Fig. 13.1
Principle of array comparative genome hybridization (aCGH)
Prominent African-Americans aberrations were chromosome X amplification in men and deletions in chromosomes 4, 8, and 18. In African-Americans several CAN genes were altered at high frequencies. EXOC4, EPHB6, GNAS, MLL3, and TBX22 were the most frequently deleted genes and HAPLN1, ADAM29, SMAD2, and SMAD4 underwent the most amplifications. One of the most deleted genes was EPHB6 that is known to slow breast cancer cell lines invasiveness. Chromosome X amplification in men with CRC should be monitored. The observed CIN may have a key role in CRC in this population (Ashktorab et al. 2010; Brim et al. 2012). Among the ethnic groups, Ashktorab and colleagues found that African -Americans possessed the highest CRC the age-standardized incidence and mortality rates compared with other ethnic groups. Most CRCs originate from pre-existing adenoma. 30 % of the US adult population has adenomas (Nouraie et al. 2010). In African-Americans, CRC is more advanced and right-sided. African-Americans continue to have higher CRC death rates despite the reduced mortality rates as a result of screening (Nouraie et al. 2010; Lipkin and Higgins 1988; Kinzler and Vogelstein 1996; Jemal et al. 2007, 2014).
In a study, 1753 CRC cases were diagnosed from 1959 to 2006 in Howard University Hospital in America. The rate of in situ tumor reached a peak of 8.5 % in the 1990s (p = 0.0001). Ashktorab and co-workers observed a decade-to-decade increasing rate of right-sided tumors, beginning with 36 % during 1959–1970 and reaching a peak of 60 % during 2001–2006 (p = 0.0001). The recent increased rate of advanced and right-sided tumor were demonstrated which is consistent with SEER data and is highly important in developing strategies for CRC prevention and treatment for African-Americans (Nouraie et al. 2010). On the other hand, 5,013 colorectal polyps were diagnosed during 1959–2006, with tubular adenoma being the most frequent pathology (73 %). Right-sided polyps were mostly seen in the 1990s. Left-sided polyps were younger (p < 0.0001), more hyperplasic (23 vs. 5 %; p < 0.0001), and more frequent in women (56 vs. 52 %; p = 0.02) compared with right-sided polyps. The frequency of right-sided adenoma significantly increased from 18 % in the 1960s to 51 % during 2001–2006 (p < 0.0001). A higher neoplastic to hyperplastic polyps ratio (8:1) than what had been reported in Caucasians (7:1) was observed. This implies a recent shift in polyps from the left side to the right side of the colon, which is in accordance with the increased colon cancer incidence in African-Americans. This, the researchers further emphasized on the key role of screening for reducing the incidence of colon cancer in African-Americans (Nouraie et al. 2010).
African-Americans microsatellite instability-high (MSI-H) cancers was high compare to microsatellite stable (MSS) and microsatellite instability-low (MSH-L) cancers. Moreover, most MSI-H tumors were well differentiated, proximal, and highly mucinous. MSI-H colorectal tumor was 2–3-folds more prevalent, while the defect in the percentage expression of mismatch repair (MMR) genes (hMLH1 and hMSH2) was similar in African-American patients compared with American Caucasians. Here, the role of environmental or genetic factors that are more common in the development of CRC in African-Americans should not be underestimated (Ashktorab et al. 2005).
In this way, Brim et al showed the different DNA aberrations processes which can cause colorectal cancer (CRC). They conducted a comprehensive molecular characterization of 27 CRCs from Iranian patients. Array CGH was performed. The MSI phenotype and the methylation status of 15 genes were established using MSP. The CGH data was compared to two established lists of 41 and 68 cancer genes, respectively, and to CGH data from African Americans. A maximum parsimony cladogram based on global aberrations was established. The numbers of aberrations seem to depend on the MSI status. MSI-H tumors displayed the lowest number of aberrations. MSP revealed that most markers were methylated, except RNF182 gene. P16 and MLH1 genes were primarily methylated in MSI-H tumors. Seven markers with moderate to high frequency of methylation (SYNE1, MMP2, CD109, EVL, RET, LGR and PTPRD) had very low levels of chromosomal aberrations. All chromosomes were targeted by aberrations with deletions more frequent than amplifications. The most amplified markers were CD248, ERCC6, ERGIC3, GNAS, MMP2, NF1, P2RX7, SFRS6, SLC29A1 and TBX22. Most deletions were noted for ADAM29, CHL1, CSMD3, FBXW7, GALNS, MMP2, NF1, PRKD1, SMAD4 and TP53. Aberrations targeting chromosome X were primarily amplifications in male patients and deletions in female patients. A finding similar to what were reported for African American CRC patients. Therefore, this first comprehensive analysis of CRC Iranian tumors reveals a high MSI rate. The MSI tumors displayed the lowest level of chromosomal aberrations but high frequency of methylation (Brim et al. 2014). The MSI-L was predominantly targeted with chromosomal instability in a way similar to the MSS tumors. Chromosome 20 consistently in 20q13 region and its corresponding genes were amplified as shown in Fig. 13.2. The global chromosomal aberration profiles showed many similarities with other populations but also differences that might allow a better understanding of CRC’s clinico-pathological specifics in this population.
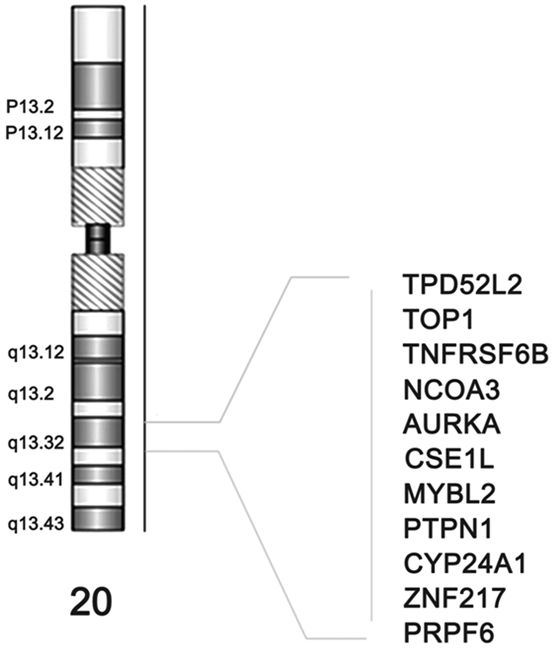
Fig. 13.2
Chromosome 20’s consistently amplified 20q13 region and its corresponding genes
In spite of the characteristics of CRC ethnicity, differences in clinical presentation and surgical management of right- and left-sided large bowel cancer are well known (Fig. 13.2). Tumors that are on the right side usually present at a more advanced stage and are accompanied by symptoms such as weight loss and anemia. On the contrary, left-sided tumors often present with rectal bleeding, change in bowel habit, and tenesmus. However, the molecular pathology of carcinomas might differ with respect to the side they are one in the large bowel. These variations and differences will become more significant with advancements in systemic treatments (Richman and Adlard 2002; Fig. 13.3).
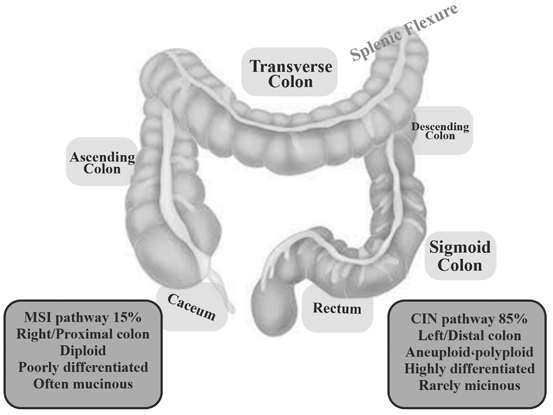
Fig. 13.3
Right-sided tumors are classified as splenic flexure (caecum, ascending colon and transverse colon), whereas left-sided tumors arise distantly to this site (descending colon, sigmoid colon, rectum). Two separate mutational pathways (MSI, CIN) have been shown
In a study conducted by Ellidokuz and colleagues, polyps and cancers were more prevalent in the left colon. Right-sided SCRCs are more fatal than left-colon cancers (Ellidokuz et al. 2003). Most SCRCs are located proximally in women and distally in men (Potter et al. 1993). Researchers have found that age is an important factor in the distribution of colorectal polyps and cancers, and a proximal trend is observed with increasing age (Samowitz et al. 2007).
A different embryological origin has been assumed for proximal and distal colons suggesting that tumors originating at any of these sites could develop along different pathways. The midgut eventually develops into distal duodenum, jejunum, ileum, caecum, appendix, ascending colon, and proximal two thirds of the transverse colon. The hindgut develops into the distal third of the transverse colon, the sigmoid colon, rectum, and upper two thirds of the anal canal. The different origins of the proximal and distal colons also lead to different vascular supplies. The proximal colon is served by the superior mesenteric artery and the distal colon by the inferior mesenteric artery. Moreover, proximal and distal colons differ in expression of several antigens, metabolism of glucose, polyamines and butyric acid, as well as in bile acid consumption, and composition and density of the bacterial population (Bufill 1990; Pocard et al. 1995; Distler and Holt 1997). Bufilland co-workers found different gene expressions in 87 genes in the ascending and descending fetal colon, indicating the occurrence of additional changes in gene expression in postnatal development (Bufill 1990). However, significant anatomic differences have already been found in the embryonic colon (Glebov et al. 2003). Gene expression was studied in fetal (17–24 weeks gestation) proximal and distal colon. More than 1000 genes were expressed differentially in adult ascending versus descending colon, with 165 genes showing > 2-fold and 49 genes showing > 3-fold differences in expression (Table 13.1).
Table 13.1
Representative gene that show more than three-fold differences in expression in ascending versus descending adult colon
Gene | Fold changes |
|---|---|
Home box DB | 8.15 |
Retinoic acid receptor responder | 11.83 |
Alpha actin 2 associated LIM protein | 14.46 |
acyl coenzyme A oxidase 1 Palmitoyl | − 13.1 |
Megrin A, B | − 15 |
Ethanol amine kinase | − 19.74 |
Therefore, developmental and biologic differences in proximal and distal colon may reflect differing susceptibilities to neoplastic transformation. Insights on the mechanisms of CRC can be provided by comparing patterns of gene or protein expression in different populations or in proximal and distal tumors. One of the most important clinical applications is related to the better selection of patients needing chemotherapy. Chemoresponsive patients will be identified more accurately through molecular profiling. For example: Patients with MSI-H tumors had a modestly better prognosis than those with microsatellite-stable (MSS) or MSI-low (MSI-L) cancers, yet also did not seem to benefit from adjuvant fluorouracil (FU)-based chemotherapy. This resistance to FU is presumably due to incorporation of FU metabolites into DNA rather than inhibition of its effective target, thymidylate synthase (Kimmie and Schrag 2010).
In one study, sporadic CRCs were analyzed for microsatellite instability, expression status of mismatched repair genes (hMLH1, hMSH2), and presence of the BRAF (V600E) in Omani, Iranian, African-Americans patients to analyze the difference between molecular genetics of CRC in different populations. Tumors with BRAF mutations were on the left side in Omanis while 88 % of such mutations were found on the right side of the colon in African-American patients. The highest and lowest rates of microsatellite instability tumors at two or more markers (MSI-H) were seen in African-Americans and Omanis (31 vs. 13 %), most of which were located in the proximal colon in African-American and Iranian patients. hMLH1 gene expression defects were seen approximately two times more in African-Americans and Iranians compared with Omanis (77 vs 38 %). Tumors in all patients had BRAF mutations and mains had the most mutations (19 %). These findings suggest that Omani and Iranian patients experience CRC at a younger age. The incidence of MSI-H was lower in these groups compared with the African-American patients. This emphasizes on the key role of hMLH1 expression and BRAF mutation in MSI-H CRC in these populations compared with other populations (Brim et al. 2008).
