Molecular Biology and Prognostic Markers in Bladder Cancer
Anirban P. Mitra
Dan Theodorescu
Seth P. Lerner
Richard J. Cote
Ram H. Datar
INTRODUCTION
Significant advances in our understanding of the molecular biology of bladder cancer and its response to therapy presents the possibility that existing and new therapies can be tailored for patients based on their risk of progression and individual molecular alterations. Targeted therapy—a treatment that specifically targets molecular deregulations and individual risk—can now be conceived for bladder cancer. We review here the potential for rational therapeutic intervention utilizing available knowledge of the molecular biology of cell-cycle regulation, apoptosis, signal transduction, angiogenesis, and tumor cell invasion in bladder cancer.
BLADDER CANCER: THE DISEASE
Bladder cancer comprises a spectrum of tumors that include urothelial carcinomas (UCs, also known as transitional cell carcinomas), squamous cell carcinomas, adenocarcinomas, and a few other subtypes. UC is the most prevalent subtype and represents nearly 90% of all bladder cancers in the Western world (1). UCs are subdivided into noninvasive papillary and invasive tumors that harbor different genetic alterations. Papillary tumors correspond to 70% of all UCs and usually are generally low grade and noninvasive at initial presentation. They begin as areas of hyperplasia that later undergo dedifferentiation (2). Invasive tumors are more poorly differentiated (i.e., higher grade) having a greater tendency to invade and metastasize. Papillary UCs are characterized by multiple recurrences, and 10% to 30% of these will progress to invasive disease. Most invasive carcinomas are believed to develop from carcinoma in situ (CIS), a flat lesion with a high propensity to progress to a high-grade invasive tumor. Added to the complexity of UCs is the fact that high-grade lesions can undergo metaplasia (squamous and/or glandular) and epithelial-mesenchymal transitions (EMTs). The variable manifestation and histology of UCs underscore the need to study the molecular biology of the disease.
BLADDER TUMORIGENESIS AND PROGRESSION: A MOLECULAR MODEL
Studies over the last two decades have identified many genetic alterations in UC leading to a better understanding of its evolution and progression. Alterations in chromosomes 9 and 17 play important roles in this process (3). Approximately 60% to 65% of all UCs are characterized by loss of heterozygosity (LOH) on chromosome 9. Allelic loss on chromosome 9 is common in early-stage, well-differentiated tumors, while other genetic changes are observed in more advanced lesions. LOH on chromosome 9 is thus considered to be an early event in bladder tumorigenesis (4).
Two categories of genes are considered to be responsible for malignant transformation—oncogenes and tumor suppressor genes. Mutations, gene amplifications, promoter methylation, or insertion of viral genetic material into human DNA can transform a protooncogene into an oncogene resulting in overexpression. Protooncogene activation results in derangement of cell-cycle control, causing malignant transformation. Tumor suppressor genes are generally recessive, thereby requiring inactivation of both alleles to induce tumorigenesis. This can occur by loss of or mutation in both alleles, or the LOH in one allele and a loss of function by either mutation or methylation-mediated silencing of the remaining allele (5). Tumor suppressor genes on chromosome 9 (marked by allelic losses in both 9p and 9q, and deletions between 9p12-9q34.1 that spans the p16INK4A locus) have been implicated in UC formation. These tumors generally have a good prognosis, and a low propensity to invade and metastasize. Similarly, low-grade papillary tumors frequently show a constitutive activation of the receptor tyrosine kinase (RTK)-Ras pathway, exhibiting activating mutations in the HRAS and fibroblast growth factor receptor 3 (FGFR3) genes. Approximately 70% of noninvasive papillary tumors harbor FGFR3 mutations compared with 10% to 20% of invasive tumors (6,7,8). In contrast, loss of genetic material on chromosome 17p (the location of TP53, which encodes for the p53 tumor suppressor protein) is associated with the more aggressive CIS (9). There thus exist at least two divergent pathways of bladder tumor progression that identify morphologically and biologically distinct subsets of UC that can be defined by unique patterns of molecular alterations. Based on these genetic defects, a sophisticated model of important molecular events in UC has been established (Fig. 17.1).
