TABLE 50.1 HISTOLOGIC CLASSIFICATIONS OF NEOPLASMS OR TUMOR-LIKE LESIONS OF ADRENAL CORTEX AND MEDULLA | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
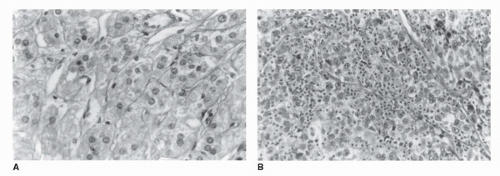 FIGURE 50.1. Histologic comparison of a benign adrenocortical adenoma and a carcinoma. (A) Benign adenoma. Adenomas are characterized by nests and cords of clear cells with abundant cytoplasm separated by fibrovascular trabeculae. Mitoses are absent or rare, and cells have an orderly, monotonous appearance. (B) Adrenocortical carcinoma (ACC). This cancer retains the fibrovascular trabeculae, but the cells demonstrate marked pleomorphism, enlarged and hyperchromatic nuclei, and obvious nucleoli. |
(a marker of cell proliferation) may be overexpressed in carcinomas compared to normal adrenal tissue, and benign adenomas (27). Immunostaining for chromogranin is positive in pheochromocytomas and negative in ACC. Similarly, the monoclonal antibody D11 has utility in distinguishing adrenocortical tumors from other tumors, but not benign from malignant. It therefore appears that immunohistochemical studies are useful in distinguishing adrenal neoplasms from other tumors but are of limited value in differentiating benign and malignant adrenal tumors.
TABLE 50.2 HISTOLOGIC CRITERIA OF MALIGNANCY IN ACC | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
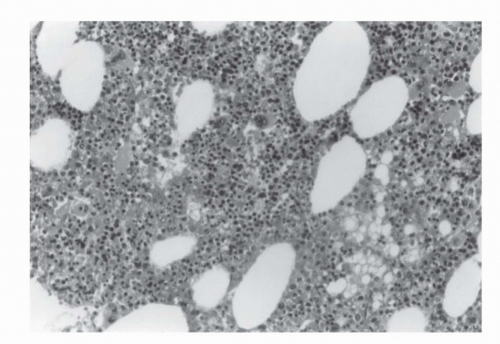 FIGURE 50.2. Histologic appearance of a myelolipoma showing fat (white areas) intermixed with myeloid tissue. |
is not well characterized and its impact on long-term morbidity of patients with benign adrenal masses is unclear. The reader is referred to an article by Boscaro et al. (37) for a detailed review of the diagnosis and management of Cushing syndrome. A finding of hypokalemia suggests an aldosterone-secreting tumor. This suspicion should be confirmed by a plasma aldosterone concentration to plasma renin activity (PAC:PRA) ratio. A ratio >30 and a PAC of >0.5 nmol/L are highly suggestive of autonomous aldosterone production. For a detailed review of the diagnosis and management of adrenal aldosterone-secreting tumors, the reader is referred to an outstanding review (38). Elevated 24-hour urinary catecholamines are indicative of pheochromocytoma. For diagnosis of pheochromocytoma, plasma-free metanephrines (normetanephrine and metanephrine) should be measured. These combined tests have a sensitivity of 99% and specificity of 89%.
