Fig. 14.1
Cancer associated amino acid substitution of hSNF5 (a) Schematic representation of hSNF5 depicting the two repeats (RPT1 and RPT2) and cancer-associated amino acid substitution mutations. (b) Conservation of cancer associated amino acid substitution mutations from Human to Yeast
14.3 Epigenetic Mechanism of SNF5-Tumorogenesis
14.3.1 SNF5 is a Tumor Suppressor Gene
The SWI/SNF complex is involved in various cellular processes that are potentially associated with tumor formation including DNA synthesis, virus integration, DNA repair, and mitotic gene regulation. Numerous studies to dissect the connection between these activities and tumor formation are currently in progress. The molecular mechanisms underlying tumor development in mice with inactivation of BRG1 or SNF5 are still unclear.
The ability of SNF5 to function as a tumor suppressor has been confirmed in studies utilizing SNF5-deficient mice. Mice strain carrying reversibly inactivating SNF5/INI1 allele by applying LoxP-Cre recombination system has been generated to find the mechanisms of tumorogenesis caused by loss of SNF5/INI1 (Roberts et al. 2002). All of the resultant mice develop short latency highly aggressive tumors such as CD8+ T cell lymphomas and rare rhabdoid tumors (Roberts et al. 2002).
Although the molecular mechanisms for SNF5/INI1 function in cell survival in normal cells is not yet known for mammals, in recent years significant progress has been made in understanding of SNF5/INI1 role in tumor suppression. It appeared, that re-expression of SNF5/INI1 in human MRT cell lines leads to an accumulation in G0/G1 phase, cellular senescence and in some cases apoptosis (Oruetxebarria et al. 2004; Vries et al. 2005; Ae et al. 2002; Versteege et al. 2002; Betz et al. 2002; Zhang et al. 2002). Cellular senescence is largely the result of direct transcriptional activation of the tumor-suppressor p16INK4a by hSNF5 (Ae et al. 2002; Betz et al. 2002; Oruetxebarria et al. 2004; Versteege et al. 2002; Vries et al. 2005; Zhang et al. 2002).
14.3.2 SNF5 Can Function in Transcription Activation and Repression of the Genome
To study the function of hSNF5 in MRT cells, two distinct strategies to re-express hSNF5 in G401 and Mon, two different MRT cell lines, has been established. Induction of the hSNF5 gene in G401 cell line was under control of the Lac repressor-operator system; in parallel hSNF5 or GFP has been transduced to Mon cells using lentiviral transfection. The expression level of SNF5 in both system were comparable.
Moreover, the levels of induced exogenous hSNF5 expression were comparable to the endogenous levels in a variety of cell lines (Oruetxebarria et al. 2004; Doan et al. 2004; Moshkin et al. 2007).
To elucidate the pathways controlled by hSNF5 in rhabdoid tumor cells, we performed cDNA microarray analysis. Our gene expression profiling results suggested that majority of the up-regulated genes encode proteins with functions in, extra cellular matrix remodeling, adhesion or cell migration (SERPINE2, ITGB5, MAP1B), apoptosis (DR6,FAS, CASP4, GAS6, ADAM19), and cancer related pathway or other specialized functions (CDKN2A, ETS2 and TRIM22). The majority of the genes down-regulated by hSNF5 encoded proteins that play key roles during cell cycle, like, CENPE, POLD3,CENPA, CDC25A, CCNF and MAD2 but remain unchanged upon induction of mutant hSNF5. These results suggested that mis-expression of mitotic checkpoint components might cause the abnormal ploidy of MRT cells. For example, over-expression of MAD2 and its regulator E2F1 was implicated in mitotic defects leading to aneuploidy (Hernando et al. 2004). Interestingly, in our microarray experiments, both genes were down-regulated following hSNF5 induction. We used QRT-PCR to corroborate our microarray results (Fig. 14.2). We found that MAD2, E2F1 and E2F1 target genes are highly expressed in MRT cells, but are strongly down-regulated following hSNF5 induction.