Disruption of the normal cell cycle is a crucial alteration in UC (10). These disruptions can occur singly or in combination, suggesting the importance of studying cell-cycle regulation to understand the molecular mechanisms leading to bladder tumorigenesis. Furthermore, the cell cycle is guided by a variety of signals from apoptotic, cell growth, signal transduction, and gene regulation pathways. Hence, understanding cell-cycle alterations in the context of other cellular pathways is critical in elucidating mechanisms underlying the development of UC.
MOLECULAR STAGING OF BLADDER CANCER
Pathological assessment of UC is based on tumor grade and stage. Pathologic staging systems are based on depth of invasion and assessment of regional and systemic spread of tumor.
For UC, stage is based on depth of tumor invasion and is the basis for the tumor/nodes/metastasis (TNM) system (11,12,13,14). However, while such criteria can provide reliable information on patient populations, they are unable to specify risk for progression or response to treatment for an individual patient. UC management is currently based on the TNM system, and the treatment can result in significant morbidity. Thus, it is essential to develop a staging system that can optimize the therapy administered to patients based on the molecular alterations of individual tumors (15).
For UC, stage is based on depth of tumor invasion and is the basis for the tumor/nodes/metastasis (TNM) system (11,12,13,14). However, while such criteria can provide reliable information on patient populations, they are unable to specify risk for progression or response to treatment for an individual patient. UC management is currently based on the TNM system, and the treatment can result in significant morbidity. Thus, it is essential to develop a staging system that can optimize the therapy administered to patients based on the molecular alterations of individual tumors (15).
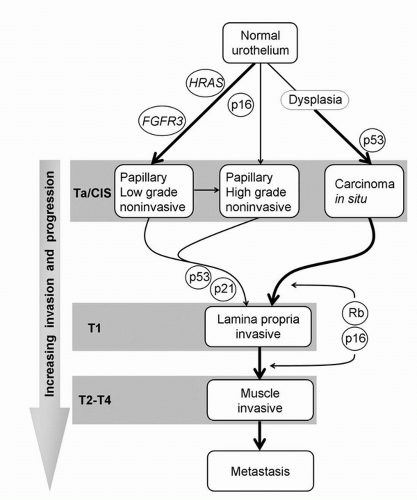 FIGURE 17.1. A molecular model for urothelial tumorigenesis and progression. Noninvasive and invasive tumors have unique molecular profiles and arise from distinct pathways. The locations of molecules indicate events that pose a risk for progression of a particular phenotype. The rare papillary carcinomas that invade are more likely to have genetic alterations at crucial loci. The thickness of arrows represents the relative frequency of occurrence. (Adapted from Mitra AP, Datar RH, Cote RJ. Molecular pathways in invasive bladder cancer: new insights into mechanisms, progression, and target identification. J Clin Oncol 2006;24:5552-5564, with permission.) |
New understanding of the molecular basis for UC progression is enabling translation of this information to develop novel molecular detection methods (3,16). This is also advancing new interventions targeting cellular mechanisms. Several molecular pathways that are altered in UC and can be potentially targeted therapeutically are depicted in Figure 17.2. The studies described below show that understanding molecular alterations in UC can facilitate more refined tumor staging and better predict clinical outcome. This can also identify molecular classes that may respond better to specific chemotherapeutics. This will eventually permit therapy tailored toward specific molecular defects, thereby lowering treatment morbidity. The ultimate goal is to approach each tumor as a separate entity and to identify its unique molecular characteristics. Specific management most efficacious for each individual patient may then be determined.
ALTERATIONS IN CELL-CYCLE REGULATION
Cell-cycle alterations are the most extensively investigated molecular aspects of UC. The cell cycle is primarily controlled by the p53 and retinoblastoma (Rb) pathways, which, in turn, are associated with apoptosis, cell growth, signal transduction, and gene regulation (Fig. 17.2).
p53 Pathway Alterations
Located on chromosome 17p13.1, the TP53 tumor-suppressor gene encodes for a 53-kDa nuclear phosphoprotein, p53, the central protein of the p53 pathway (17). They play a critical role in regulating the normal cell cycle and apoptotic response. Functional loss of TP53 is implicated in a complex array of aberrations involving cell cycle, apoptosis, senescence, and angiogenesis. p53 mediates growth arrest and DNA damage repair by inhibiting cell-cycle progression at the G1-S transition. This is mediated through the transcriptional activation of p21WAF1/CIP1. Once the DNA damage is repaired, accumulated p53 levels are abrogated by Mdm2. Alternatively, when cell-cycle arrest cannot be mediated, p53 can execute an apoptotic response (18). This dual function in maintaining cellular homeostasis has earned p53 the title, “guardian of the genome” (19).