On the other hand, 36 (23.8 %) tumors out of 151 were MSI+ in Iranians, especially in those with proximal tumors (OR = 10.4; 95 %CI = 3.9–27.8) and in smokers (OR = 2.9; 95 %CI = 1.3–6.7). MTHFR 677CT+ TT genotype was strongly associated with MSI (OR = 2.6; 95 %CI = 1.3–5.3) and a positive relationship was found between the hypermethylation of mismatch repair genes and the incidence of MSI (p = 0.001) Therefore, the researchers suggested the MTHFR 677CT+ TT variant genotype as a risk factor for MSI+ cancer (Naghibalhossaini et al. 2010). A study on the Iranian population provided insights into the mechanisms of colorectal tumor development. K-ras, p53 mutations were significantly more common in left-sided than in right-sided tumors, indicating differences in the carcinogenesis pathways in these tissues. This finding is consistent with previous reports (Russo et al. 2005; Calistri et al. 2005; Toribara and Sleisenger 1995). This could be explained by the fact that the left-sided bowel lumen exposed to ingested carcinogens and mutagens more than the right-sided bowel lumen. Moreover, simultaneous p53 and K-ras mutations were rarely seen in the same tumor. In the mentioned study, the researchers evaluated the hot spot genetic changes leading to the development of CRC in three key genes (APC, K-ras, and p53) in a CRC series from southern Iran for the first time. Mutations in exons 5 and 7 of p53, and exons 1 or 2 of K-ras genes were investigated in 151 sporadic CRC tumors by PCR-SSCP, and K-ras results were confirmed by pyrosequencing. p53 was the most frequently mutated gene, with a frequency of 62/151 (41.1 %). This finding is consistent with previous studies on CRC, with reported frequencies ranging from 40 to 50 % of p53 alterations in CRC (Soong et al. 2000; Soussi et al. 2000; Leroy et al. 2014). K-ras gene mutations were identified in 46 (30.5 %) of 151 cases which is in line with the reported frequency of 27.4 % (Smith et al. 2002). Mutations of K-ras result in specific amino acid substitutions that lead to permanent activation of the encoded p21 ras protein (Barbacid, 1987). It is well known that the activating K-ras mutations cluster at codons 12/13 (GTP-binding domain). Similar with previous reports, most samples of K-ras mutations were found in codon 12, with a smaller number of nucleotide substitutions in codon 13. No K-ras mutations were detected in codon 61 in Iranian patients. This confirms that codons 12 and 13 are also the hot spots of mutations in the Iranian population. K-ras gene mutations were localized more in distal (41.8 %) than proximal (13.3 %) tumors (p = 0.001). Most K-ras mutations were base pair transitions which mostly occurred at the second bases of codons 12 and 13. All mutations in codon 12 and 13 were G→A transitions (data not published). Ras proteins are key components of signal transduction pathways leading from cell surface receptors to the control of cell proliferation, differentiation or death. Active Ras as well as mutated Ras stimulates the RAS-RAF-MEK-ERK-MAP kinase signaling pathway through tyrosine kinase receptors (RTKs) and EGF ligand. Therefore, anti-EGFR therapies via monoclonal antibody or EGFR-tyrosine kinase inhibitors are able to block cell proliferation, survival, invasion and metastasis. When the EGFR is blocked wild type K-ras doesnot signal and the tumor cells do not proliferate. However, mutated K-ras is permanently turned on, tumor cells proliferate.
On the other hand, Mokarram and colleagues found that one of the critical factors for cancer progression is the hypermethylation of the specific locus near the MGMT start codon. In patients with CRC, the assessment of MGMT-B which is associated with K-ras mutation has high prognostic value (Mokarram et al. 2012). Moreover, 41.1 % of Iranian cases carried a pathogenic p53 mutation, which is similar to those reported for CRC in other populations (Mcdermott et al. 2002; Iacopetta 2003; Russo et al. 2005). The type of p53 mutations was not determined in the aforementioned study. However, in other populations (Russo et al. 2005; Béroud and Soussi 2003; Olivier et al. 2003; Soussi and Beroud 2003), most detected p53 mutations were G→A transitions at CpGs (Pfeifer 2006). This type of mutation could be the result of spontaneous deamination or preferential adduct for mutation at methylated CpGs (Soussi and Beroud 2003; Pfeifer 2006; Greenblatt et al. 1994), followed by G→A transitions at non-CpGs that might be associated with hypemethylation of MGMT in our population (Mokarram et al. 2012). However, since simultaneous mutations in K-ras and p53 were very rare (p = 0.007) and the frequency of p53 mutations in tumors differed in the left and right colon, it seems that MGMT does not follow the same mechanism in two genes.
Other study showed when neoplasia gains K-ras mutations, the neoplasia would transform into tubular adenoma with villous architecture and accumulate dense methylation in the SFRP2/MGMT promoter, consequently becoming non-MSI cancer (Takeda 2011; Fig. 13.4).

Fig. 13.4
Methylation pattern of polyp to cancer
21/62 (33.8 %) mutations in left-sided tumors were in exon 5, and 12/62 (19.4 %) were in exon 7. In the right colon, only 6/62 (9.6 %) mutations occurred in exon 5, while 12/62 (19.4 %) of mutations occurred in exon 7. 11/62 (17.7 %) of the simultaneous mutations in both exon 5, 7 were localized in the left colon. Mokarram and co-workers did not find alterations in p53 (exons 5 or 7) or K-ras genes in 35.7 % of Iranian patients with cancer. Therefore, these mutations possibly lie on alternate pathways in the development of CRC which depends on epigenetic mechanisms (data not published).
The researchers found the MSI-H phenotype in 23.8 % of the patients which is higher than reports from Western countries but not in African-Americans (Gryfe and Gallinger 2001; Peltomaki 2001). Their finding was consistent with the results reported from the North of Iran (Bishehsari et al. 2006). The high prevalence of MSI-H could be related to the relatively high number of right-sided tumors found in the studied group (43.3 %). Most MSI-H tumors in the mentioned study were right sided (72.2 %) which is consistent with results from developed countries indicating a relatively high rate of MSI-H in proximal colon cancer (Ashktorab et al. 2005; Urso et al. 2008). A higher percentage of K-ras gene mutations were detected in non-MSI-H tumors (40/46, 87 %) compared with MSI-H tumors (6/46, 13 %, p = 0.04). Similar results were obtained for total mutations (individuals with mutation in any of two genes, p53 or K-ras) and MSI status (p = 0.04). This finding is consistent with another report showing that K-ras mutation is less frequent in patients with sporadic MSI-H with hMLH1 hypermethylation compared with non-MSI-H CRC without hMLH1 methylation and MSS CRC (Oliveira et al. 2004). There is an inverse relationship between microsatellite instability and K-ras, p53 mutations regardless of the type of microsatellite. However, the existence of such a relationship is controversial among different studies (Fujiwara et al. 1998).
A previous report from Iran showed that sporadic MSS and sporadic MSI-H CRCs shared similar K-ras mutation frequencies. However, the type of K-ras mutations differed between MSI-H and MSS subgroups, suggesting a link between this specific type of mutation and MMR defect in MSI-H CRCs. Defects in other DNA repair systems, such as O6-methylguanine-DNA methyltransferase (MGMT) activity might be related to inability to protect from G→A transition in K-ras induced by alkylating agents (Esteller et al. 2000; Lees et al. 2004; Qi et al. 2005), which was shown in a recent study (Mokarram et al. 2012).
The relatively low frequency of tumors with mutations in both genes (K-ras and p53) as well as the fact that 57 % of tumors had mutations in only one gene (p53, or K-ras), implies that the progressive accumulation of multiple mutations in these genes is not a prerequisite for tumor development and does not represent a synergistic evolutionary pathway. Therefore, the simultaneous alterations in K-ras and p53 are not frequent, suggesting that the sequential K-ras-p53 module is not obligatory in the progression of CRC in Iranian population. While some studies have shown that K-ras mutations may be lost through selection in the progression of tumors from adenoma to carcinoma, (Pretlow et al. 1993; Ines et al. 2014) we found no statistically significant difference in K-ras mutations between stage I and II. However, a significant difference was found in the frequency of p53 mutations between stage I and II, suggesting that p53 inactivation may be an important determinant of tumor progression. In contrast to other reports suggesting that MSS and MSI-L tumors have a common molecular background (Bouzourene et al. 2000; Laiho et al. 2002). Mokarram and co-workers observed more p53 mutations in the MSI-L tumors in lower stages (I, II). It seems that MSI-L CRCs were distinct from both MSI-H and MSS cancers in our study population. Correlations between p53 mutations and tumor stage correspond with the notion that p53 mutations accumulate during CRC progression (Bouzourene et al. 2000; Akkiprik et al. 2007). An important aspect of this study is that two major gene mutations were analyzed in Iranian patients with CRC in relation to prognosis for the first time. Although we and other researchers showed that there are genetic variations between population and proximal and distal CRCs, the issue of whether proximal and distal CRC should be considered as a single entity or two distinct entities is still debated. Further studies addressing the heterogeneity of the pathogenetic pathways leading to sporadic CRC in different populations as well identifying biological and/or molecular differences between proximal and distal CRCs should be done in this regard. With respect to the obtained data, future therapies must be targeted on individual patients, based on the detailed understanding of their genetic background and the nature of the mutation present in individual tumors.
13.2 Epigenetic and CRC
Epigenetics include variations in chromatin structure that modulate the use of the genome by histone modification, chromatin remodeling, histone variant composition, DNA methylation, and noncoding RNAs. Most of these modifications and chromatin changes are reversible and, therefore, are unlikely to be propagated through the germ line. In today’s modern terms, epigenetics can be molecularly defined as “the sum of the alterations to the chromatin template that collectively establish and propagate different patterns of gene expression (transcription) and silencing from the same genome that is associated with alteration in cancers (Roloff and Nuber 2005; Zoratto et al. 2014; Wolffe and Matzke 1999). Multiple, cumulative epigenetic and DNA sequence changes in the cells’ genome would lead to tumor. Sequence changes include deletions in chromosome regions accompanied with gene loss that may be associated with negative cell- cycle regulation (tumor suppressor genes), mutations that may activate or inactivate a number of proteins, gene amplifications entailing an over expression of specific genes, and even loss or gain of entire chromosomes.
The etiology of human cancer changing from a benign neoplasm to a malignant tumor has been explained by pathways involving the accumulation of genetic and/or epigenetic alterations (Kinzler and Vogelstein 1996; Vogelstein et al. 2013). Mutations in oncogenes and tumor suppressor genes comprise classic genetic alterations. The “classic” genotype comprises only about 10 % of CRCs (Smith et al. 2002, Rennie and Nelson, 1998). Besides these genetic alterations, the initiation and promotion of cancer could occur by epigenetic mechanisms. These mechanisms can be defined as heritable changes in gene function that are not due to changes in DNA sequence (Jones and Baylin 2002). Three widely accepted mechanisms for the “epigenetic regulation” of genes are (a) changing patterns of DNA methylation, (b) histone acetylations/deacetylations, and (c) alterations in stable circuits and regulatory feedback loops for growth and transcription factors (Rennie and Nelson 1998).
Ever since Fearon and Vogelstein defined their colorectal carcinogenesis model in 1990, a deeper understanding of this process is constantly developing (Fearon and Vogelstein 1990; Alberto Morán et al. 2010). Currently, only 60 % of CRCs emerge from adenomas via the suppressor route, also called chromosome instability (CI), which is initiated by a mutation in gene APC—this is the classical description of colorectal carcinogenesis corresponding to the adenoma- carcinoma sequence (Fearon and Vogelstein 1990; Alberto Morán et al. 2010). Moreover, two additional alternative, non–excluding routes, are now considered; the microsatellite instability (MSI) pathway, associated with Lynch syndrome and a small proportion of sporadic cases, and the methylator phenotype pathway, most recently identified and referred to as CIMP (CpG Island Methylator Phenotype) in the English-language literature. Therefore, CRC is being increasingly classified into various phenotypes according to its molecular profile (Fearon and Vogelstein 1990; Vilar et al. 2014). Therefore, from a molecular viewpoint it is classified based on the predominant cell event (CIN, MSI, CIMP) or, equivalently, according to the event-initiating factor (suppressor pathway for CIN; mutation pathway for MSI; methylator pathway for CIMP). Mutual exclusion can be seen in some colorectal carcinogenesis pathways such as MSI and CIN, whereas in CIMP there may be some overlap. Since MSI arises through a process of methylation of the hMLH1 promoter, it is not surprising that a significant overlap between CIMP and sporadic MSI has been reported (Toyota et al. 1999). With respect to the CIMP phenotype and mutation overlapping, the methylation status of three cancer-related genes (APC2, p14ARF, and ECAD) were evaluated in colorectal carcinogenesis and their association with the mutational status of BRAF and K-ras among Iranian patients with CRC (Naghibalhossaini et al. 2011). The frequencies of APC2, E-CAD, and p14 methylation were 92.6, 40.4, and 16.7 %, respectively. However, V600E mutation was not found in the BRAF gene in any of the samples and gene methylation was not related to K-ras mutations. These findings implied a distinct molecular pathway for APC2, p14, and ECAD methylation different from those previously described for CRCs with BRAF or K-ras mutations (Naghibalhossaini et al. 2011).