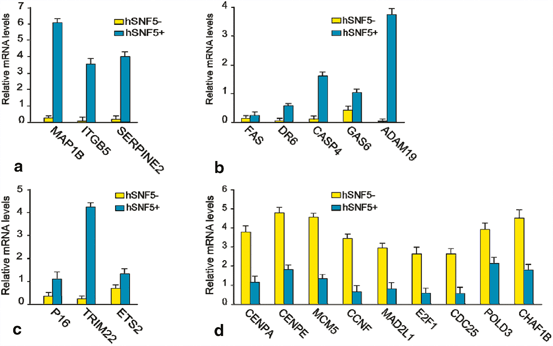
Fig. 14.2
QRT-PCR analysis of gene regulated by hSNF5 identified by whole-genome expression profiling. RT-qPCR analysis of gene expression in MRT cells reveals hSNF5-dependent induction of genes involved in (a) apoptosis pathway (b) Cell migration and invasion (c) Other pathway and (d) depletion of E2F and E2F target genes affected. Cells were collected 48 h following transduction with lentiviruses expressing either GFP (yellow bars) or hSNF5 (blue bars). mRNA levels were plotted as percentage of GAPDH mRNA. The bar graphs represent the mean of three independent biological replicates, each analyzed by three separate qPCR reactions. Standard deviations are indicated
Further we transduced GFP or SNF5 in MRT cells (G401 and MON), Hela cell and U2OS cell with lentivirus (Fig. 14.3a). Western immunoblot analysis of extracts from cells transduced by GFP (lanes 1, 3 and 5) or cells transduced with lentiviruses expressing SNF5 (lanes 2, 6 and 8) revealed expression of hSNF5 in G401 and Mon cells (lane 2,4) and over-expression of hSNF5 in Hela and U2OS cells (lane 6 and 8). Antibodies directed against histone H3 were used as a loading control. QRT-PCR revealed induction of FAS, ETS2, TRIM22, GAS6, ADAM19 and SERPINE2 in MRT cells upon induction of SNF5 while they remain unchanged or down-regulated in either Hela or U2OS cells (Fig. 14.3b–14.3g).
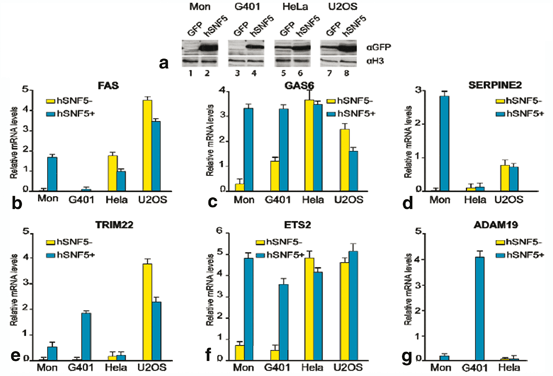
Fig. 14.3
RTQ-PCR analysis of gene regulated by hSNF5 in different cells (a) Western immunoblotting analysis of hSNF5 expression in MON. G401, Hela and U2OS cells transduced with lentiviruses expressing either GFP (lanes 1, 3, 5, 7) or hSNF5 (lanes 2, 4, 6, 8). Cell lysates were resolved by SDS-PAGE and analyzed by Western immunoblotting using antibodies directed against hSNF5. Histone H3 serves as a loading control. (b–e) RTQ-PCR analysis of genes upon induction or over expression of hSNF5 in two MRT cells (Mon, G401) Hela and U2OS cells. PCR revealed induction of (b) FAS, (c) GAS6, (d) SERPINE2, (e) TRIM22, (f) ETS2 and (g) ADAM19 in MRT cells upon induction of SNF5 while they remain unchanged or down-regulated in either Hela or U2OS cells. Procedures were as described in the legend to Fig. 14.2
To identify hSNF5 direct target genes of hSNF5 we performed chromatin immuneprecipitation on selected set of genes identified as potential hSNF5 targets upon hSNF5 transduced MRT cells. MRT cells were transduced with lentivirus (or were induced with IPTG). Chromatin was prepared from cells at 48 h post-stimulation and subjected to chromatin immunoprecipitation with an antibody specific for SMARCB1 (hSNF5). As shown in Fig. 14.4a our results indicate that, while initially absent from the TRIM22, ETS2, ANGPT2 and MAD2L genes, SNF5 is specifically recruited to these promoters in response to SNF5 expression. hSNF5 expression does not result in recruitment of SNF5 to the adjacent P14, ADAM19 and INTGB5 promoters. SNF5 is also recruiting BRG1 to the promoter of TRIM22, ETS2, ANGPT2 and MAD2L but not ADAM19 and INTGB5 (Fig. 14.4). Taken together, these results demonstrate that re-expression of hSNF5 in MRT cells results in induction and specific recruitment of SNF5 and BRG1 to the TRIM22, ETS2, ANGPT2 and MAD2L promoters but not P14, ADAM19 and INTGB5 promoters.