Although most UCs exhibit loss of a single 17p allele, mutation in the remaining allele can inactivate TP53, leading to loss of its tumor-suppressor function (20,21). However, LOH on chromosome 17 occurs during later stages of UC and is usually associated with aggressive phenotypes (22). The short half-life of wild type p53 (6-30 minutes) prevents its accumulation in the nucleus (23). However, TP53 mutations result in an altered protein that is resistant to normal ubiquitin-mediated degradation. This increases intranuclear accumulation of the protein, which can be detected by immunohistochemistry (IHC). Hence, nuclear p53 accumulation has been hypothesized to correlate with TP53 mutations and thus a loss of p53 function. Such p53 nuclear immunoreactivity is prognostic particularly for patients with invasive, organ-confined, node-negative (T1-2bN0) UC (24,25,26). This has been recently confirmed in a multi-institutional retrospective cohort (27). Specifically, nuclear p53 accumulation is significantly associated with an increased risk of recurrence and a decreased survival (24). In UC patients, p53 immunoreactivity was significantly associated with increasing stage, increased risk of recurrence (p < 0.001), and decreased survival (p < 0.001) independent of stage and grade (Table 17.1 and Fig. 17.3). The 5-year recurrence rates in bladder-confined UC patients with wild type p53 were significantly lower than for those with organ-confined tumors that had p53 immunoreactivity. This indicates that patients with bladder-confined, p53-altered UC may need adjuvant treatment besides surgery.
While TP53 mutations are at least partially responsible for nuclear accumulation of p53, this is not always the case (28). Absent nuclear p53 accumulation does not rule out TP53 alterations. In fact, nonsense mutations may produce unstable transcripts or truncated proteins that lack the nuclear localization signal, thereby giving negative IHC results. These events can contribute to approximately 20% of TP53 mutations in head and neck cancers (29). Furthermore, not all cases with p53 nuclear accumulation have a loss of function. Studies examining p53 immunoreactivity and corresponding mutations in primary UC suggest that although both factors are independent prognostic indicators, tumors with a mutated TP53 and an altered protein phenotype exhibit the worst prognosis and those with a wild-type gene and an unaltered protein perform the best (Fig. 17.4) (30). It also appears that the site of TP53 mutation may be an important prognostic factor.
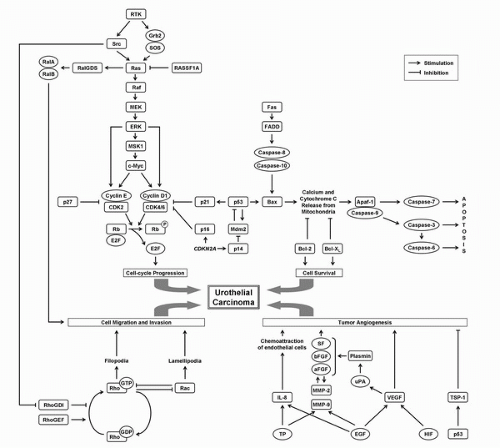 FIGURE 17.2. Major molecular pathways prone to alteration during bladder cancer progression, and which can be targeted therapeutically. |
The importance of TP53 mutations in UC progression is irrefutable. However, despite numerous retrospective studies indicating association between p53 alterations with adverse outcomes, there is some ambiguity about its clinical impact. Some studies have failed to show this relationship as independent from known prognostic factors. Meta-analyses highlight these differences in the conclusions on outcome, which may be ascribed to technical aspects of p53 IHC (such as antibody selection, varying cutoff values), study design, and patient selection (31,32).
The p53-targeted therapy trial tested the hypothesis that p53 alterations determined by IHC were associated with worse prognosis in patients with organ-confined node-negative urothelial carcinoma treated with radical cystectomy and bilateral pelvic lymphadenectomy (33). Patients with p53-altered tumors were randomized to three cycles of adjuvant MVAC (methotrexate, vinblastin, doxorubicin, cisplatin) chemotherapy versus observation, testing the hypothesis that high-risk patients with p53-altered tumors would have a more favorable outcome with chemotherapy. The recurrence probability in the trial was much more favorable compared to the retrospective cohort and there was no difference in recurrence-free and overall survival between the p53-wild type and p53-altered cohorts, and adjuvant chemotherapy was not associated with a reduced risk of recurrence in patients with p53-altered tumors.