In another study, the MTHFR CT genotype was associated with an increased risk of tumor methylation (OR = 2.5; 95 % CI, 1.1–5.6). Methylated tumors were more frequent in the high methyl donor, especially in those who had the MTHFR CT and CT/TT genotypes (p = 0.01, p = 0.002, respectively). Hence, the risk of promoter methylation in tumor-specific genes was higher when serum folate/vitamin B(12) levels were higher. Such a relationship is modified by MTHFR C677T genotypes (Mokarram et al. 2008).
The aberrant methylation profile of four genes (APC, Axin1, Axin2, and GSK3β) was studied in an unselected series of 112 sporadic CRCs using methylation specific PCR to better understand the methylation silencing of the WNT pathway during colorectal carcinogenesis. The Axin2 C148T genotype was assessed in patients with CRC as well as healthy controls with PCR-RFLP. Among 18.75 % of CRCs at least one had methylated gene and 7.1 and 11.9 % of tumors had experienced promoter methylation in Axin2 and APC genes, respectively (Naghibalhossaini et al. 2012). Gsk3β and Axin1 gene were not methylated in these tumor series. Promoter methylation of Axin2 was seen in women more than men implying that this type of methylation is sex-related (p = 0.002). Patients with distal tumors had a lower risk of developing CRC compared with proximal ones (OR = 0.3; 95 % CI 0.1–0.9, p = 0.04). These findings depict the minor role of Axin1 and GSK3β methylation in carcinogenesis of CRC (Naghibalhossaini et al. 2012). In general, there is a strong correlation between polymorphism in specific genes and CIMP profile (Curtin et al. 2009; Slattery et al. 2009).
The Wnt signaling pathways are a group of signal transduction pathways made of proteins that pass signals from outside of a cell through cell surface receptors to the inside of the cell. Dysregulation of WNT signaling pathway was first identified for its role in carcinogenesis.
13.2.1 The Role of DNA Methylation and Chromatin Modification in CRC
Deviant changes in DNA methylation and histone modification are two of the most common types of epigenetic alterations in cancer. Such alterations direct gene expression through maintaining restricted and permissive chromatin states and occur at multiple layers of regulation. They can also be controlled by cancer cells for oncogenic gain (Eden et al. 2003; Ashktorab et al. 2009). Hypo or hyper- DNA methylation and loss of cytosine methylation results in genome instability (Fig. 13.5).
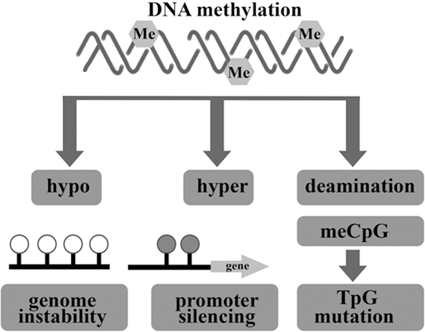
Fig. 13.5
Epigenetic alteration involving DNA methylation can lead to cancer by various mechanisms
DNA methylation (an enzyme-induced chemical modification to the DNA structure) comprises the major form of epigenetic information in mammalian cells. A methyl (CH3) group is covalently bonded to the 5-carbon on the cytosine base of CpG island in promoter. The methyl group is provided by S-adenosyl methionine (SAM), and this is converted to S-adenosyl homocysteine (SAH) in the process. In a pathway dependent on folate and cobalamin this is recycled back to SAM (Wajed et al. 2001).
CpG methylation occurs in vast areas in the genome in which the sequences are repeated such as centromeres and transposon elements (Wang and Leung 2004) CpG island shores (Irizarry et al. 2009), noncoding regions (ie, enhancer regions and miRNAs; Varambally et al. 2008), and gene bodies (silencing alternative transcription start sites; Guenther et al. 2007). CpG sites are seen in about 60 % of gene promoters (Bird 2002).
In mammals, the interplay of three independently encoded DNMT’S (DNMT1, DNMT3A, and DNMT3B) is influential in global cytosine methylation patterns. The role of fourth DNMT (DNMT2) which has been cloned needs to be proven in vitro or in vivo (Okano et al. 1998; Robertson 2001; Yoder and Bestor 1998; Subramaniam et al. 2014). The enzyme Dnmtl is specific for CpG and has significant activity against non-methylated DNA. As a maintenance enzyme, DNMT1 preserves existing methylation patterns after cell replication, and its deletion leads to apoptosis (Bird 2002) as well as death in mice if lost during embryonic development (Endres et al. 2001). In contrast, DNMT3A and -3B are de novo methyltransferases that methylate previously unmethylated DNA. Although their enzymic class is the same and their catalytic domains are similar to some extent, they might play different roles in tumorigenesis; cancer progression may be promote by DNMT3A deletion (Gao et al. 2011), while deletion of DNMT3B might inhibit oncogenesis (Nosho et al. 2009; Linhart et al. 2007). Transition between active and non- methylated gene promoter due to DNA methylation is mediated by Methyl-CpG binding proteins (MeCP) and Sin3A.
Ashktorab and colleagues studied the effect of gene methylation in the development of CRC (Wang et al. 2013). Their results showed that EDN2 and EDN3 genes were hypermethylated in primary human colon cancers and in a panel of human colon cancer cell lines. Therefore, epigenetic inactivation of ET-2 and ET-3 occurs frequently in both rat and human colon cancers (Wang et al. 2013). Sojblom et al and Schuebel et al also introduced CAN genes (including 13 genes, GPNMB, SYNE1, APC2, EVL, MMP-2, ICAM-5 PTPRD, CDH5, LGR6, STARD8, CD109, RNF182 and showed that genetic and epigenetic alterations of these markers are important in the progression of CRC (Schuebel et al. 2007; Sjoblom et al. 2006). They described a microarray of whole human transcriptome screen for identifying the genes that are silenced by promoter hypermethylation in human CRC. Therefore, candidate cancer genes are identified in single tumors with high validation efficiency. Moreover, as shown in Fig. 13.6, the important relationship the altered tumor genome and the gene hypermethylome was identified by comparing candidate hypermethylated genes with recently identified mutated genes in CRC (Schuebel et al. 2007; Sjoblom et al. 2006).
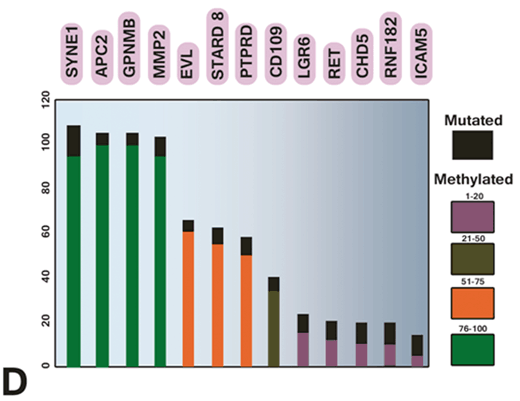
Fig. 13.6
Comparing methylation and mutation frequency of cancer genes in CRC tumor samples. (Schuebel et al. 2007)
Mokarram and co-workers studied CAN gene methylation and CHD5 protein expression CRC tissue microarrays (TMA) using immunohistochemical staining. Methylation-specific PCR was used to study the status of promoter methylation in CAN genes in Iranians and African-Americans (Mokarram et al. 2009). The researchers also studied microsatellite instability (Mokarram et al. 2009). More than 65 % of SYNE1, MMP2, APC2, GPNMB, EVL, PTPRD, and STARD8 genes were methylated, whereas the methylation was < 50 % for LGR6, RET, CD109, and RNF in both populations (Mokarram et al. 2009). However, a significant difference was observed for chromodomain-helicase-DNA-binding protein 5 (CHD5), ICAM5, and GPNMB methylation among the two studied populations (Mokarram et al. 2009). MSI-H was respectively found in 31 and 28 % of tumors in African-Americans and Iranians. African-Americans had a significantly higher methylation rate with respect to GPNMB, ICAM5, and CHD5 genes (Mokarram et al. 2009). The low expression of CHD5 in CRC was correlated with CHD5 promoter CpG island hypermethylation. CHD5 is a tumor suppressor which is frequently downregulated in a variety of human cancers.
The effect of microRNA-211 (miR-211)-regulated CHD5 expression was investigated on colorectal tumorigenesis. MiR-211 was predicted to target CHD5 using Target Scan software analysis. An exogenous miR-211 CRC cell line (HCT-116[miR-211]) was generated using lentiviral transduction and used as a model for in vitro and in vivo studies. The expression level of miR-211 in HCT-116(miR-211) cells had a 16-fold upregulation rate compared with vector control cells (HCT-116[vector]). The CHD5 protein level in HCT-116(miR-211) cells was decreased by 50 % as a result of the direct binding of exogenous miR-211 to the 3’-untranslated region (3’-UTR) of CHD5 mRNA. HCT-116 (miR-211) cells had a higher cell proliferation level, tumor growth, and cell migration compared with HCT-116 (vector) cells under in vitro and in vivo conditions, using MTT, colony formation, flow cytometry, scratch assay, and tumor xenografts, respectively. Moreover, p53 pathway-associated regulatory proteins (MDM2, Bcl-2, Bcl-xL, and Bax) were induced changes as a result of the enforced expression of miR-211 in HCT-116 cells. Ashktorab’s results show that CHD5 is a direct target of miR-211 regulation (Cai et al. 2012). Tumor cell growth is promoted by this enforced expression, partially by downregulating the expression level of the CHD5 tumor suppressor. Therefore, a better understanding of the association of between miR-211-regulated CHD5 expression and CHD5 function in colorectal tumorigenesis is provided (Cai et al. 2012).
Epigenetic alterations occur early in the progression process and often precede malignancy (Wang and Tang 2008). The adenoma to carcinoma progression sequence and the hyperplastic polyp-serrated adenoma to carcinoma sequence are concurrent molecular changes that are influential in the development of colon cancer (Petko et al. 2005). These sequences result from the progressive accumulation of genetic and epigenetic alterations that transform normal colonic epithelium to colon adenocarcinoma (Grady et al. 2001). Two types of hyperplastic polyp associated with CRC can be defined through methylation patterns (Wynter et al. 2004).
According to the classic model for genetic alteration of colon cancer, the altered DNA methylation is shown to occur very early. One alternative mechanism includes proteins that bind selectively to methylated DNA. MeCP2 is one of these proteins which were initially identified in 1992. Structurally, it has a methyl-CpG binding domain (MBD) which recognizes a symmetrically methylated CpG dinucleotide via contacts in the major groove of the double helix, and a transcriptional repression domain (TRD) that interacts with other regulatory proteins.
Another mechanism by which DNA methylation could inhibit transcription is through blocking the access of transcription factors through MBD binding. It was found that MeCP2 could recruit histone deacetylase (HDAC). Therefore, DNA methylation was able to repress transcription and result in a chromatin structural change by recruiting MBD’s and their associated HDAC’s to methylated DNA. As a result, core histone tails would be locally deacetylated, in turn leading to tighter DNA packaging. This would ultimately reduce the transcription factors’ access to their binding sites. Recent studies have shown a link between four of the MBD-containing proteins (MeCP2, MBD1, MBD2, and MBD3) and aspects of the chromatin remodeling machinery in addition to HDAC. An understanding of the mechanism of repression came from the realization that MeCP2 associates with the Sin3a corepressor complex and depends on histone deacetylation for its action. This finding showed that DNA methylation could be read by MeCP2 and provides a signal to alter chromatin structure. (Wynter et al. 2004; Laird 2005; Hall et al. 2013; Pancione et al. 2010; Cowley et al. 2005; Curradi et al. 2002).
By treating cells with a combination of the DNA methyltransferase inhibitor 5-azaCdR and the histone deacetylase inhibitor trichostatin A (TSA) a link between DNA methylation and histone deacetylation can be determined. When the doses of 5-azaCdR were low, re-expression was low and the demethylation of hypermethylated CpG-island-associated genes were minimal. However, the same genes were robustly activated when a combination of 5-azaCdR and TSA were used, while TSA per se had no effect. This showed that DNA methylation and histone deacetylation work together to silence transcription. Moreover, DNA methylation was dominant over the histone acetylation status (Robertson 2001; Laird 2005; Cowley et al. 2005; Curradi et al. 2002; Toiyama et al. 2014).
A considerable amount of DNA methylation in embryonic stem cells seems to occur independent from CpG sites (Lister et al. 2009). The Ten-Eleven-Translocation (TET) oxidase family converts 5-methylcytosine to 5-hydroxymethylcytosine which is a step towards demethylation. This process does not seem to be limited to CpG islands (Ito et al. 2011). The exact roles of these phenomena in epigenetic regulation are still not fully understood.