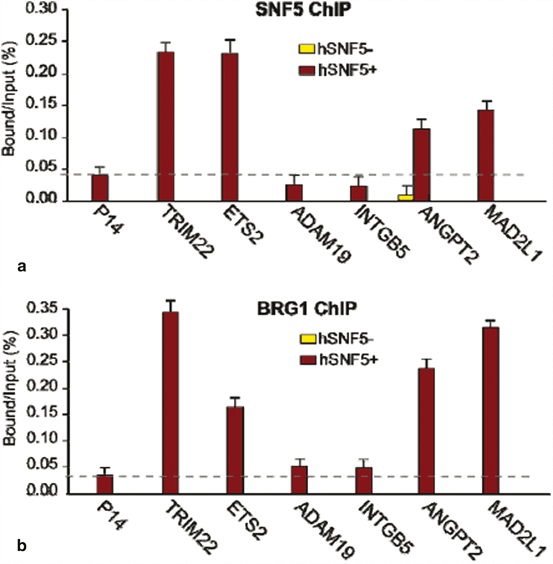
Fig. 14.4
hSNF5 Mediates BRG1 Recruitment to the MAD2, TRIM22, ETS2 and ANGPT2 promoters (a) ChIP-qPCR analysis of hSNF5 binding to the MAD2,TRIM22, ETS2 and ANGPT2, p14Arf, INTGB5 and ADAM19 revealed that hSNF5 binds directly to the MAD2, TRIM22, ETS2 and ANGPT2 promoters, but not to p14Arf. Cross-linked chromatin was isolated from MRT cells that either lack- (light bars) or express hSNF5 (dark bars). (b) RG-1 binding to the MAD2, TRIM22, ETS2 and ANGPT2 promoters is hSNF5-dependent, as revealed by ChIP-qPCR using antibodies directed against BRG-1
All ChIP data presented in this study are the result of at least three independent experiments. The abundance of specific DNA sequences in the immuneprecipitates was determined by qPCR and corrected for the independently determined amplification curves for each primer set. Background levels were determined by ChIP using species and isotype-matched immunoglobins directed against an unrelated (GST) protein. ChIPs with antibodies directed against SNF5 and BRG1 were analyzed by qPCR using primer sets corresponding to MAD2, TRIM22, ETS2, ANGPT2, p14 Arf , INTGB5 and ADAM19 promoters.
ChIP signal levels for each region are presented as percentage of input chromatin.