Located on chromosome 6p21, the p21WAF1/CIP1 tumor suppressor gene encodes for p21, a cyclin-dependent kinase inhibitor (CDKI) that is transcriptionally regulated by p53. Loss of p21 expression and subsequent loss of cell-cycle control is thought to be a mechanism by which p53 alterations may influence tumor progression. However, p21 expression may also be regulated in a p53-independent manner (34). p21 expression can thus be maintained despite p53 alterations. Patients with p53-altered UC with p21 underexpression show increased recurrence and decreased survival probabilities compared to those who maintained p21 expression (Fig. 17.5) (35). In fact, maintenance of p21 expression appeared to abrogate the deleterious effects of p53 alterations.
TABLE 17.1 ASSOCIATION OF p53 IMMUNOREACTIVITY WITH GRADE AND PATHOLOGIC STAGE OF BLADDER CANCER AND PRESENCE OR ABSENCE OF LYMPH-NODE METASTASES IN 190 PATIENTS NOT PREVIOUSLY TESTED FOR p53 ALTERATION | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
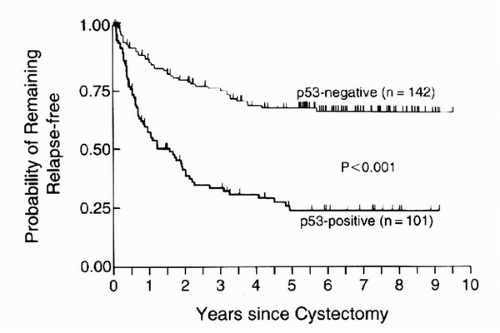 FIGURE 17.3. Probability of remaining relapse-free in 243 patients with bladder cancer and either p53-positive or p53-negative tumors (log-rank p < 0.001). (Adapted from Esrig D, Elmajian D, Groshen S, et al. Accumulation of nuclear p53 and tumor progression in bladder cancer. N Engl J Med 1994;331:1259-1264, with permission.) |
Encoded by MDM2 on chromosome 12q14.3-q15, the Mdm2 protein is involved in an autoregulatory feedback loop with p53, thus controlling its activity (36). Increased p53 levels upregulate the MDM2 promoter. The translated protein mediates proteasomal degradation of p53. The lowered p53 levels then reduce Mdm2 levels (37). The frequency of MDM2 amplification increases with increasing UC stage and grade (38). Alternatively spliced MDM2 transcripts have been reported in UC and appear to be more common in muscle-invasive disease (39). A single nucleotide polymorphism in the MDM2 promoter region, SNP309, has also been reported to predict younger age at disease onset and poorer survival, and it can provide an enhanced prognostic value when coupled with TP53 mutation status (40).
Retinoblastoma Pathway Alterations
The retinoblastoma gene (RB), located on chromosome 13q14, was the first tumor suppressor gene to be identified. It encodes the Rb protein, which interacts with multiple proteins involved in the G1/S transition (41). Active, dephosphorylated Rb binds and sequesters the transcription factor E2F. The G1/S transition is regulated by cyclin-dependent kinases (CDKs) that can bind to cyclins (42). During late G1, Rb is phosphorylated and inactivated by cyclin D1-CDK4/6 complex, which releases E2F. Free E2F can transcribe genes required for DNA synthesis. CDKIs such as p16, p21, and p27 inhibit CDK activity and regulate Rb phosphorylation.
The most common mechanism of RB inactivation in UC is by deletion of chromosome 13q. The frequency of RB inactivation in invasive UC varies considerably between different studies (between 14% and 80%). Miyamoto et al. (43) reported RB mutations in 21% of Ta and T1 tumors, and 36% of ≥T2 tumors. This suggests that RB mutations are involved across all UC stages. A combined examination of p53 and Rb in 45 cases with T1 UC revealed that patients with alteration of either or both proteins had increased risk of progression (p < 0.04 and p = 0.005, respectively), while no patient with normal expression of both proteins progressed (44).