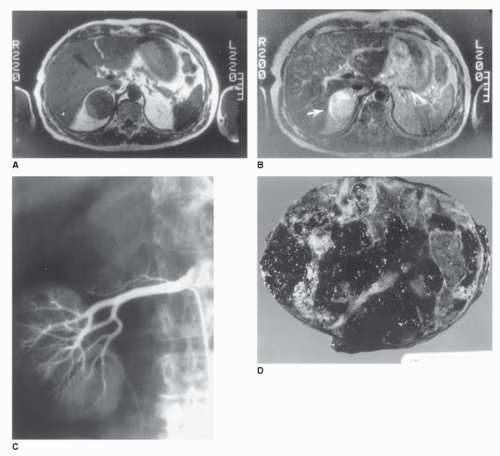 FIGURE 50.3. Radiographic and gross pathologic characteristics of a cystic benign adrenal adenoma. (A) T1-weighted magnetic resonance image in a 57-year-old woman performed after incidental binding of an adrenal mass on CT scan obtained because of vague abdominal discomfort. A 5-cm homogeneous mass isodense with the liver replaces the right adrenal gland. (B) T2-weighted MRI in the same patient. The tumor (arrow) appears hyperintense compared with the liver, a finding suggestive of malignancy. (C) Selective right renal angiogram in the same patient. The tumor is hypovascular and displaces branches of the right adrenal artery. The upper pole of the right kidney is displaced interiorly and laterally. (D) Gross specimen revealing multiple hemorrhagic pseudocysts arising from a benign cortical adenoma. |
TABLE 50.3 DIFFERENTIAL DIAGNOSIS OF AN ADRENAL MASS | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
TABLE 50.4 CLINICAL FEATURES OF HYPERCORTISOLISM | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||
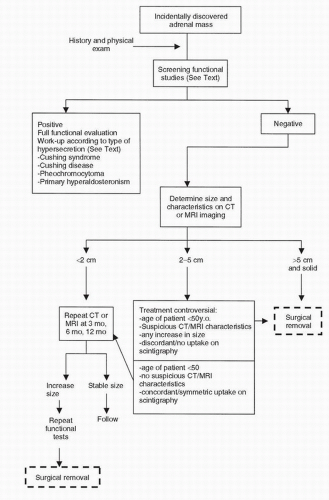 FIGURE 50.4. Algorithm illustrating the stepwise clinical evaluation and treatment of a patient with an incidentally discovered adrenal mass. (Modified from Moinzadeh A, Libertino JA. Asymptomatic adrenal mass. In: Resnick MI, Elder JS, Spirnak JP, eds. Critical decisions in urology, 3rd ed. Hamilton, ON: BC Decker Inc., 2004:381.) |
 FIGURE 50.5. Nomogram illustrating the stepwise clinical evaluation of patients with suspected hypercortisolism. |
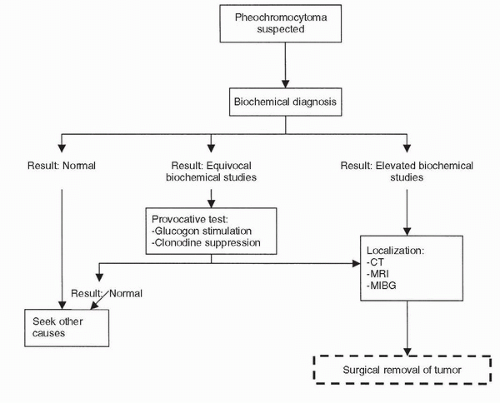 FIGURE 50.6. Algorithm illustrating the stepwise clinical evaluation and treatment of a patient with pheochromocytoma. (Modified from Moinzadeh A, Libertino JA. Pheochromocytoma. In: Resnick MI, Elder JS, Spirnak JP, eds. Critical decisions in urology, 3rd ed. Hamilton, ON: BC Decker Inc., 2004:381.) |
TABLE 50.5 DEXAMETHASONE SUPPRESSION TESTS: INTERPRETATION OF RESULTS | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
4.2 years. The adrenal masses enlarged in 8% and decreased in size in 1.3%, and none of the lesions proved to be malignant.