14.3.3 SNF5 is an Activator of P16 Tumor Suppressor Gene
SNF5/INI1 binds to the promoter of p16 ink4a tumor suppressor gene and recruits BRG1-containing SWI/SNF complex resulting in transcription activation (Oruetxebarria et al. 2004; Kia et al. 2008). p16 ink4a gene encodes a specific cyclin-dependent kinase (CDK)4/CDK6 inhibitors, which targets retinoblastoma (pRb) protein for phosphorylation (Lowe and Sherr 2003). Hyperphosphorylated pRb dissociates from E2F transcription factor allowing S phase specific genes expression and promoting cell proliferation. Therefore, increased p16 ink4a expression upon SNF5/INI1 induction in MRT cells results in pRb hypophosphorylation and inhibits cell cycle progression (Fig. 14.5). Interestingly, MRT cell lines lacking p16 ink4a activity or expressing p16 ink4a -insensitive mutant of CDK4 continue to grow after SNF5/INI1 re-expression supporting a role of SNF5/INI1 in p16ink4a-CDK4/Cyclin D1-pRB/E2F pathway (Oruetxebarria et al. 2004; Vries et al. 2005). Several groups have also shown that SNF5/INI1 can repress Cyclin D1 expression in ATRT/MRTs by recruiting HDACs to the promoter, or it can function in the repression of E2F target genes via direct association with pRb, thereby causing cell-cycle arrest in G1 phase (Zhang et al. 2000; Zhang et al. 2002). In agreement with a role of SWI/SNF ATP-dependent chromatin remodelers in the regulation of pRb/E2F transcriptional circuitry, genome-wide expression profiling revealed up-regulation of some of the E2F-target genes in MRT cell lines, including mitotic checkpoint gene MAD2 (Vries et al. 2005). Over-expression of Mad2 leads to chromosomal instability and tumorogenesis (Sotillo et al. 2007; van Deursen 2007).
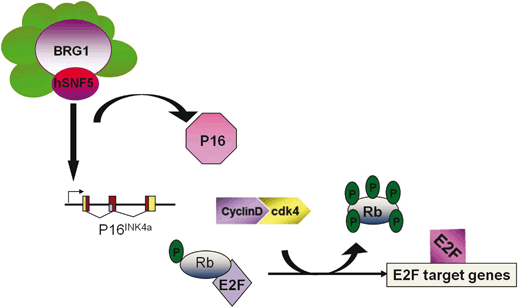
Fig. 14.5
Mammalian ATRT/MRT tumor cells lack SNF5/INI1 function resulting in increased CDK4/cyclin D1 activity
The SWI/SNF complexes lack sequence specific DNA binding, and are therefore thought to be recruited to specific promoters via interactions with DNA binding proteins. It is still of interest to identify other DNA binding regulators that activate p16 INK4a through association with the hSNF5 chromatin-remodeling factor. ETS2 is a transcriptional activator of p16 INK4a which has been shown to bind to the p16 INK4a promoter (Ohtani et al. 2001). ChIP data revealed that ETS2 and SNF5 are co-recruited to the P16 promoter in MRT cells (Fig. 14.6). shRNA knock down analysis of ETS2, and studies of p16 INK4a activation upon induction of hSNF5 will be informative useful to address the recruitment to and role of hSNF5 in activation of p16INK4a.
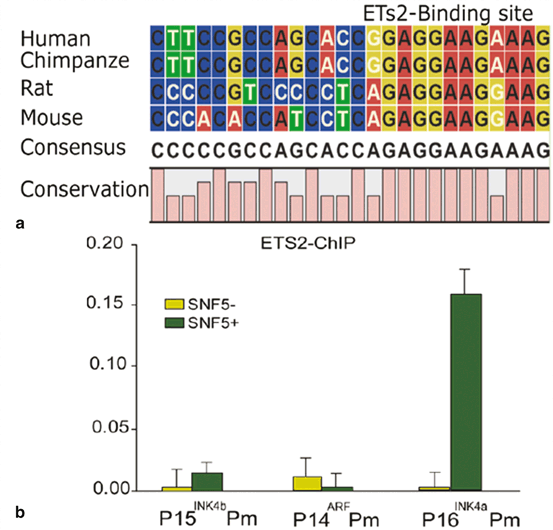
Fig. 14.6
hSNF5 and ETS2 are co-recruited to the p16INK4a Promoter. (a) Conservation of ETS2 binding site at p16INK4a promoter during evolution (b) ETS2 binding to the p16INK4a promoter is hSNF5-dependent, as revealed by ChIP-qPCR using antibodies directed against ETS2. ChIPs using antibodies directed against ETS2. Cross-linked chromatin was prepared from MRT cells lacking hSNF5, but expressing GFP (light green bars), or cells expressing hSNF5 (dark green bars). ChIPs were analyzed by qPCR using primer sets specific for the Ink4-Arf locus, revealing that ETS2 binding peaks at the p16INK4a promoter. Upon induction of hSNF5, ETS2 is co-recruited
This triggers pRb hyperphosphorylation and unleashes transcription of E2F-dependent genes causing increased proliferation, chromosomal instabilities and cancer. Re-expression of SNF5/INI1 reverses cell proliferation and leads to G0/G1 arrest, senescence and apoptosis primarily due to activation of P16 INK4a tumor suppressor gene expression and repression of Cyclin D1 gene expression.