It was originally believed that only inactivating RB mutations could result in loss of Rb function; such loss is important in UC progression (45,46). However, a significant proportion of tumors expressing Rb show clinical consequences of loss of Rb function (47). Patients with tumors expressing the highest levels of Rb have clinical outcomes virtually identical to those with no detectable Rb (Fig. 17.6). These patients have lower recurrence-free and overall survival compared with patients with tumors expressing moderate Rb levels. The tumors with the highest Rb expression levels demonstrate constitutively hyperphosphorylated Rb, which results from loss of p16 expression and/or cyclin D1 overexpression (Fig. 17.7) (48). RB alterations causing loss of a functional protein result in increased p16 through feedback mechanisms, which attempt
to “brake” the cell cycle. In this situation, cyclin D1 expression appears to be redundant. On the other hand, loss of p16 through genetic or epigenetic mechanisms in the presence of an intact RB results in inactivation of the Rb protein through constitutive hyperphosphorylation. This change in Rb function is reflected as cells having a high Rb phenotype. Cyclin D1 overexpression can result in similar inactivation of Rb even when p16 expression is maintained. This is the biologic basis for the reliability of IHC evaluation of Rb as an indicator of loss of RB function; not only loss of Rb expression, but aberrant Rb overexpression also indicates a disrupted pathway.
to “brake” the cell cycle. In this situation, cyclin D1 expression appears to be redundant. On the other hand, loss of p16 through genetic or epigenetic mechanisms in the presence of an intact RB results in inactivation of the Rb protein through constitutive hyperphosphorylation. This change in Rb function is reflected as cells having a high Rb phenotype. Cyclin D1 overexpression can result in similar inactivation of Rb even when p16 expression is maintained. This is the biologic basis for the reliability of IHC evaluation of Rb as an indicator of loss of RB function; not only loss of Rb expression, but aberrant Rb overexpression also indicates a disrupted pathway.
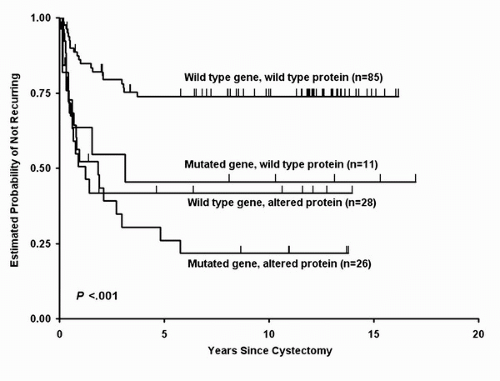 FIGURE 17.4. Estimated probability of remaining recurrence-free, based on combined expression of TP53 gene status and p53 protein status with single exon-5 mutations added to the tumors with no mutations, for all patients combined (log-rank p < 0.001). (Adapted from George B, Datar RH, Wu L, et al. p53 gene and protein status: the role of p53 alterations in predicting outcome in patients with bladder cancer. J Clin Oncol 2007;25:5352-5358, with permission) |
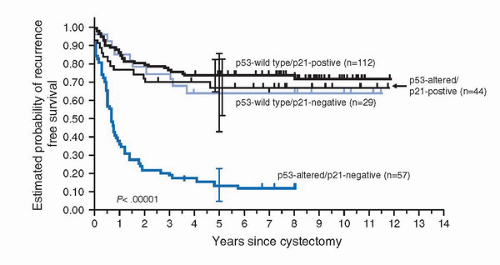 FIGURE 17.5. Probability of remaining recurrence-free in 242 patients with bladder tumors stratified by p53 and p21 status: p53 wild type, no detectable p53 nuclear reactivity; p53-altered, nuclear p53 reactivity detected; p21-positive, p21 nuclear expression detected; p21-negative, no detectable p21 nuclear expression. The patients were stratified into four groups on the basis of p53 status and p21 status of their tumors. Patients with p53-altered/p21-negative tumors had a significantly higher rate of recurrence (log-rank p < 0.00001) in comparison with other patients. (Adapted from Stein JP, Ginsberg DA, Grossfeld GD, et al. Effect of p21WAF1/CIP1 expression on tumor progression in bladder cancer. J Natl Cancer Inst 1998;90:1072-1079, with permission.) |
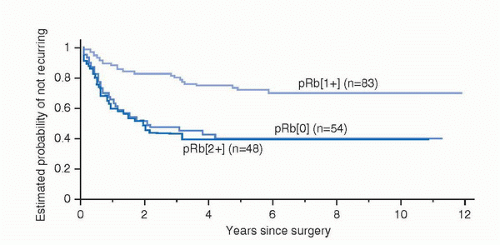 FIGURE 17.6. Probability of remaining relapse-free in 185 patients with invasive urothelial cancer of the bladder according to Rb status. pRb 0, no detectable nuclear reactivity; pRb 1+, 1% to 50% nuclear reactivity; pRb2+, >50% nuclear reactivity. (Adapted from Cote RJ, Dunn MD, Chatterjee SJ, et al. Elevated and absent pRb expression is associated with bladder cancer progression and has cooperative effects with p53. Cancer Res 1998;58:1090-1094, with permission.) |
Rb phosphorylation is facilitated by the cyclin/CDK complexes. CDK4 amplification frequency increases with UC stage and grade (38). Translocation and amplification of the cyclin D1 gene (CCND1) were only observed in peripheral blood lymphocytes of patients with UC and not in normal controls (49). Reports suggest that cyclin D1 protein expression can be used as an inverse indicator for the level of UC invasiveness, but not as an independent prognostic factor (50).