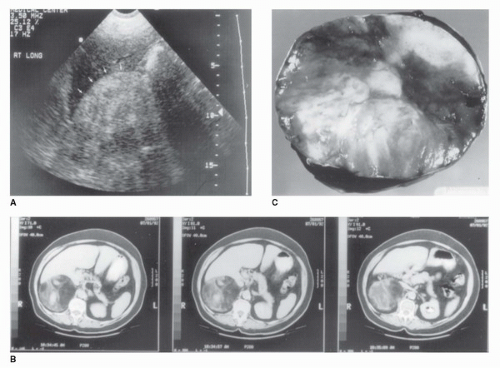 FIGURE 50.7. Radiographic and pathologic appearance of an adrenal myelolipoma. (A) Renal ultrasound in a 47-year-old woman with right flank pain. A large, solid tumor with the hyperechoic appearance typical of fat is demonstrated (arrows). (B) CT scan demonstrating a heterogeneous tumor in the right adrenal gland with areas of low attenuation also typical of fat. Myelolipoma was suspected based on these studies. The tumor measured 10 cm in diameter. (C) Gross appearance of a myelolipoma after removal, demonstrating heterogeneous nature. |
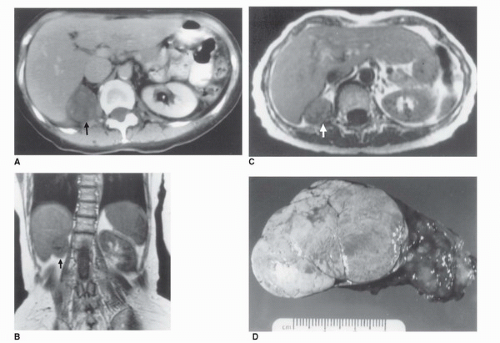 FIGURE 50.8. Radiographic and gross pathologic appearance of a benign functioning adrenocortical adenoma. (A) CT scan in a 64-year-old woman with hypercortisolism. A 4.5-cm mass replaces the right adrenal gland (arrow). (B) T1-weighted magnetic resonance image in the same patient in the coronal plane. The right upper quadrant mass (arrow) appears isodense with liver. (C) T2-weighted MRI in the same patient in a transverse plane. The tumor appears homogeneous and remains isodense with liver (arrow). (D) Gross specimen after surgical removal, with the typical smooth, lobulated, and homogeneous appearance of a benign cortical adenoma. |
mutations in p53 are responsible for autosomal dominant Li-Fraumeni syndrome, and 5% of such patients develop ACCs (63). Germline mutations of p53 have been found in children with ACCs without classic family history of Li-Fraumeni syndrome, as well (63). The complete loss of one insulin-like growth factor type II (IGF-II) allele, which maps to 11p15.5 locus, and a duplication of the remaining allele in association with IGF-II overexpression has been demonstrated in tumors with Beckwith-Wiedemann syndrome and in sporadic adrenocortical tumors (63,64). In a report by Gicquel et al. (65), IGF-II was overexpressed in 27 of the 29 malignant adrenocortical tumors compared to 3 of 35 benign adenomas. Carney complex is another hereditary syndrome that is associated with primary pigmented nodular adrenocortical disease in 20% to 30% of patients (66). Two chromosomal loci have been identified (2p16 and 17q22-17q24); however, no deletions at these loci have been identified, implying that the responsible gene may be an oncogene rather than a tumor- suppressor gene (66,67). Approximately 35% of patients with MEN I have adrenal nodules, and the syndrome is related to germline mutations in the tumor-suppressor menin gene (11q13). In a series of 33 patients with MEN I, 12/33 had adrenocortical tumors, with loss of constitutional heterozygosity reported in 1 patient with ACC and not in 11 patients with benign adenomas (68). The genes involved in familial adenomatous polyposis coli (APC gene) and McCune Albright syndrome (activating mutations in the GNAS1 gene) have also been investigated as possible contributors to the presence of adrenal lesions in patients with these genetic syndromes.