Core histone modifications (ie, two copies of H2A, H2B, H3, and H4 proteins) are also crucial in epigenetic regulation apart from direct DNA manipulation. Epigenetic silencing of gene transcription does not occur only by promoter methylation, but is mediated by a complex series of molecular events that remodel chromatin configuration (Gronbek et al. 2007; Goossens et al. 2014). Recent studies show chromatin changes occurring during tumorigenesis. Cancer cells display palmate changes in histone methylation and histone acetylation patterns (Sharma et al. 2010). Histone methylation and acetylation for gene activation or deactivation depend on the modified residue of the histone (Vo and Millis 2012).
To moderate transcription, histones bind selectively and release DNA as nucleosomes. DNA is regulated by adding acetyl, methyl, phosphoryl, ubiquityl, or sumoyl groups, producing a dynamic epigenetic histone code (Rodríguez-Paredes and Esteller 2011). These histone marks are deposited or removed by a plethora of proteins and clinical targets, including histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methyltransferases (HMTs), histone demethylases (HDMs), kinases, phosphatases, and others (Campos and Reinberg 2009; Beck et al. 2011). The positive histone charge is contradicted by the acetylation of lysines at specific sites (such as on H3K4) by HATs, and permits negatively charged DNA to form a configuration ready for transcription. However, acetyl groups are removed from histones by HDACs, and consequently DNA is shielded from expression because the oppositely charged histone is bind to the DNA. Although there are some exceptions, gene expression is usually silenced by DNA methylation. Moreover, histone marks can activate or silence genes, depending on the target residues, the targeted histone, and extent of the alteration (Sawan and Herceg 2010). With respect to histone modification and cancer incidence, the status of global histone acetylation (by measuring H3, H4 acetylation of lysine residues, which also occur over large regions of chromatin including coding regions and non-promoter sequences) and the expression of histone deacetylase 2 (HDAC2) was assessed in CRC tissues. Global histone H4 acetylation and HDAC2 expression in colon adenoma and carcinoma (Ashktorab et al. 2009). High levels of HDAC2 nuclear expression were detected in 81.9, 62.1, and 53.1 % of CRC, adenoma, and normal tissues, respectively (p = 0.002). In moderate to well differentiated tumors increased global expression levels were seen for H4K12 and H3K18 acetylation as opposed to poorly differentiated tumors (p = 0.02). A significant correlation was found between HDAC2 expression and progression of adenoma to carcinoma, when comparing cancer and non-cancer tissues. Thus, a significant association between HDAC2 expression and CRC progression is implied (Ashktorab et al. 2009).
According to Fig. 13.7 many combinations of histone modifications are known to establish specific states for activating or inhibiting expression. For example, histone H3 marks such as trimethylation of lysine (K) 4 over the promoter and K36 over the gene body (H3K4me3 and H3K36me3, respectively) establish permissive chromatin states (Barski et al. 2007; Rosenfeld et al. 2009). For example, histone H3-K9 methylation induces gene silencing and histone H3- K4 induces gene activation.
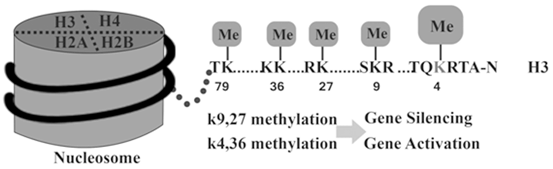
Fig. 13.7
Histone Methylation can lead to gene expression activation or inhibition
Monoacetylation of H3K9 and H3K14 (H3K9ac1 and H3K14ac1) (Li et al. 2007), the presence of variant histones such as H2A.Z (Hu et al. 2012), and methylation of enhancer elements downstream (H3K4me1) are among other enabling marks (Heintzman et al. 2009). An additional contributory layer is represented by nucleosome positioning. This is determined by the intrinsic binding affinity of the DNA sequence, competitive binding of surrounding proteins, and translocation activity by adenosine triphosphate-dependent remodeling complexes (Gaffney et al. 2012).
However, classically H3K27me3, H3K9me3, and H3K9me2 indicate the heterochromatin configurations that compact nucleosomes (Baylin and Jones 2011). They also indicate the presence of inhibitory proteins such as CTCF and HP1 (Wen et al. 2012; Keller et al. 2012) in addition to dense, localized DNA methylation. Polycomb group (Polycomb repression complex [PRC]) proteins can initiate repression, which methylate the histones in promoter regions. Such PRC regulators, including zeste 2 (EZH2) enhancer (Asangani et al. 2012), are themselves regulated by noncoding RNAs such as miRNA-101 to inhibit expression (Banerjee et al. 2011; Sakurai et al. 2012). EZH2 induces methylation of H3K27 which leads to the transcriptional repression.
Conclusively, the importance of the epigenome in coordinating transcription and downstream biological events have been underestimated considering the interplay between systems that previously thought to be independent. The complexity of such a system enables flexible conditions for guiding development and physiology, as well as guiding states such as tumorigenesis.
13.2.2 Hypermethylated Gene Promoters in CRC
Mammalian DNA contains 5-methylcytosine, the genomic distribution of which is specific for each cell type and is largely established during embryonic development. In normal tissues, DNA methylation patterns partially depend on the relative levels and activities of DNMT’S and DNA demethylases whose expression is regulated at both the transcriptional and posttranscriptional level (Turker and Bestor 1997; Jost and Bruhat 1997). Mammalian DNMT’S recognize CpG dinucleotides at specific sites within the genome called “CpG Island,” a CpG-rich region often encompassing the promoter and transcription start site of the associated gene possessing de novo methylation activity (Bock et al. 2007; Jones and Takai 2001). Approximately half of all human genes contain CpG islands in the 5′ area of their gene promoter (Bock et al. 2007; Jones and Takai 2001). Tumor suppressor or oncogenes are regulated through epigenetic marks. It is widely accepted that the activation of oncogenes or the hypermethylation of tumor suppressors cause pre-neoplastic lesions that in turn trigger SCRCs (Tanaka et al. 2006).
In nearly 35 % of CRCs the mechanism by which methylation occurs in the promoter regions of various genes plays is highly important (Snover 2011). CRCs can be differentiated from other tumors by features such as proximal colon preference, occurring more in women and in older age, and poor differentiation (Van Rijnsoever et al. 2002; Samowitz et al. 2005; Ogino et al. 2006). A number of studies have clearly indicated that the pattern of DNA methylation in tumors is strongly influenced by age, sex, and anatomic site (Wiencke et al. 1999; McMichael and Potter 1985; Vaiserman et al. 2014; Yamashita et al. 2014; Mcmichael and Potter 1983).
So far, three types of altered DNA methylation patterns have been identified in human cancer:hypomethylation, hyper-methylation, and loss of imprinting (LOI) (Schulz 1998; Bressan et al. 2014). Loss of the differential expression of parental allele is mostly seen in embryonal tumors (Reik and Surani 1989; Rainier et al. 1993). Many tumors, especially those in advanced stages experience DNA hypomethylation, which is widely known to be a genome wide event (Rainier et al. 1993; Bedford and Van Helden 1987). Promoter hypomethylation with an associated increase in gene transcription has also been defined. The hypermethylation of DNA takes place at special regulatory sites in the promoter regions or repetitive sequences (Sakai et al. 1991; Okada et al. 2010; Herman et al. 1994; Graff et al. 1995) and has tumor specificity (Baylin et al. 1997, 2012; Tanaka et al. 2006; Fan and Beck 2004; Kwabi-Addo et al. 2007). A heavy density of cytosine methylation in the CpG islands of the tumor suppressor gene promoters can lead to a complete block of transcription (Fan and Beck 2004), and many types of cancer use this mechanism to inactivate tumor suppressor genes and initiate cancer formation and progression (Sakai et al. 1991; Graff et al. 1995; Baylin et al. 1997, 2012).
Studies show that the earliest steps in tumorigenesis are depicated as abnormal clonal expansion which evolves during the stress of cell renewal. Factors such as aging, chronic injury, inflammation and epigenetic alteration could drive tumor progression via abnormal clonal expansion. Also, there is strong association between DNA methylation and histone modification to regulate gene expression. CpG island methylation affects a number of genes in colon cancer, and many studies have reported the significance of the epigenetic alterations in the pathogenesis of colon cancer.
13.2.3 Classification of Cancer Candidate Genes
Mokarram et al divided Cancer candidate genes into 5 classes which are classified in Fig. 13.8 (Mokarram et al. 2009).
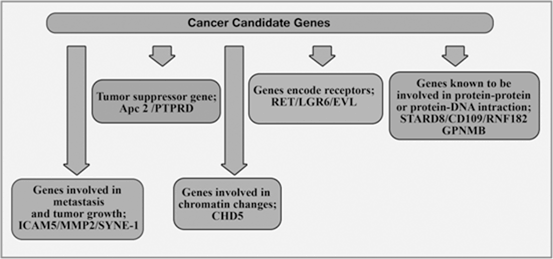
Fig. 13.8
Cancer candidate genes in CRC
The methylation profile of 13 genes in African-Americans and Iranian CRC tissue was investigated. Fresh (n = 51) sporadic primary CRC tumor samples were collected from surgical patients at several hospitals affiliated to Shiraz University of Medical Sciences, Shiraz, Southern Iran from July 2003 to September 2005. Also, 51 African-Americans CRC samples (19 fresh and 32 formalin fixed and paraffin embedded, from Howard University) sex and age-matched patients were also collected (Mokarram et al. 2009). The two groups of samples were analyzed using MSP analysis of 13 genes. It was hypothesize that in different populations CAN genes might show a different profile. It should also be mentioned that these genes could be inactivated through epigenetic silencing. In both populations, most analyzed genes were highly methylated. However, their levels of methylation differed in the two study populations, and from one gene to another. The synaptic nuclear envelope encoding protein (SYNE-1 gene) was methylated in all analyzed samples; while the RNF182 promoter, that encodes a ring finger protein, was not methylated in any of the samples (Mokarram et al. 2009). Function has been known for RNF182, SYNE-1 protein is involved in the process of cytokinesis (Fan and Beck 2004; Liu et al. 2008). The latter protein and KIF3B protein facilitate the accumulation of membrane vesicles at the spindle midbody. How this gene’s methylation is involved in a carcinogenic process is currently unknown. More than 65 % methylation was achieved for SYNE1, MMP2, APC2, GPNMB, EVL, PTPRD, and STARD8, while it was less than 50 % for LGR6, RET, CD109, and RNF.
13.2.3.1 Methylation and Age
Although there is a global methylating process that is age dependent in other studies, (Kwabi-Addo et al. 2007), the methylation pattern of 13 genes was independent from the patients’ age. The results obtained from the 13 CAN genes in the mentioned study reflect show that these genes are important in carcinogenic processes that are dependent on methylation.
Four genes (CD109, CHD5, LGR6, and ICAM-5) showed different levels of methylation depending on tumor locations. These genes were less frequently methylated in the left colon than in the right-side, which could be associated with the descending methylation gradient from the proximal to the distal colon.
13.2.3.2 EVL Methylation
Only the EVL gene promoter had a higher level of methylation from well differentiated to poorly differentiated tumors. Bournier and colleagues found that the co-expression of the EVL protein along with alpha-II spectrin reinforce the cell to cell interaction (Bournier et al. 2006). The down regulation of the EVL gene by hypermethylation in all poorly differentiated tumors and more than 75 % of well and moderately differentiated tumors possibly make the cells contact loose and ready to become invasive. This finding could be justified by the fact that this gene’s methylation increases by tumor stage. While EVL methylation was found only in 56 % of stage I tumors, this gene was methylated in 86 % of stage IV tumors.
13.2.3.3 RET Methylation
A similar stage dependent increase in methylation was observed for RET gene where 44 % of tumors were methylated at stage I, while 71 % were methylated at stage III and IV. No explanation for this result can be currently suggested, knowing that RET gene is a proto-oncogene (Herman et al. 1998) activated by gene translocations and mutations (Hedayati et al. 2006). However, the low methylation of RET, may be consistent with its tumorgenetic function in colon cancer progression. The LGR6 gene (encoding a Leucin-rich repeat-containing G-protein-coupled receptor) seemingly displayed a stage-dependent methylation process. Therefore, it is thus involved in cell proliferation via signal transduction (Shu et al. 2000). However, this gene’s function could not explain the methylation decrease from stage I to IV (78 to 29 %). The methylation profile was similar for MSI-H as well as non-MSI-H tumors for all genes, except the PTPRD gene which encodes a protein that is a member of the protein tyrosine phosphatase (PTP) family. This finding confirms dissociation between the microsatellite instability (MIN) phenotype and the CIMP in colon cancer tumors which has been previously established.