Low cyclin E1 expression has been associated with advanced pathologic stage, lymphovascular invasion, lymph node metastases, and altered expression of Rb, cyclin D1, and p27Kip1 (51). By multivariate analysis, low cyclin E1 expression was significantly associated with disease-specific mortality. Cyclin E1 expression may be associated with aggressive tumor growth and may be associated with p27Kip1 expression in regulating cell-cycle progression in UC (52).
Low cyclin E1 expression has been associated with advanced pathologic stage, lymphovascular invasion, lymph node metastases, and altered expression of Rb, cyclin D1, and p27Kip1 (51). By multivariate analysis, low cyclin E1 expression was significantly associated with disease-specific mortality. Cyclin E1 expression may be associated with aggressive tumor growth and may be associated with p27Kip1 expression in regulating cell-cycle progression in UC (52).
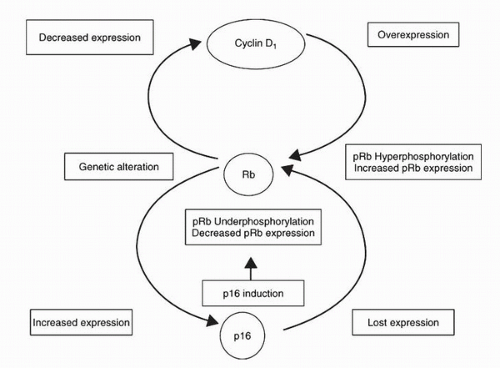 FIGURE 17.7. Proposed model showing the interrelationships between Rb, p16, and cyclin D1 in bladder cancer. Genetic alterations in RB result in increased p16 expression and decreased cyclin D1 expression. In cells with an intact RB gene, loss of p16 expression and/or increased cyclin D1 expression leads to hyperphosphorylation of Rb, resulting in high Rb expression on immunohistochemistry. This phenotype is associated with increased cell proliferation and poor clinical outcome. Induction of p16 in cells with an intact RB gene leads to a decrease in the levels of hyperphosphorylated Rb, resulting in moderate Rb expression. This phenotype is associated with decreased cell proliferation and improved clinical outcome. (Adapted from Chatterjee SJ, George B, Goebell PJ, et al. Hyperphosphorylation of pRb: a mechanism for RB tumor suppressor pathway inactivation in bladder cancer. J Pathol 2004;203:762-770, with permission.) |
The major CDKIs relevant to UC include p16INK4a, p27Kip1, and p21. Of these, p21 has been described in context with p53. The locus 9p21 harbors CDKN2A and CDKN2B. LOH analysis established that CDKN2A microdeletion is the predominant mechanism of 9p21 inactivation in UC (53). Using an alternative reading frame, CDKN2A encodes for p16INK4a and p14ARF, both of which inhibit cell-cycle progression (54). p16INK4a prevents Rb phosphorylation (55). p14ARF functions upstream of p53 by promoting Mdm2 degradation (56). This facilitates regulation of two key cell-cycle regulatory pathways—Rb and p53—from a single locus (54). Deletions and methylation of INK4A occur frequently in noninvasive UC. However, only those deletions that affect both p16INK4a and p14ARF have been shown to correlate with clinicopathologic parameters of worse prognosis (57). Homozygous deletion of p16INK4a correlates with a lower recurrence-free survival. Other reports indicate that p14ARF is a target of homozygous deletion, whereas p16INK4a is the hot spot for hypermethylation in UC (58). (See the section “Role of DNA Methylation.”) A significant correlation has also been reported between low p27Kip1 expression and decreased disease-free and overall survival in UC (59). Low p27Kip1 expression is more common in poorly differentiated muscle-invasive UCs and is a major player in cell-cycle control in these neoplasms (60). Loss of p27Kip1 expression has been associated with subsequent development of invasive disease; this predictive value was enhanced significantly when combined with caspase-3 overexpression (61).