14.3.4 The Mechanism of SNF5- Dependent Transcriptional Control of the INK4b-ARF-INK4a Locus in MRT Cells
14.3.4.1 The INK4-ARF locus
The INK4B-ARF-INK4A locus, spanning an approximately 40 kb stretch of the human chromosome 9p21 (chromosome 4 in mouse), encodes three distinct tumor suppressors, P15iINK4B, P16INK4A and P14ARF( p19 ARF in mice) whose expression enhances the growth-induction and -suppressive functions of the retinoblastoma (RB) and p53, respectively. Both p15 and p16 are able to induce cell cycle arrest in G1 by inhibiting cyclin dependent kinase CDK4 and CDK6 to inactivate retinoblastoma (RB) (Ortega et al. 2002). Since p16 can only form stable, binary complex with both CDK4 and CDK6, p16 is likely the most effective inhibitor of CDK4 and CDK6 (Parry et al. 1999). The unrelated p14 protein acts via MDM2 to activate the key check point protein TRP53, thereby inducing either cell cycle arrest (both in G1 and G2) or apoptosis (Pomerantz et al. 1998). The locus p14 ARF (named ARF because it uses the second exon of INK4A in an alternative translational reading frame) is implicated in various types of cancer (reviewed in (Gil and Peters 2006; Sharpless 2005)). In mammalian cells, products of the INK4A-ARF locus play major roles in senescence and tumor suppression in different contexts, whereas the adjacent INK4B gene is generally associated with transforming growth factor beta (TGF-beta)-mediated growth arrest (Lowe and Sherr 2003). Mouse models indicate that both p16 INK4a and p14 ARF are tumor suppressors while mice deficient for p15 Ink4b show only a very subtle tumor predisposition (Latres et al. 2000). Mice specifically defective for p14 ARF are highly tumor-prone but mice deficient for all Ink4b-Arf-Ink4a locus genes have are more tumor prone and develop a wider spectrum of tumor than p16 Ink4a mutant mice, with skin tumor and soft tissue sarcomas (Kamijo et al. 1997; Sharpless et al. 2004). It is reported that p15Ink4b serves as a backup of p16Ink4a (Krimpenfort et al. 2007). In chicken cells, which lack p16 Ink4a , p15 Ink4b has the major role rather than p14 Arf in senescence (Kim et al. 2006). In a variety of tumors, P16 INK4a is inactivated through epigenetic silencing, involving PcG (Polycomb groups) proteins and DNA methylation (Gil and Peters 2006; Jones and Baylin 2007; Sparmann and van Lohuizen 2006). Significantly, the PcG protein BMI1 promotes oncogenesis in mice through silencing of the Ink4a-Arf locus (Jacobs et al. 1999). Both the Polycomb-repressive Complex1 (PRC1) and Polycomb-repressive Complex2 (PRC2) directly bind and silence the Ink4a-Arf locus (Bracken et al. 2007; Kotake et al. 2007; Kia et al. 2008). In MRT cells, human embryonic fibroblast (TIG3) and Human neonatal fibroblast, both PRC1 and PRC2 bind to INK4A and INK4B (Kheradmand Kia et al. 2009; Kia et al. 2008). It has been shown that depletion of EZH2 subunit of the Polycomb-repressive Complex2 (PRC2) in response to stress causes the loss of H3K27me3, displacement of BMI1 subunit of the Polycomb-repressive Complex1 (PRC1) and transcription activation of INK4A (Bracken et al. 2007). Depletion of EZH2 during aging and differentiation causes displacement of BMI1 and activation of INK4A and INK4B (Kheradmand Kia et al. 2009; Kia et al. 2008). Numerous studies showed that promoter hypermethylation of CpG island (CGI) sequences is the most frequent pathway for inactivation of P 16 INK4A in human carcinomas,including those that arise in the lung, oropharynx, bladder,cervix, liver, colon, pancreas, and other sites (Baylin et al. 1998). The H3K36 demethylase JmjC domain-containig histone demethylase 1b in primary MEFs regulates cell proliferation and senescence through Ink4b (He et al. 2008). The Ink4/Arf locus is normally expressed at very low levels in most tissues