13.2.3.4 Different Methylation Profile was Found Between Iranians and African-Americans
Cell growth, differentiation, mitotic cycle, and oncogenic transformation are among the various cellular processes that are regulated by PTPs which are signaling molecules. Mori et al. have shown that a similar gene PTPRO is highly methylated in MSI-H tumors supporting this gene’s increased methylation status in the MSI tumors (Mori et al. 2004). Different methylation profile was found between Iranians and African-Americans for only four genes, GPNMB (89 vs 100 %), CHD5 (47 vs 78 %), LGR6 (31 vs 49 %), and ICAM-5 (7.5 vs 40 %).
13.2.3.5 GPNMB Gene Could Delay Growth and Reduce Metastatic Potential
GPNMB, a type I transmembrane glycoprotein, shows expression in the low metastatic human melanoma cell lines. However, it does not show such expression in high metastatic cell lines (Kuan et al. 2006). Therefore, since this gene is methylated more in African-Americans, it could account for the high aggressiveness and fast progression of colon tumors in this population.
13.2.3.6 ICAM-5 Gene Encodes a Type I Transmembrane Glycoproteins that is a Member of the Intercellular Adhesion Molecule (ICAM) Family
This finding is also reinforced by the fact that another gene involved in metastasis, ICAM-5, is highly methylated in African-Americans compared with Iranians (40 vs 7.5 %). ICAM-5 gene encodes a type I transmembrane glycoproteins that is a member of the intercellular adhesion molecule (ICAM) family. The high methylation rate of ICAM-5 leads to reduced cell-to-cell adhesion in corresponding tumor cells and in turn increased their invasive potential. This finding is consistent with the results of the GPNMB product, is similar to the ICAM-5 gene product, and leads to cumulative effects that all increase the invasiveness and metastatic potential of the colon tumor cells in African-Americans (Mokarram et al. 2009).
13.2.3.7 CHD5 Gene Methylation in African-American CRC Tumors Could Explain the High Incidence and Aggressiveness of CRC
Unlike GPNMB and ICAM-5, CHD-5 is involved in early tumorigenic processes at the chromatin remodeling and controls events such as that proliferation, apoptosis, and senescence via the p19Arf/p53 pathway (Bagchi et al. 2007). The higher level of methylation of this gene in African-Americans compared with Iranians (78 vs 47 %) might indicate the higher level of colon cancer in African-Americans (Mokarram et al. 2009).
The expression profiles of many genes are affected by chromatin modification. Chromatin modification affects tumor progression rate and aggressiveness. Colon tumors from African-Americans showed increased global Histone H4 acetylation and HDAC2 expression. Race might be the driving force for the high profile gene methylation since MSI in both African-Americans and Iranians had little difference. Environmental factors including smoking and drinking may also play a role in the distinct MSI and methylation level in these two populations.
The mentioned methylation study confirmed the biomarker status of many of the CAN genes that were shown to be highly methylated in the African-American and Iranian populations despite similarities in the MSI level.
A thorough analysis of both populations might need to be performed on the patients’ established cell lines using agents targeting both whole genome methylation (5-azacytidine) and/or the chromatin modification inhibitor (TSA) followed by differential microarray expression experiments to gain a global insight of all epigenetic processes taking place in the tumorigenesis within these two groups of patients.
13.2.4 Inactivation of MMR Genes in CRC
The most common cause of the inactivation of MMR genes (especially MLH1 and MSH2) in CRC is epigenetic silencing due to methylation in the promoter region. MMR deficiency via mutation or epigenetic silencing display MSI phenotype in hereditary or sporadic CRC and hMLH1 imperfection is more common in CRC.
In one study, 77 % of MSI-H tumors in both African-Americans and Iranians and 38 % of tumors in Omanis were found to have defects in hMLH1 gene expression (Brim et al. 2008). The aberrant methylation of MLH1 occurs in > 80 % of sporadic microsatellite instability (MSI) CRC, and the restoration of MLH1 expression and function by demethylating the MLH1 promoter in MSI CRC cell lines suggests that such aberrant methylation is a cause rather than a consequence of colorectal carcinogenesis (Herman et al. 1998; Kane et al. 1997).
On the other hand, Promoter methylation of p14ARF could be a significant alteration leading to CRC with MSI-L. Target CpG sites in GABRA1 and LAMA2 experienced aberrant with high frequency in tumor tissues compared with matched tumor-adjacent normal tissues in another study (Lee et al. 2012).
UNC5C and DCC share the ability to transmit cell death signals in the absence of their ligand (Ackerman et al. 1997; Hong et al. 1999; Llambi et al. 2001; Sanchez-Cespedes et al. 2000) and thus act as tumor suppressors in CRC (Ackerman et al. 1997; Mazelin et al. 2004). The netrin-1 receptors are aberrantly methylated in primary CRC (Kim et al. 2009; Shin et al. 2007) and are significantly correlated with Dukes’ stage C (Hibi et al. 2009b; Hibi et al. 2009a). UNC5C inactivation occurs early, whereas DCC loss is seen in advanced CRC more often (Chen et al. 2005 and Han et al. 2012). Therefore, it is implied that epigenetic alterations in the netrin-1 receptors do not occur randomly in CRC and may be related to its malignant potential. In 82 and 69 % of primary colon cancers, aberrant methylation was observed in netrin-1 receptor and UNC5C genes, respectively (Hibi et al. 2009a; Hibi et al.2009b).
A study showed that methylation at the p16INK4A promoter is higher in colon cancer tumors when compared with the normal tissue of the same individuals (Yoruker et al. 2012). CDKN2A promoter hypermethylation has been described in 12–51 % of CRCs and is often included in the panel of markers used to assess the CIMP phenotype (Shima et al. 2011).
Frequent APC hypermethylation is also found in CRC tissue but a promoter that is repressed in normal gastric tissue has been shown to be the site of hypermethylation in gastric cancers. Hence, frequent APC hypermethylation at major promoter sites in these tissues is important (Sproul et al. 2012). The following genes displayed alterations in methylation pattern from that of the mucosa of the non-cancer tissues compared with neoplasmic mucosa: MGMT, hMLH1, p16 INK4a, MINT1, MINT31 and with a great level of changes: COX2, cyclin A1 and CDX1, RAR, MYOD1, p15 INK4b, CDH13, CXX1, p73 and WT1 (Xu et al. 2004). One study investigated the methylation of 12 genes (APC, COX-2, DAP-kinase, E-cadherin, GSTP1, hMLH1, MGMT, p14, p16, RASSF1A, THBS1, and TIMP3) in normal colon mucosa, colon adenoma, and CRC. All the mentioned 12 genes were methylated in CRC and colon adenoma, except for GSTP1 gene. Also, normal colon mucosa was methylated for APC only (Lee et al. 2004).
Some studies also indicate an association between MGMT and CDKN2A methylation and K-ras mutations (Hawkins et al. 2009). Higher methylation levels were observed in tumor samples from patients with multiple lesions compared with those with solitary tumors regarding the following genes: MGMT, CDKN2A, SERF1, TMEFF2, HS3ST2, RASSF1A, and GATA4 (Gonzalo et al. 2010).
The frequencies of APC2, E-CAD, and p14 methylation were reported to be 92.6, 40.4 and 16.7 %, respectively, in another study (Gonzalo et al. 2010). NDRG2, THBS4, and Desmocollin 3 (DSC3) were also downregulated in CRC compared to benign colorectal tissues. Other studies showed that the vimentin gene (VIM), usually activated in mesenchymal cells, was highly methylated in colorectal carcinoma (Shirahata et al. 2010) and all connexins including GJA1, GJA9, GJB1, GJB2, GJC1 and GJD3 were hypermethylated in colon cancer cell lines (Sirnes et al. 2011).
Other researchers investigated the methylation level of several genes using tissue samples that included normal mucosa, adenomas, and carcinomas of the colorectum. The eleven studied genes were ADAMTS1, CDKN2A, CRABP1, HOXA9, MAL, MGMT, MLH1, NR3C1, PTEN, RUNX3, and SCGB3A1. ADAMTS1, CDKN2A, CRABP1, MLH1, NR3C1, RUNX3, and SCGB3A1 genes showed increased methylation levels from adenomas to carcinomas; while HOXA9, MAL, and MGMT were similarly methylated in all tumor stages. PTEN was not methylated in carcinomas, and therefore the researchers did investigate this gene in any of the tumor stages (Ahlquist et al. 2008).
A new hypermethylated gene called Phosphatase and Actin Regulator 3 (PHACTR3) was found that showed increased DNA methylation levels by more than 70-fold in advanced neoplasia (Bosch et al. 2012). Researchers evaluated the methylation level of two different regions from the RASSF2 and SFRP2 promoters in DNA from various tissue samples. Normal colonic mucosa specimen was mostly unmethylated, colorectal adenoma samples were partially methylated, and a CRC specimen was extensively methylated (Nagasaka et al. 2009). In primary CRC tissues, oncostatin M receptor-β (OSMR) and β-1,4-galactosyltransferase-1 (B4GALT) are highly methylated. Such methylation levels are rarely seen in corresponding normal adjacent mucosa or in non-malignant normal colon tissues. PAPSS2, TUBG2, NTRK2, B4GALT1 and SFRP4 are methylated in colon cancer tissues samples (Kim and Deng 2007). Methylation of BMP3, EYA2, ALX4, WIF-1, EGFR, SFRP-1, OST2 and vimentin was detected in tissue specimens of patients with colon cancer (Shirahata et al. 2010; Zou et al. 2007; Tokuyama et al. 2010). The hypermethylation of the KEAP1 promoter region leads to increased nuclear Nrf2 and downstream ARE gene expression in CRC cell and tissues (Hanada et al. 2012).
Conclusively, one of the major epigenetic modifications influential in the physiological control of genome expression is DNA methylation. As shown in Table 13.2 methylation patterns are extensively changed in cancer cells, thus leading to better differentiation of cancer cells from normal tissues. Measuring the extent of DNA methylation can help cancer diagnosis by identifying methylated genes that are commonly expressed.
