerbel R (1998) von Hippel-Lindau gene-mediated growth suppression and induction of differentiation in renal cell carcinoma cells grown as multicellular tumor spheroids. Cancer Res 58:4957–4962PubMed
25.
Pause A, Lee S, Lonergan KM, Klausner RD (1998) The von Hippel-Lindau tumor suppressor gene is required for cell cycle exit upon serum withdrawal. Proc Natl Acad Sci U S A 95:993–998PubMedCentralPubMed
26.
Iliopoulos O, Jiang C, Levy AP, Kaelin WG, Goldberg MA (1996) Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci 93:10595–10599PubMedCentralPubMed
27.
Krieg M, Marti H, Plate KH (1998) Coexpression of erythropoietin and vascular endothelial growth factor in nervous system tumors associated with von hippel-lindau tumor suppressor gene loss of function. Blood 92(9):3388–3393PubMed
28.
Maxwell P, Weisner M, Chang G-W, Clifford S, Vaux E, Pugh C, Maher E, Ratcliffe P (1999) The von Hippel-Lindau gene product is necessary for oxygen-dependent proteolysis of hypoxia-inducible factor a subunits. Nature 399:271–275PubMed
29.
Siemeister G, Weindel K, Mohrs K, Barleon B, Martiny-Baron G, Marme D (1996) Reversion of deregulated expression of vascular endothelial growth factor in human renal carcinoma cells by von Hippel-Lindau tumor suppressor protein. Cancer Res 56:2299–2301PubMed
30.
Kibel A, Iliopoulos O, DeCaprio JD, Kaelin WG (1995) Binding of the von Hippel-Lindau tumor suppressor protein to elongin B and C. Science 269:1444–1446PubMed
31.
Duan DR, Humphrey JS, Chen DYT, Weng Y, Sukegawa J, Lee S, Gnarra JR, Linehan WM, Klausner RD (1995) Characterization of the VHL tumor suppressor gene product: localization, complex formation, and the effect of natural inactivating mutations. Proc Natl Acad Sci U S A 92:6495–6499
32.
Duan DR, Pause A, Burgress W, Aso T, Chen DYT, Garrett KP, Conaway RC, Conaway JW, Linehan WM, Klausner RD (1995) Inhibition of transcriptional elongation by the VHL tumor suppressor protein. Science 269:1402–1406PubMed
33.
Lonergan KM, Iliopoulos O, Ohh M, Kamura T, Conaway RC, Conaway JW, Kaelin WG (1998) Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau protein requires binding to complexes containing elongins B/C and Cul2. Mol Cell Biol 18:732–741PubMedCentralPubMed
34.
Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, Lane WS, Kaelin WGJ, Elledge SJ, Conaway RC, Harper JW, Conaway JW (1999) Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science 284:657–661PubMed
35.
Kishida T, Stackhouse TM, Chen F, Lerman MI, Zbar B (1995) Cellular proteins that bind the von Hippel-Lindau disease gene product: mapping of binding domains and the effect of missense mutations. Cancer Res 55:4544–4548PubMed
36.
Lisztwan J, Imbert G, Wirbelauer C, Gstaiger M, Krek W (1999) The von Hippel-Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity. Genes Dev 13:1822–1833PubMedCentralPubMed
37.
Iwai K, Yamanaka K, Kamura T, Minato N, Conaway R, Conaway J, Klausner R, Pause A (1999) Identification of the von hippel-lindau tumor-suppressor protein as part of an active E3 ubiquitin ligase complex. Proc Natl Acad Sci U S A 96:12436–12441PubMedCentralPubMed
38.
Ohh M, Park CW, Ivan M, Hoffman MA, Kim T-Y, Huang LE, Chau V, Kaelin WG (2000) Ubiquitination of HIF requires direct binding to the von Hippel-Lindau protein beta domain. Nat Cell Biol 2:423–427PubMed
39.
Kamura T, Sato S, Iwain K, Czyzyk-Krzeska M, Conaway RC, Conaway JW (2000) Activation of HIF1a ubiquitination by a reconstituted von Hippel-Lindau tumor suppressor complex. Proc Natl Acad Sci U S A 97:10430–10435PubMedCentralPubMed
40.
Cockman M, Masson N, Mole D, Jaakkola P, Chang G, Clifford S, Maher E, Pugh C, Ratcliffe P, Maxwell P (2000) Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem 275:25733–25741PubMed
41.
Tanimoto K, Makino Y, Pereira T, Poellinger L (2000) Mechanism of regulation of the hypoxia-inducible factor-1alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J 19:4298–4309PubMedCentralPubMed
42.
Krieg M, Haas R, Brauch H, Acker T, Flamme I, Plate K (2000) Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 19:5435–5443PubMed
43.
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara J, Lane W, Kaelin WJ (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292:464–468PubMed
44.
Jaakkola P, Mole D, Tian Y, Wilson M, Gielbert J, Gaskell S, Kriegsheim A, Hebestreit H, Mukherji M, Schofield C, Maxwell P, Pugh C, Ratcliffe P (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292:468–472PubMed
45.
Yu F, White S, Zhao Q, Lee F (2001) HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci U S A 98:9630–9635PubMedCentralPubMed
46.
Masson N, Willam C, Maxwell P, Pugh C, Ratcliffe P (2001) Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO 20(18):5197–5206
47.
Chan DA, Sutphin PD, Denko NC, Giaccia AJ (2002) Role of prolyl hydroxylation in oncogenically stabilized hypoxia-inducible factor-1alpha. J Biol Chem 277(42):40112–40117PubMed
48.
Epstein A, Gleadle J, McNeill L, Hewitson K, O’Rourke J, Mole D, Mukherji M, Metzen E, Wilson M, Dhanda A, Tian Y, Masson N, Hamilton D, Jaakkola P, Barstead R, Hodgkin J, Maxwell P, Pugh C, Schofield C, Ratcliffe P (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107:43–54PubMed
49.
Related posts:
 Genetic Heterogeneity of Kidney Cancer
Angiogenesis Inhibitor Therapy in Renal Cell Cancer
Presurgical Therapy in Renal Cell Carcinoma
Genetic Heterogeneity of Kidney Cancer
Angiogenesis Inhibitor Therapy in Renal Cell Cancer
Presurgical Therapy in Renal Cell Carcinoma
 The Role of mTOR Inhibitors and PI3K Pathway Blockade in Renal Cell Cancer
The Role of mTOR Inhibitors and PI3K Pathway Blockade in Renal Cell Cancer
 Renal Cell Carcinoma: Clinical Presentation, Staging, and Prognostic Factors
Renal Cell Carcinoma: Clinical Presentation, Staging, and Prognostic Factors
 Hereditary Kidney Cancer Syndromes
Hereditary Kidney Cancer Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