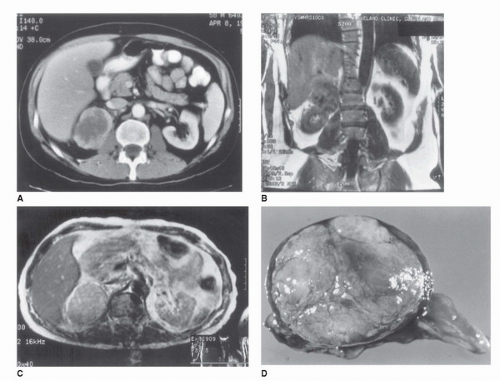 FIGURE 50.9. Radiographic and pathologic characteristics of an adrenocortical carcinoma (ACC). (A) CT scan in a 46-year-old man performed because of malaise and fever. A 7 × 5 cm heterogeneous mass with an irregular border replaces the right adrenal gland. (B) T1-weighted MRI in the same patient in a coronal plane. The tumor is isodense with liver and displaces right kidney. (C) Transverse T2-weighted MRI in the same patient demonstrating a slight increase in the tumor image intensity compared with the liver. (D) Adrenal tumor after excision, demonstrating an irregular lobulated surface and areas of hemorrhage suggestive of ACC. |
shown equal distributions of chromosomal gains and losses in benign and malignant adrenocortical tumors, although the specific genetic events have been different (15). Bernard et al. (71) suggested multistep tumorigenesis. They studied a patient with malignant and benign sections in a tumor. This was confirmed by molecular analysis (expressions of IGF II and allelic status of 11p15 and 17p13 loci) and CGH. Using the “candidate gene approach,” several studies have looked at putative oncogenes and tumor-suppressor genes such as p53, IGF II, APC, MEN I, and Ras; however, a low prevalence of mutations were found (72). Mutations of ACTH receptors and constitutive activations of regulatory proteins of cAMP such as G-protein-coupled receptors have also been implicated in adrenal tumorigenesis. Mutational analysis of the coding region of the receptor in 41 adrenocortical tumors, however, did not demonstrate presence of any mutation (73).
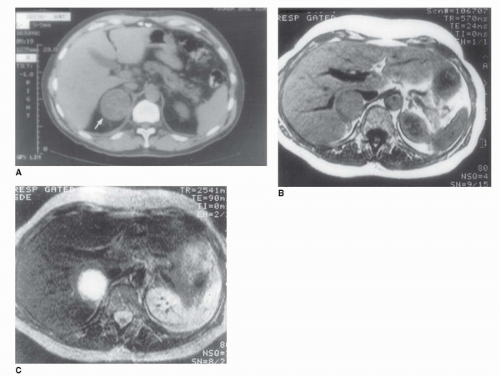 FIGURE 50.10. CT and MRI of a pheochromocytoma. (A) An abdominal CT in a 45-year-old patient with hypertension and elevated serum catecholamine level demonstrates a rounded homogeneous tumor replacing the right adrenal gland (arrow). (B) T1-weighted MRI of the 45-year-old patient illustrated in (A). The tumor is isodense with the liver. (C) T2-weighted MRI in the same patient. The tumor has a very high signal intensity and appears bright white, which is pathognomonic of a pheochromocytoma. |
Feminization or aldosterone excess is rarely reported (76,77). Both functional and nonfunctional adrenal cancers have been described. Reviews of various case series in patients with adrenal carcinoma demonstrate that clinical presentation varies based on whether the report comes from an oncologic or endocrine clinic. Functional carcinomas are more frequent in the endocrinologic literature. In a review of 1,480 patients in which functional status was mentioned, Wooten and King (60) noted that 60% of the cases were associated with evidence of function. In large retrospective series of patients with adrenal cancers, functional tumors have been reported in 34% to 72% (78).
TABLE 50.6 STAGING SYSTEM FOR ACC | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||
Related posts:
 Androgen Receptor Signaling in Castration-Resistant Prostate Cancer
Management of High Risk Prostate Cancer
Bladder Cancer Staging
Testicular Cancer: Clinical Signs and Symptoms
Late Events and Long-Term Complications After Treatment for Testicular Cancer
Radiologic Imaging of Renal Cell Carcinoma: Its Role in Diagnosis
Androgen Receptor Signaling in Castration-Resistant Prostate Cancer
Management of High Risk Prostate Cancer
Bladder Cancer Staging
Testicular Cancer: Clinical Signs and Symptoms
Late Events and Long-Term Complications After Treatment for Testicular Cancer
Radiologic Imaging of Renal Cell Carcinoma: Its Role in Diagnosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