Table 13.2
Methylation of the genes in CRC
Gene | Name | Histological type (% methylation, cases) | References |
|---|---|---|---|
3OST2 | 3-O-Sulfotransferase 2 | CRC (71.4 %) | (Tokuyama et al. 2010) |
ALX4 | Aristaless-like homeobox-4 | CRC (64 %, 30/47), adenoma (85 %, 11/13), normal colon mucosa (0 %, 0/21) | (Ebert et al. 2006) |
APC | Adenomatosis polyposis coli | MSS CRC (28 %, 7/25), MSI CRC (36 %, 10/28) | (Miotto et al. 2004) |
APC | Adenomatosis polyposis coli | Normal tissue (0 %, 0/21), CRC (21,%, 10/47), liver metastasis (42 %, 10/24) | (Miotto et al. 2004) |
AXIN 2 | AXIN 2 (CONDUCTIN) | MSI CRC (50 %, 5/10), MSS CRC | (Miotto et al. 2004) |
BNC1 | Basonucleolin | Colon tumor (92 %, 22/24), normal colon tissue (54 %, 13/24) | (Shames et al. 2006) |
BNIP3 | BCL2/adenovirus E1B interacting protein 3 | CRC (66 %, 40/61) | (Tan et al. 2007) |
CD133 | Prominin 1 | CRC (62 %, 10/16), normal colon control (0 %, 0/19) | (Lenhard et al. 2005) |
CDH1 | E-cadherin | MSS CRC (42 %, 10/24), MSI CRC (39 %, 11/28) | (Miotto et al. 2004) |
CDH13 | H-cadherin | CRC (38 %, 23/61) | (Jensen et al. 2008) |
CDH13 | H-cadherin | CRC (32 %, 27/84) | (Hibi et al. 2004) |
CDH13 | H-cadherin | CRC (65 %, 42/65) | (Xu et al. 2004) |
CDH4 | R-cadherin | CRC (78 %, 38/49), normal tissue (29 %, 5/17), CRC or adenoma (100 %, 10/10), normal tissue (0 %, 0/10) | (Miotto et al. 2004) |
CDKN2A/p14 | Cyclin-dependent kinase inhibitor 2A, alternated reading frame | Adenoma from FAP-Pts (41 %, 13/32), multiple adenoma Pts (69 %, 20/29), MSI-H CRC Pts (86 %, 12/14), MSS/MSI-L CRC Pts (88 %, 14/16) | (Wynter et al. 2006) |
CDKN2A/p14 | Cyclin-dependent kinase inhibitor 2A, alternated reading frame | MSS CRC (12 %, 3/24), MSI CRC (39 %, 17/28) | (Miotto et al. 2004) |
CDKN2A/p14 | Cyclin-dependent kinase inhibitor 2A alternated reading frame | CRC (32 %, 61/188) | (Kang et al. 2008) |
CDKN2A/p16 | Cyclin-dependent kinase inhibitor 2A | Adenomas (34 %, 14/41) | (Petko et al. 2005) |
CDNK2A/p16 | Cyclin-dependent kinase inhibitor 2A | Adenoma from FAP-Pts (52 %, 17/33), multiple adenoma Pts (45 %, 13/29), MSI-H CRC Pts (64 %, 9/14), MSS/MSI-L CRC Pts adenoma (63 %, 10/16) | (Wynter et al. 2006) |
CDNK2A/p16 | Cyclin-dependent kinase inhibitor 2A | MSS CRC (28 %, 7/25), MSI CRC (36 %, 10/28) | (Miotto et al. 2004) |
CDX1 | Audal type homeobox transcription factor 1 | CRC (100 %, 65/65) | (Xu et al. 2004) |
COX2 | Prostaglandin-endoperoxide synthase 2 | CRC (72 %, 47/65) | (Xu et al. 2004) |
CXCL12 | Chemokine (C-X-C motif) ligand 12 | CRC (76.2 %, 16/21), AP matched normal mucosa (0 %, 0/19) | (Wendt et al. 2006) |
Cyclin A1 | Cyclin A1 | CRC (100 %, 65/65) | (Xu et al. 2004) j |
DCC | Deleted in colorectal carcinoma | CRC (56 %, 28/50) | (Hibi et al. 2009a) |
EphA7 | Eph receptor A7 | CRC (49 %, 37/75) | (Wang et al. 2005) |
ESR1 ESR1 | Estrogen receptor-α Estrogen receptor-α | Lymph node from stage I & II CRC Pts (UICC stage) (31 %, 15/49), disease-free (86 %, 13/15), local recurrence (14 %, 2/15) UC with neoplasia UC without neoplasia | (Harder et al. 2009) (Tominaga et al. 2005) |
FLBN3 GATA4 GATA5 GJA4 | GATA binding protein 4 GATA binding protein 5 Gap junctions A4 | CRC (38.8 %) CRC (70 %, 63/90), noncancerous colon mucosa (6 %, 5/88) CRC (79 %, 61/77), noncancerous colon mucosa(13 %, 13/100) CRC (60 %, 29/48) | (Grady et al. 2008) (Hellebrekers et al. 2009) (Hellebrekers et al. 2009) (Sirnes et al. 2011) |
GJB6 | Gap junctions B6 | CRC (25 %, 12/48) | (Sirnes et al. 2011) (Sirnes et al. 2011) |
GJD2 ITGA4 LAMA2 | Gap junctions D2 Integrin, alpha 4 | CRC (96 %, 46/48) Adenoma (75 %, 27/36), adenocarcinoma (92 %, 69/75), colon mucosa (6 %, 2/32) CRC (80.6 %), normal tissue (13.4 %) | (Ausch et al. 2009) (Lee et al. 2012) |
MAL MGMT | T cell differentiation protein O-6-methylguanine-DNA | CRC (80 %, 49/61), adenomas (71 %, 45/63), normal mucosa (4 %, 1/23) Adenomas (49 % 19/39) | (Lind et al. 2008) (Petko et al. 2005) |
MGMT | methyltransferase O-6-methylguanine-DNA | MSS CRC (40 %, 10/25), MSI CRC(39 %, 11/28) | (Miotto et al. 2004) |
MGMT MLH1 MSX1 MYOD | methyltransferase O-6-methylguanine-DNA methyltransferase MutL homolog 1, colon cancer, nonpolyposis type 2 Msh homeobox 1 Myogenic factor 3 | Adenoma from FAP-Pts (66 %, 22/33), multiple adenoma Pts (41 %, 12/29), MSI-H CRC Pts (43 %, 6/14), MSS/MSI-L CRC Pts (53 %, 8/15) MSS (0 %, 0/25) CRC, MSI CRC (39 %, 11/28) Colon tumor (87 %, 21/24), normal colon tissue (42 %, 10/24) CRC (69 %, 45/65) | (Wynter et al. 2006) (Miotto et al. 2004) (Shames et al. 2006) (Xu et al. 2004) ,mj |
NDRG2 NDRG4 NMDAR2 NTRK2 OSMR p15INK4 p73 | N-myc downstream-regulated gene 2 N-myc downstream-regulated gene 4 N-methyl-D-aspartate receptor-2A Neurotrophin tyrosine kinase receptor type 2 Oncostatin M receptor-β Cyclin-dependent kinase inhibitor 2B Tumor protein p73 | CRC (27 %, 8/30), matched normal colon (0 %, 0/30) CRC (1st, 86 %, 71/83), CRC (2nd, 70 %, 84/128), noncancerous colon mucosa (4 %, 2/48) CRC (82 %, 82/100), matched normal colon mucosa (15 %, 15/100), noncancerous normal colon (9 %, 1/11) CRC (83 %, 25/30), noncancerous normal colon (8 %, 1/13) CRC (80 %, 80/100), matched normal colon mucosa (4 %, 4/100), noncancerous normal colon (0 %, 0/13) CRC (68 %, 44/65), CRC (63 %, 41/65) | (Piepoli et al. 2009) (Melotte et al. 2009) (Kim et al. 2007b) (Kim et al. 2009) (Kim et al. 2009) (Xu et al. 2004) (Xu et al. 2004) |
PAPSS2 PTG RARβ | 3′-phosphoadenosine 5′-phosphosulfate synthase 2 Prostaglandin I2 (prostacyclin) synthase Retinoic acid receptor-β | CRC (70 %, 21/30), noncancerous normal colon (8 %, 1/13) Adenoma (30, 3/10), CRC (43 %, 43/100) CRC (85 %, 55/65) | (Kim et al. 2009) (Frigola et al. 2005) (Xu et al. 2004) |
RASSF1A RASSF1A | Ras association (RalGDS/AF-6) domain family 1 Ras association (RalGDS/AF-6) domain family 1 | MSI + CRC (52 %, 16/31), HNPCC (30 %, 6/20) CRC (81 %, 39/48), normal colon mucosa (49 %, 19/39) | (Frigola et al. 2005) (Sakamoto et al. 2004) |
RASSF1A | Ras association (RalGDS/AF-6) domain family 1 | CRC (21 %, 14/64) | (Brandes et al. 2005) |
RASSF2A | Ras association (RalGDS/AF-6) domain family 2 | CRC (73 %, 106/146), matched normal Colon (12 %, 2/17) | (Park et al. 2007) |
RASSF2A | Ras association (RalGDS/AF-6) domain family 2 | CRC (42 %, 51/122) | (Akino et al. 2005) |
RASSF2A | Ras association (RalGDS/AF-6) domain family 2 | CRC (70 %, 121/30), matched normal colon (0 %, 0/30) | (Hesson et al. 2005) |
RASSF2A | Ras association (RalGDS/AF-6) domain family 2 | Adenoma (43 %, 21/49), CRC (42 %, 51/122) | (Akino et al. 2005) |
RECK | Reversion-inducing-cysteine-rich protein with kazal motifs | CRC (44 %, 11/25) | (Cho et al. 2007) |
RIL | A LIM domain gene mapping to 5q31 | CRC (70 %, 30/43), adenoma (85 %, 11/13), normal colon (5 %, 1/22) | (Boumber et al. 2007) |
RUNX3 | Runt-related transcription factor 3 | CRC (34 %, 31/92) | (Imamura et al. 2005) |
RUNX3 | Runt-related transcription factor 3 | Polyps (74 %, 64/87), normal colon (16 %, 2/12), HP (89 %, 17/19), SA (86 %, 12/14), sTAs (44 %, 7/17), FAP polyps (74 %, 28/38) | (Subramaniam et al. 2009) |
SPARC | Osteonectin | CRC (100 %, 20/20), normal colon mucosa (15 %, 3/20) | (Yang et al. 2007) |
TFPI2 | Tissue factor pathway inhibitor-2 | (62 %) primary colon colon carcinomas | (Hibi et al. 2010) |
TPEF/HPP1 | Transmembrane protein with EGF-like and two follistatin-like domains 2 | Normal tissue (5 %, 1/21), CRC (77 %, 36/47), liver metastasis (79 %, 19/24) | (Ebert et al. 2005) |
TPEF/HPP1 | Transmembrane protein with EGF-like and two follistatin-like domains 2 | Adenoma from FAP-Pts (31 %, 10/32), multiple adenoma Pts (65 %, 17/26), MSI-H CRC Pts (64 %, 9/14), MSS/MSI-L CRC Pts (54 %, 7/13) | (Wynter et al. 2006) |
TUBG2 | γ-Tubulin gene 2 | CRC (97 %, 29/30) | (Kim et al. 2009) |
UNC5A | unc-5 homolog A | CRC (68 %, 34/50) | |
UNC5C | unc-5 homolog C | CRC (62 %, 112/147), AP (63.5 %, 33/52), matched normal colon mucosa (6 % 9/147) | (Shin et al. 2007) |
Vimentin | Vimentin | CRC (65 %, 31/45) | (Shirahata, 2010) |
Vimentin | Vimentin | CRC Pts (62 %, 95/153), HD (2 %, 1/46) | (Chen et al. 2005) |
WNT5A | Wingless-type MMTV integration site family, member 5A | CRC (48 %, 14/29), matched normal colon mucosa (13 %, 2/15) | (Ying et al. 2008) |
DFNA5 | Autosomal dominant 5 | CRC (65 %, 65/100), matched normal colon mucosa (3 %, 3/100), noncancerous normal colon (9 %, 1/11) | (Kim et al. 2009) |
DKK-3 | Dickkopf homolog 3 | CRC (52.3 %, 67/128), adjacent nontumor tissues (5 %, 1/20) | (Yu et al. 2009) |
EphA1 | Eph receptor A1 | CRC (49 %, 26/53) | (Herath et al. 2009) |
13.2.5 Epigenetic Gene Silencing Role in the Evolution of CRC—Importance for Early Tumor Progression Stages (ie; IBD, Polyp)
Inflammatory bowel disease (IBD) is a heterogeneous disease strongly associated with chronic uncontrolled inflammation of the intestines, and has two distinct disease categories, including Crohn’s disease (CD) and ulcerative colitis (UC) (Hartnett and Egan 2012). Each year 30,000 people worldwide are diagnosed with IBD (Hanauer 2006). Chronic Inflammation is thought to be the root cause of tumor development and associated with about 20 % of all human cancers (Hartnett and Egan 2012; Hanauer 2006; Ullman and Itzkowitz 2011). CRC is a serious complication in patients with IBD. These patients are 60 % more likely to develop CRC than the general population (Herrinton et al. 2012; Goel et al. 2011). IBD-associated CRC (IBD-CRC) accounts for about 10–15 % of mortality rates in patients with IBD (Munkholm 2003). Screening of such patients using colonoscopy and intestinal biopsy might be difficult and inefficient as a cancer surveillance method (Chambers et al. 2005; Mattar et al. 2011). In order to overcome such difficulties and improve diagnosis and surveillance, noninvasive diagnostic tool and well-validated molecular markers for the early detection, prognosis and monitoring of IBD patients seems necessary. Epigenetic alterations, particularly alteration in DNA methylation are believed to play an early etiopathogenic role in IBD (Hartnett and Egan 2012; Petronis and Petroniene 2000; Tahara et al. 2009; Lin et al. 2011; Olaru et al. 2012; Cooke et al. 2012). Table 13.3 shows the association of some epigenetic alterations in IBD.
Gene ID | Gene ID |
|---|---|
BCL6 | HIF1A |
TJP1 | APC |
TNFSF | BGN |
EYA4 | GAS1 |
PTPN6 | LAT |
PURA | EGF |
GATA6 | RIPK4 |
TCOF1 | FANCC |
THRAP2 | GFPT1 |
GBGT1 | ANKRD9 |
PAQR6 | TNFSF12–13 |
HFE | EVI1 |
IFNGR2 | THRAP2 |
GATA4 | ABCC2 |
GATA5 | EFNB3 |
HACE1 | IL1B |
MAS1 | FLJ20712 |
IL18BP | GABRA5 |
LMTK2 | HOXB2 |
MMP3 | MAPK10 |
RHOH | MAS1 |
ITGA4 | PITPNM2 |
LRRC3B | MS4A4A |
DOK2 | SERPINA5 |
CHML | FABP3 |
VWF | NEU1 |
MGMT | S100A4 |
FUT7 | GPR116 |
TMEM116 | GPX3 |
TNFSF4 | MAGEL2 |
FCGBP | SLC22A18 |
SPATA22 | STAT5A |
TNFRSF1A | ASCL2 |
NOTCH1 | IL16 |
PDE1B | RUNX1T1 |
PDGFRB | SP11 |
MLH1 | ICAM3 |
MSX1 | MMRN2 |
MYOD | GCET2 |
CYP2D6 | RBM13 |
COG8 | KCNK4 |
FOLR1 | AKAP2 |
HTR2A | CD28 |
Chronic inflammation is associated with infections or autoimmune disease precedes tumor development and can contribute to it through induction of oncogenic mutations, genomic instability, early tumor promotion, and enhanced angiogenesis.