ALTERATIONS IN APOPTOSIS
The coordinated cell death events that occur during normal development and in response to a several initiation stimuli are referred to as apoptosis. It is quantified by the apoptotic index, which is the ratio of apoptotic cells to the total number of tumor cells, and has been correlated to tumor progression, recurrence-free and overall survival in patients with superficial UC (62). Apoptosis can be initiated by two pathways. The extrinsic pathway involves activation of death receptors such as Fas, while the intrinsic pathway is mediated by the mitochondria. Both pathways activate caspases that lead to the characteristic apoptotic changes (63). UC acquires mechanisms to escape Fas-mediated apoptosis during malignant transformation (64), potentially via circulating soluble Fas that antagonizes cell-surface Fas function (65). This has been corroborated in the urine of superficial UC patients (66). Decreased Fas expression has been associated with increased stage and grade, and poor prognosis (67). The death receptors activate caspase-8 and -10, which initiate the apoptotic cascade. In vitro caspase-8 expression can induce apoptosis in UC cell lines (68). Initiator caspases may activate “effector” caspase-3, -6, and -7, which results in apoptosis (17).
The Bcl-2 protein family plays an important role in the intrinsic pathway; it includes antiapoptotic members such as Bcl-2 and Bcl-XL and proapoptotic members such as Bax. p53 exerts its control on apoptosis through the regulation of Bcl-2 and Bax expression (69). Localized on the outer mitochondrial membrane, Bcl-2 regulates the formation of megapores that control ionic flux, thereby inhibiting caspase activation (70,71). Bcl-2 overexpression is associated with poor prognosis in UC patients treated with radiotherapy (72) or synchronous chemoradiotherapy (73). Bcl-2 expression can also identify patients with advanced UC undergoing radiotherapy who may benefit from neoadjuvant chemotherapy (74). Bcl-2 has been associated with decreased tumor-free survival in high-grade T1 UC (75) and can act as a good prognostic indicator in superficial UC in combination with p53 (76). Using the expression levels of Mdm2, p53, and Bcl-2, Maluf et al. established a prognostic index where normal expression of all markers was associated with the best survival probability and where aberrations in all markers corresponded with worst survival probability (77). Bcl-XL has been shown to interact and work with Bcl-2 in progression of low-stage UC (78), and Bcl-XL downregulation in vitro sensitizes UC cell lines to cytotoxic agents (79).
Cellular stress causes rapid opening and closing of the mitochondrial megapores, which direct Bax to the site. This alters the mitochondrial membrane potential, causing release of stored calcium and cytochrome c, thereby activating effector caspases and leading to apoptosis (Fig. 17.2) (80). Bax is an independent predictor of favorable prognosis in invasive UC (76,81,82). In the cytoplasm, cytochrome c binds to and activates Apaf-1 (83). This complex then binds ATP, forming the apoptosome that in turn activates caspase-9. This leads to the activation of the downstream effector caspases that drive apoptosis. Decreased expression of caspase-3 has been associated with an increased probability of disease recurrence in UC patients undergoing cystectomy (84).
ALTERATIONS IN CELL GROWTH AND SIGNAL TRANSDUCTION
Several factors and their associated RTKs are responsible for modulating growth signals from external cues and transducing the signals into urothelial cells’ nuclei. Aberrations in these growth factors, their receptors, and/or the signals transmitted
by them can result in abnormally increased transduction of growth signals, causing uncontrolled cellular proliferation and tumor formation.
by them can result in abnormally increased transduction of growth signals, causing uncontrolled cellular proliferation and tumor formation.