of young organisms (Krishnamurthy et al. 2004). It is well established that the Ink4/Arf locus is activated during organismal ageing in both rodents and humans, and the levels of p16 INK4a constitute an impressively good overall biomarker of ageing (Kim and Sharpless 2006).These observations point to P16INK4A both serving as a brake for the proliferation of cancer cells, and also limiting the long-term renewal of stem cells. The Ink4a-Arf locus responds to stress signals, limiting cell proliferation and modulating oncogene-induced apoptosis (Lowe and Sherr 2003). A challenging issue which remains is to understand the interplay between signaling and PcG control of INK4B-ARF-INK4A locus.
14.3.4.2 SWI/SNF and Polycomb Group Proteins Act Antagonistically on INK4-ARF Locus
Polycomb Group Complex
Polycomb group proteins were first identified in Drosophila Melanogaster as mutants deregulating Hox gene expression pattern during fly early development. The PcG are required to maintain chromatin in a repressed state while the trithorax-group (trxG) proteins (including the hSWI/SNF complex) are necessary for the maintenance of transcriptional activity of several developmental genes. The PcG is a diverse group of proteins that form at least three different complexes: Polycomb repressive complex 1 and 2 (PRC1 and PRC2) and pleiohomeotic (Pho) repressive complex (PhoRC) (Schwartz and Pirrotta 2007).
The main function of the PRC1 proteins is to inhibit chromatin remodeling and maintain the repressed state of chromatin by out-competing the TrxG protein complexes such as SWI/SNF chromatin-remodeling complex. PRC2 on the other hand is known to be the initiator of the suppression process in which chromatin and or DNA are marked for repression. The key component of PRC2 is the SET domain H3 methyltransferase protein enhancer of Zeste (E (Z)). E (Z), when assembled in the complex, methylates H3K27. In human cells, PRC2 can physically associate with Histone deacetylases (HDACs 1 and 2). HDACs can deacetylate H3K27 to make it available for methylation by PRC2 (Kuzmichev et al. 2002). In vivo, trimethylation of H3K27 is characteristic of PcG target genes (Schwartz et al. 2006). The H3K27me3 mark is thought to act as a docking site for chromo-domain of the CBX family proteins, which recruit other members of the PRC1 complex (Bernstein et al. 2006). Genome wide mapping of polycomb target genes revealed that EZH2 depletion alone causes INK4b induction (Bracken et al. 2006).
Mammalian Polycomb group (PcG) proteins are essential transcription silencers that control multiple development processes, including stem cell self-renewal, cell differentiation and have been implicated in several types of cancers (Boyer et al. 2006; Lee et al. 2006; Lund and van Lohuizen 2004; Valk-Lingbeek et al. 2004).
Polycomb Group Complex and INK4-ARF Locus
A number of reports have demonstrated a role for Polycomb silencers, including PRC1 and PRC2 upstream of P15INK4B, P14ARF and P16INK4A (Core et al. 2004; Gil et al. 2004; Kheradmand Kia et al. 2009; Bracken et al. 2007; Martin et al. 2013; Jacobs et al. 1999). The SWI/SNF and PcG proteins act antagonistically on INK4-ARF locus. It is clearly established that PRC1 and PRC2 mainly act upstream of P16INK4A and specifically localize within a region spanning approximately 800 bp. Interestingly, restoration of SWI/SNF in MRT cells causes removal of Polycomb silencers (PRC1 and PRC2) from the P16INK4A promoter (Kia et al. 2008). However it is not consistent with a model in which PRC1 blocks SWI/SNF mediated chromatin remodeling as has been previously reported from experiments conducted in vitro (Shao et al. 1999). Instead, the silencing by PcG complexes is a less rigid more dynamic process subject to removal in response to SWI/SNF expression (Kia et al. 2008).