Inflammation acts at all stages of tumorigenesis. It may contribute to tumor initiation through different mechanisms such as mutation, genomic instability and epigenetic modifications. Inflammation activates tissue repair responses, induces proliferation of premalignant cells, and enhances their survival and metastasis.
Genetic susceptibility is influenced by luminal microbial; stimulate immune responses in pathogenic or protective way. Environmental triggers are necessary to initiate or reactivate disease expression. As shown in Fig. 13.9 long lasting intestinal inflammation stimulates cell proliferation in the mucosal and could eventually lead to the low grade of dysplasia, high grade dysplasia and ultimately carcinoma.
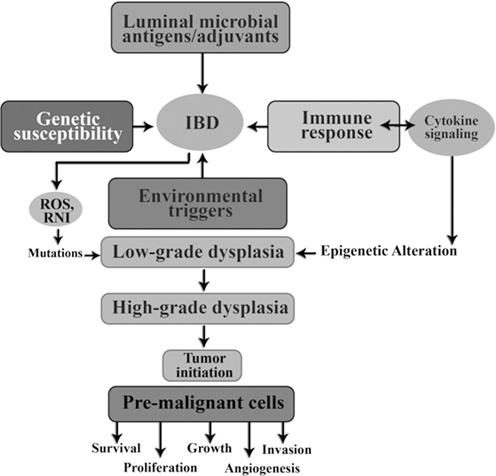
Fig. 13.9
Role of inflammation in tumor initiation
Reactive oxygen species (ROS) and reactive nitrogen intermediate (RNI) produced by inflammatory cells may cause mutations in epithelial cells. Also, cytokines produced by inflammatory cells can elevate intracellular ROS and RNI in premalignant cells. In addition, as seen in Fig. 13.9 inflammation can result in epigenetic changes that favor tumor initiation. Tumor associated inflammation contribute further Ros, RNI, and cytokine production.
13.2.5.1 DNA Methylation May Play an Important Role in IBD Susceptibility
Currently more than 32 susceptibility loci have been identified for IBD (Yamazaki et al. 2005; Duerr et al. 2006; Hampe et al. 2006; Rioux et al. 2007; Burton et al. 2007; Parkes et al. 2007). However, all these genetic risk factors can only account for approximately 20 % of the genetic risk (Xavier and Rioux 2008; Barrett et al. 2008), suggesting that other factors, including possible epigenetic factors, are involved in the pathogenesis of IBD (Petronis and Petroniene 2000). Heritable and reversible epigenetic alteration has been recognized as a factor affecting disease pathogenesis (Moss and Wallrath 2007; Shames et al. 2007). DNA methylation and histone modification are the most studied epigenetic events. DNA methylation and its crucial role has been extensively investigated in various cancers (Wilson et al. 2007; Robertson 2005) and several other human diseases including IBD. Abnormal DNA methylation has been observed in UC patients in the estrogen receptor (ER), P14ARF, E-cadherin gene and other genes (Maeda et al. 2006). Moreover, there is evidence that epigenetic factors are involved in the regulation of gene activity as a factor in the etiopathogenesis of IBD (Tahara et al. 2009).
DNA hypermethylation is associated with gene silencing and is often observed in CpG islands of cancer-related genes in IBD. Aberrant cancer specific methylation of genes such as p16, E-cadherin, hMLH1, and p14 has been reported in IBD associated neoplasia (Dhir et al. 2008). These epigenetic changes may accelerate the development of IBD-CRC (Levin, 1992; Petronis and Petroniene 2000; Kellermayer 2012). CpG island methylation phenotype and global DNA methylation with LINE-1 assay were also observed in 17 and 58 % of UC-related cancers, respectively (Konishi et al. 2007). Loss of hMLH1 protein expression associated with gene hypermethylation is common in Microsatellite instability (MSI) positive UC cancers (Fleisher et al. 2000). Methylation of hyperplastic polyposis gene 1 (HPP1) was observed in 50 % UC adenocarcinoma and 40 % of dysplasias but not in normal mucosa (Sato et al. 2002). Also, methylation of E-cadherin (CDH1) was detected in 93 % of the patients with dysplasias (Azarschab et al. 2002). Moreover, the increased level of methylation is widespread in the colon affected by inflammation and occurs early in the process of carcinogenesis (Issa et al. 2001).
DNA methylation in CRC has been previously studied leading to the identification of several tumor-associated DNA methylation patterns (Ahmed 2007; Wong et al. 2007). Previous IBD-related DNA methylation studies have focused on the development of CRC in patients with UC. Initial studies reported that four genes were highly methylated in high-grade dysplastic epithelium from patients with UC (Toyota et al. 2002). Subsequent observations of disease-related methylation status were made for the ER, HPP1, MLH1, RAB32, MGMT, and P14ARF (Issa et al. 2001; Fujii et al. 2005; Schuebel et al. 2007) and E-cadherin genes from patients with UC (Azarschab et al. 2002).
Inflammatory rectal mucosa from patients with UC showed increased methylation level and frequency with respect to the MDR1 gene promoter, especially in patients with shorter disease duration with a younger onset (Tahara et al. 2009). Recently, the expression of the MD-2, a critical TLR4 coreceptor that is upregulated in IBD but not in normal or diverticulitis patients, is upregulated by demethylation of the MD-2 promoter region (Vamadevan et al. 2010). Multiple genetic risk loci are common in both CD and UC, indicating a significant shared genetic component in the etiopathophysiology of these diseases (Anderson et al. 2009; Fisher et al. 2008).
13.2.5.2 Aberrant Wingless Signaling Pathways (WNT) in Patients with IBD
Aberrant wingless signaling pathways (WNT) have crucial roles for cancer progression as early progression events (Anastas et al. 2012). Epigenetic alteration of WNT signaling genes has been reported in up to 80 % of patients with CRC (You et al. 2008; Barker and Clevers 2006). Differential methylations were identified in tissues of patients with IBD, suggesting that IBD-associated changes in DNA methylation may be disease subtype specific.
WNT Ligands interact with the Fz receptor and LRP co-receptor and thus initiate the WNT signaling cascade (Anastas et al. 2012). These interactions subsequently lead to the formation of a complex composed of APC, AXIN, and Glycogen synthase kinase-3β (GSK-3β). Together, APC/AXIN/GSK-3β makes up the β-catenin destruction complex. In the absence of WNTs stimulation, the APC/AXIN/GSK-3β complex phoshphorylate β-catenin. Phosphorylated β-catenin is degraded by proteosome system (Baylin and Ohm 2006; Fyang et al. 2014) β-catenin is the cytoplasmic protein and the key component of the WNT signaling pathway. In the presence of WNTs stimulation, or loss of APC due to mutation, the destruction complex is destabilized, resulting in excessive amounts of β-catenin. β-catenin then translocates to the nucleus, where it associates with the Tcf family of transcription factors, and promotes the transcription of many target genes including c-MYC and cyclin D1 whose products increase cell proliferation (Noah et al. 2013). Secreted Frizzled-related proteins (SFRPs) are tumor suppressor genes that function as antagonists of WNT signaling. SFRP2 bind to WNTs and prevent their interaction with Fz and LRP (Baylin and Ohm 2006). Dysregulation of WNT signaling pathway may be involved in IBD-CRC owing to epigenetic silencing of key tumor suppressor genes such as APC and SFRP2 (Dhir et al. 2008).
Although, there are few studies addressing the effects of WNT gene methylation in IBD, little is known about the role of WNT related genes in IBD initiation and progression in this region. Also, since the mutation of WNT signaling genes is rare in neoplasia associated with IBD (Dhir et al. 2008),
Mokarram and et al showed the association of promoter methylation APC1A, SFRP2 genes from WNT pathway occur during the IBD-associated carcinogenesis (personal communication). Our study related to IBD patients demonstrates for the first time hypermethylation of the SFRP2 not APC1A gene promoter region was involved in IBD developing lesions.
13.2.6 The Role of Kras and MGMT Methylation in CRC
On the other hand, the activation of the Raf/ MEK/ ERK (MAPK) kinase pathway through either K-ras or BRAF mutation was detected in 30 % of UC related cancers. Non-dysplastic UC mucosa of patients with UC cancers show K-ras, but not BRAF mutation, indicating that K-ras mutations are initiating events in UC carcinogenesis (Aust et al. 2005) Several studies have also reported that reduced expression and function of DNA repair enzyme induced by oxidative stress during inflammation disease (Switzeny et al. 2012). DNA Mismatch repair elements and O6-methylguanine-DNA methyltransferase (O6-MGMT) are DNA repair proteins. MGMT loss of activity associated with creating a permissive state for G→A mutations in protooncogenes such as K-ras and p53 (Mokarram et al. 2012). K-ras mutations occur in about 60 % of CRCs and are an early event in UC carcinogenesis (Aust et al. 2005). MGMT promoter has two loci (Mokarram et al. 2012) described as MGMT-A and MGMT-B. Recently, it has been shown that methylation silencing of MGMT-B was significantly associated with the K-ras gene mutation rather than MGMT-A in patients with CRC (Mokarram et al. 2012) and analyzing the promoter profile methylation of MGMT-B could be of high prognostic value for patients with CRC. Because of this correlation, it is hypothesized that methylation of MGMT-B as a caretaker gene may be responsible for dysregulation of signaling through K-ras mutation in CRC lesions. On the other hand, our study related to IBD patients demonstrates for the first time hypermethylation of the MGMT gene promoter region was involved in IBD developing lesions (personal communication). Overall, methylation of MGMT and SFRP2 in IBD patients may provide a method for early detection of IBD-associated neoplasia which will be independent of the pathologist.
13.2.7 DNA Methylation in Polyp
CRCs develop as a result of the transformation of normal colonic mucosal epithelium to cancer through a series of precursor lesions with genetic and epigenetic changes termed the adenoma to carcinoma sequence and the hyperplastic polyp-serrated adenoma to carcinoma sequence. Epigenetic events have been involved in the stepwise histological progression such as adenoma-carcinoma and hyperplastic polyp/serrated adenoma-carcinoma sequences in the development of CRC (Ashktorab et al. 2014; Dhir et al. 2011). These precursor lesions experience CGI methylation of tumor suppressor genes as well as genetic changes (APC and K-ras mutations). Aberrant crypt foci (ACF), known as the earliest precursor lesions, experienced aberrant CGI methylation. Such methylation was also seen in adenomas; although it was less frequent than in adenocarcinoma. However, CGI methylation increased as early adenomas progressed towards advanced adenomas. In different stages of the adenoma-carcinoma progression sequence the methylation frequency gene-specific. The MGMT, p16INK4a, and HLTF genes had the highest methylation rate during this sequence whereas adenocarcinoma had the highest methylated MLH1 and TIMP3. Therefore, depending on the gene, the tumor initiation or progression process is affected by the inactivation of tumor suppressor genes caused by aberrant DNA methylation.
Recently, colorectal polyps have been classified into two major groups; serrated polyps and conventional adenomas (Spring et al. 2006; Pereyra et al. 2014; Spring et al. 2006). More CGI methylation and BRAF mutations were seen in serrated polyps (including hyperplastic polyps), sessile serrated adenomas, sessile adenomas, and mixed polyps.
BRAF mutations occur rarely in tubular adenomas. Hyperplastic polyps are mostly diminutive lesions are less likely to develop into cancer. However, sessile serrated adenomas, serrated adenomas and mixed polyps have a higher risk of progressing into the cancer. Sessile serrated adenomas were prevalent in about 9 % of patients who were undergoing colonoscopy compared with sessile adenomas (0.7 %) and mixed polyps (1.7 %). In the aforementioned study, sessile serrated adenomas were linked to BRAF mutations, proximal location, female sex, and presence of multiple polyps. In other studies, associations were found between serrated adenomas and CIMP and MSI-H cancers, tending to show near diploid DNA indices, more frequent allelic imbalance at 18q, and less frequent allelic imbalance at 5q or K-ras mutations. Serrated polyps and tubular adenomas also differed with respect to the proportions of specific methylated genes (Spring et al. 2006; Kim et al. 2006; Kim et al. 2014).