There are three ubiquitously expressed receptors of TGF-β, a pleiotropic growth factor—TβR-I, TβR-II, and TβR-III. TβR-I and TβR-II are required for TGF-β signal transduction (85,86). TGF-β is a potent growth inhibitor; however, malignant cells are frequently resistant to this effect (87). Alteration in the expression of TβRs may likely play a role in this scenario (88,89). IHC analysis of 59 UC specimens demonstrated loss of TβR-I and TβR-II expression in 31% and 44% of cases, respectively (90). Loss of expression correlated with tumor grade, pathologic stage, and nodal status. Loss of TβR-I expression was also associated with progression and decreased survival. Another study on 80 UC patients showed that TGF-β was an independent predictor of progression, whereas TβR-I was an independent predictor of disease progression and disease-specific survival (91). Furthermore, preoperative plasma levels of TGF-β can strongly predict potential for invasion, nodal and distant metastases, recurrence, and disease-specific survival (92).
The FGFR family consists of four receptors (FGFRs 1-4). Activating FGFR3 mutations have been most extensively studied in this receptor class. Approximately 70% of low-grade Ta tumors have FGFR3 mutations, and this is strongly associated with the development of low-grade papillary tumors (6,7,8). FGFR3 mutations activate the Ras-mitogen-activated protein kinase (MAPK) pathway. FGFR3 and Ras gene mutations are generally mutually exclusive in papillary UC, which probably reflects activation of the same pathway by either event (93). Approximately 82% of grade 1 tumors and Ta tumors have mutations in either a Ras gene or a FGFR3, suggesting that MAPK pathway activation may be an obligate event in most of these cases.
The epidermal growth factor receptor (EGFR) family consists of four closely related receptors that transmit signals via the Ras-MAPK pathway (Fig. 17.2). Among the best-studied receptors in this family are EGFR (ErbB-1) and ErbB-2 (Her2/neu). Unlike FGFR3, these receptors are also overexpressed in invasive tumors (94,95). Increased EGFR expression has been associated with increased probability of disease progression and death (96,97,98). Elevated ErbB-2 expression has been associated with poor disease-specific survival (95,99,100). While the combined expression of EGFR and ErbB-2 has been suggested to be a better outcome predictor for UC than each individual marker alone (101), this finding was not supported by another study (102).
Vascular endothelial growth factor (VEGF) is an important signaling protein involved in angiogenesis. VEGF family members elicit responses by binding to VEGF receptors (VEGFRs). VEGFR2 (KDR/Flk-1) mediates most cellular responses to VEGF. VEGFR2 expression has been correlated with increasing tumor stage (103). In addition, VEGFR2 levels may be an important determinant for prediction of nodal metastasis in UC patients (104).
Activation of growth receptors results in transduction of signals to transcription factors in the nuclei. Ras-MAPK is the most extensively studied signaling pathway in UC. Activating Ras mutations, resulting in constitutive pathway activation, are found in approximately 13% of Ta tumors (93). HRAS mutations have been observed in tumors cells that exfoliate in the urine of patients with low-grade UC (105). Evidence also indicates that HRAS is significantly overexpressed in nonprogressing Ta tumors, compared with those that eventually progress to an invasive phenotype (106).
ALTERATIONS IN SRC
c-SRC is the best-studied proto-oncogene in the Src family of kinases (SFKs) that is activated by growth factors or activating mutations resulting in a transformation (Fig. 17.2) (107). While c-Src expression is positively correlated with grade and stage in several cancers (107,108,109), its role in UC is not well defined. Early studies report low Src levels and activity in UC cell lines, and elevated tyrosine kinase activity of pp60c-Src in UCs over normal bladder mucosa (110). Increased level and kinase activity were mainly in low-grade UC cell lines and tumors compared to high-grade counterparts (111). Novel substrates were identified displaying elevated pp60c-Src kinase activity, suggesting an inverse association for the Src proto-oncogene in UC differentiation and progression (110,111). Further analysis on human tumors revealed that Src levels diminish as a function of UC stage (112). In vitro analyses also suggest that c-Src activation might correspond to an early event in growth factor-triggered signaling, diverging the signaling cascade into EMT but not to mitogenesis (113,114). This suggests that in UC, the low level and activity of c-Src may be compensated by overexpressed RTKs as EGFR or FGFRs, or more unusually, c-Src may either activate tumor or metastasis suppressors or inactivate other oncogenes or metastasis promoters.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