The neoplastic potential of ACF is still unknown. However, some ACF lesions harbor K-ras or APC mutation and methylation of specific gene loci such as RASSF1A, CRBP1, MINT31, CDH13, MINT1, SLC5A8, and MGMT. It is a precancerous condition of the colorectum such as hyperplastic polyposis (HPP), the polyps are morphologically distinguished from typical small hyperplastic polyps. Similar to adenomas, the polyps in HPP can evolve through a histological progression sequence that culminates in colon adenocarcinomas. A serrated adenoma intermediate could also give rise to hyperplastic polyps in some CRCs. The HP-serrated adenoma–adenocarcinoma sequence is linked with DNA methylation of HPP1, CDKN2A/p14, CDKN2A/p16, and MLH1 and mutations in BRAF and is more common in the proximal colon. Moreover, detection of DNA methylation in normal colorectal mucosa may serve as a useful diagnostic biomarker for high cancer risk in HPP.
Figure 13.10. represents the morphologic and genetic alterations along a putative serrated adenoma-carcinoma pathway in comparison to the classic form of adenomatous polyp progression. Similar differences are seen in proximal and distal pathway of tumorigenesis.
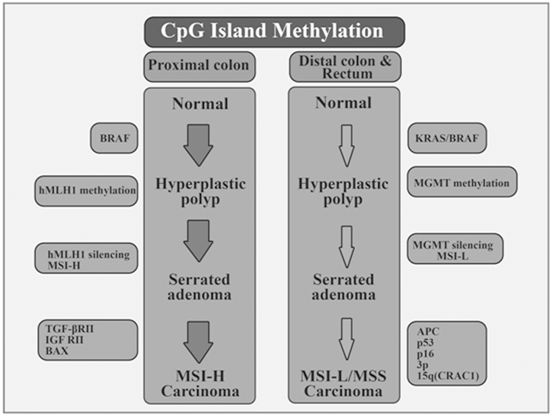
Fig. 13.10
Pattern of genetic and epigenetic alterations in polyp and CRC based on location of tumorigenesis
When SSA polyps and HP polyps are analyzed to look at mutations in key cell cycle regulatory genes, it found that HPs have a higher mutation rate in K-ras but SSAs frequently have mutation in BRAF (Pereyra et al. 2014). SSAs tend to display CpG island methylation to a greater degree than sporadic HPs. Minoo and co-workers investigated a series of serrated polyps and matched normal mucosa to find distinguishing molecular features in patients with and without HPP. They found higher DNA methylation in sessile serrated adenomas and normal colorectal mucosa in HPP. A high level of CpG island methylator phenotype (CIMP, a subset of colon tumors with accumulation of type C methylation) in the normal mucosa of patients with HPP was also observed. In some forms of HPP, the earliest manifestation of transformation might be hyper-methylation of multiple gene promoters in normal colorectal mucosa (Kim et al. 2010; Minoo et al. 2006).
13.2.7.1 Aberrant CpG Island Hypermethylation in Polyp
Since aberrant CpG Island DNA promoter hypermethylation can be detected not only in colorectal polyps and cancers, but also in sera and stool, epigenetic changes are considered as a diagnostic hallmark during the initiation and progression process of tumors. This pathway implicates the inactivation of DNA mismatch repair genes tracked by mutations in the microsatellite repeat sequences in the genes that are important in tumor progression as TGF RII and BAX, which leads to uncontrolled cellular growth and decreased apoptosis. Loss of APC functions with activation of WNT pathway is observed in most familial adenomatosis polyposis-associated and sporadic CRC. Studies have reported that promoter regions of genes that are responsible for encoding secreted frizzled related proteins (SFRPs) undergo abnormal methylation in colorectal neoplasms (Kim and Deng 2007; Papa et al. 2011; Petko et al. 2005; Belshaw et al. 2004). SFRPs are antagonists of WNT signaling in normal colon epithelial cells, by competing with WNT proteins for binding to their receptors frizzled (FRZ) on the cell surface. SFRPs act as constitutive inhibitors of WNT signaling in normal colon epithelial cells. As a result of promoter methylation of SFRP genes, SFRP expression is lost. Therefore, WNT signaling is activated by the receptor FRZ, which in turn leads to the expansion of colon epithelial stem cells. As a result, mutations occur in the downstream components of the pathway (such as APC) facilitating tumor initiation and progression. Other genes that have hypermethylated promoters with the silencing of genes are MGMT, p16INK4a, HIC1, RASSF1, ESR1, HPP1, and MSH2 (Kim and Deng, 2007, Papa et al. 2011, Petko et al. 2005, Belshaw et al. 2004). A subset of genes that included EVL, GATAs, HIN-1, SFRPs, SOX17 and SYNE1 were methylated frequently in all premalignant gastrointestinal adenomas such as tubular adenomas, villous adenomas, sessile serrated adenomas, and sessile serrated adenomas with dysplasia. However, they were not frequently methylated in non-premalignant polyps such as HPPs. CDX2, hMLH1 and TLR2 methylation may be diagnostically useful in differentiating sessile serrated adenomas from hyperplastic polyps in cases that diagnosis is histologically challenging. Methylation of CDX2, hMLH1, and TLR2 was detected in sessile serrated adenomas and sessile serrated adenomas with dysplasia but not in hyperplastic polyps (Dhir et al. 2011).
Ashktorab and colleagues evaluated the methylation status of the tumor suppressor gene dickkopf homolog 1 (DKK1) as a risk factor for colon polyp in Africans-Americans and found that in 96 % of the studied samples the DKK1 gene promoter was unmethylated (Ashktorab et al. 2011). Based on unpublished data were performed by Mokarram et al in patients with polyp, at least one of two genes (SFRP2 and MGMT) was methylated in 93.8 % of the patients. Both genes were methylated in 45.8 % of the patients, implying that half of the patients had both methylated genes. These data revealed the potential diagnostic value of these two genes in polyps and adenomas. By comparing MGMT and SFRP2 genes and considering that the extensive methylation in MGMT is associated with the development of polyp and adenoma. Therefore, MGMT has a key role in the pathologies of patients with polyp and adenoma.
13.2.8 Detection of CRC by Microarray or DNA Methylation Assays
Currently, molecular biology has turned medical oncology into a more interesting topic of study since molecular changes influential in progression can be studied in detail using molecular biology. Therefore, the carcinogenesis process is better understood and novel prognostic markers and therapeutic targets can be discovered. Although clinical and pathological parameters are at hand for the classification and prognostic stratification of cancer, inadequacy might still exist in daily practice due to the vast biological and genetic heterogeneity of cancer.
Also, since patient-tailored therapies are becoming increasingly common, it is necessary to gain a more comprehensive knowledge of downstream signaling pathways in order to discover new tumor targets and as a result develop novel biological drugs. Here, gene expression profiling analysis with microarray technologies is a newly emerging field that allows the simultaneous mapping of thousands of gene expression in as little as one tumor sample. Fig. 13.11 shows how microarray performs the gene expression evaluation.
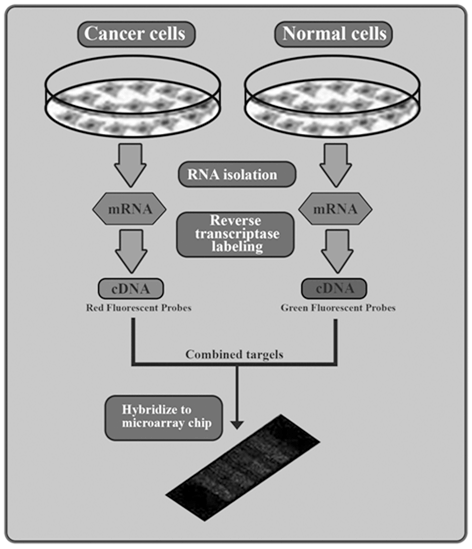
Fig. 13.11
DNA microarray to compare gene expression in cancer and normal samples
DNA microarrays can survey virtually the entire expressed genome. Studies showd that a small amount of high quality RNA from tumor or non-tumor tissues is labeled and hybridized on the surface of chip which is composed of spotted cDNA clones or probes spotted or synthesized on the surface of the chip. DNA microarrays analysis is most useful when it can be integrated with clinical, imaging and histological data. DNA microarray accompanied with clinical data provides complementary types of information, so most microarray studies use combination of these approaches.
Gene microarray analysis is becoming an increasingly valued tool in studying human cancers theoretically and practically. Researchers are now able to investigate the expression of ≤ 50,000 genes using complementary DNA or oligonucleotide arrays, which facilitates an improved understanding of the complex relationships between genes. Moreover, this approach may provide insight as to how cellular gene expression might yield important prognostic and/or predictive information.
Understanding the molecular signature of tumors could provide physicians with a highly sensitive, quantitative, reproducible, and individualized approach for making treatment decisions as well as provide a more accurate assessment of a patient’s tumor burden when compared with current methods that rely on conventional clinical criteria.
The various clinical applications of microarray technologies could be divided into three categories in CRC; [1] studies on carcinogenesis process, [2] studies on prognosis prediction, [3] studies on treatment response prediction.
One of the earliest applications for microarray in preclinical settings is examining tumor specimens versus normal tissues to differentiate the gene expression of normal and neoplastic tissue. The hypothesis was that these genes could provide insight into the changes in the subcellular machinery that eventually gives rise to neoplasia, directly or indirectly. Several studies have identified differential gene profiles in normal versus cancerous tissues (Li et al. 2004; Bandres et al. 2004; Abajo et al. 2012). Gene microarray has proven valuable in prognostic studies on hormonal and cytotoxic treatment in breast cancer. CRC is also an interesting field in microarrays research because it is a biological tumorigenesis model with distinct molecular alterations (Vogelstein et al. 1988). Moreover, for distinguishing high risk from low risk CRC, traditional clinical and pathological parameters are not always adequate due to the unavailability of validated molecular markers with prognostic value. And finally, although there is currently controversy regarding data on molecular markers that have predictive tumor response value, some cytotoxic and biological medications are extensively used in routine clinical practice (Spano et al. 2008).
In addition, CpG Island Methylator Phenotype (CIMP) is one of the underlying mechanisms in colorectal cancer (CRC). Study aimed to define a methylome signature in CRC through a methylation microarray analysis and a compilation of promising CIMP markers from the literature (Ashktorab et al. 2014). Illumina HumanMethylation27 (IHM27) array data was generated and analyzed based on statistical differences in methylation data (1st approach) or based on overall differences in methylation percentages using lower 95 % CI (2nd approach). Pyrosequencing was performed for the validation of nine genes. A meta-analysis was used to identify CIMP and non-CIMP markers that were hypermethylated in CRC but did not yet make it to the CIMP genes’ list. Our 1st approach for array data analysis demonstrated the limitations in selecting genes for further validation, highlighting the need for the 2nd bioinformatics approach to adequately select genes with differential aberrant methylation. A more comprehensive list, which included non-CIMP genes, such as APC, EVL, CD109, PTEN, TWIST1, DCC PTPRD, SFRP1, ICAM5, RASSF1A, EYA4, 30ST2, LAMA1, KCNQ5, ADHEF1 and TFPI2 was established. Array data are useful to categorize and cluster colonic lesions based on their global methylation profiles; however, its usefulness in identifying robust methylation markers is limited and rely on the data analysis method. Ashktorab et al. have identified 16 non-CIMP-panel genes for which we provide rationale for inclusion in a more comprehensive characterization of CIMP+ CRCs (Ashktorab et al. 2014). The identification of a definitive list for methylome specific genes in CRC will contribute to better clinical management of CRC patients.
Related posts:
 Reduced Risk of Cancer in Schizophrenia, a Bridge Toward Etio-Pathology and Therapy of Both Diseases
Reduced Risk of Cancer in Schizophrenia, a Bridge Toward Etio-Pathology and Therapy of Both Diseases
 Biodynamic Phenotypic and Epigenetics Changes of Circulating Tumor Cells: Their Application in Cancer Prognosis and Treatment
Biodynamic Phenotypic and Epigenetics Changes of Circulating Tumor Cells: Their Application in Cancer Prognosis and Treatment
 An Introduction to Impact of Bio-Resonance Technology in Genetics and Epigenetics
An Introduction to Impact of Bio-Resonance Technology in Genetics and Epigenetics
 Epigenetics of Thyroid Cancer
Epigenetics of Thyroid Cancer
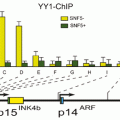 Malignant Rhabdoid Tumor: Epigenetic Mechanism of Tumorogenesis
Malignant Rhabdoid Tumor: Epigenetic Mechanism of Tumorogenesis
 Sentinel Gene Within Cell Territory and Molecular Platforms in Cancer: Methylation Diversity of p53 Gene in Brain Tumors
Sentinel Gene Within Cell Territory and Molecular Platforms in Cancer: Methylation Diversity of p53 Gene in Brain Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