Concomitant with the decrease in H3K27me3, the active H3K4me3 mark is strongly induced at P16INK4A and P15INK4B (Kia et al. 2008). The prominent H3K4me3 methyltransferase MLL1 is the human homologue of Drosophila TRX, the founding member of the trxG (Canaani et al. 2004). During hSNF5-mediated p16 INK4a activation, the trxG activators SWI/SNF and MLL1 replace the PcG silencers PRC1 and PRC2.
Here we show that YY1 and PcG silencers concomitantly bind to the INK4A/B locus. Strikingly, induction of hSNF5 leads to the removal of YY1 and PcG proteins (Fig. 14.7). YY1 is the human homolog of Drosophila Pleiohomeotic (PHO)—a key sequence specific DNA binding recruiter of PcG complex.
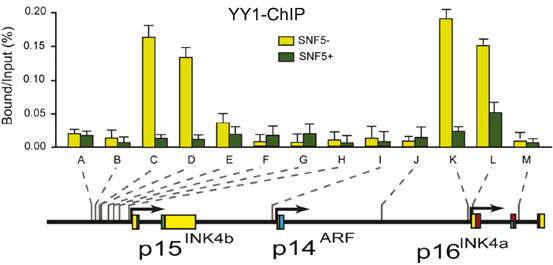
Fig. 14.7
Restoration of SWI/SNF Causes Removal of YY1 from P15INK4B and P16INK4A Promoters. (a) Restoration of SWI/SNF Causes Removal of YY1 from INK4a/b locus. ChIPs using antibodies directed against YY1. Cross-linked chromatin was prepared from MRT cells lacking hSNF5, but expressing GFP (light green bars), or cells expressing hSNF5 (dark green bars). ChIPs were analyzed by qPCR using primer sets specific for the regions indicated by A–M along the INK4B-ARF-INK4A locus, revealing that YY1 binding peaks at the p16INK4a promoter and 3 kb upstream of p15INK4b. Upon induction of hSNF5 YY1 is removed or strongly reduced. QPCR primer sets correspond to the upstream of p15INK4b promoter (A–H), the p14ARF promoter (I), an intergenic control region (J), and various regions of the p16INK4a locus (sets K–M). Primer sets K and L cover the p16INK4A promoter. The positions of the amplified regions on the INK4B-ARF-INK4A locus are indicated at the bottom
It is not clear how PcG proteins and specifically those bound to downstream regions exert their repression over target genes. Long-range chromatin interactions by chromatin looping are thought to be one of the potential mechanisms to explain PcG action over broad distances in cis (Cleard et al. 2006; Comet et al. 2006; Kadauke and Blobel 2009; Schwartz and Pirrotta 2007). The first direct evidence comes from work in Drosophila showing that all major PcG-bound elements at the BX-C multi-gene locus—including PREs and core promoters—physically associate by chromatin long-range interactions in the repressed state (Lanzuolo et al. 2007). The PcGs and histone marks help to silence target genes via the looping process in human cells (Schwartz and Pirrotta 2007; Tiwari et al. 2008b). The PcGs also bind to P16INK4A locus and 3 kb upstream of P15INK4B in the repress state and is removed upon induction of hSNF5 during aging and differentiation (Kia et al. 2008).
Methylation of INK4-ARF Locus
Hypermethylation of CpG island (CGI) sequences are a nearly universal somatic genome alteration in cancer. DNA methylation, occurring on cytosine bases in CpG dinucleotides, is an important epigenetic mechanism of gene regulation in eukaryotic cells. CpG Island of the p16 INK4A promoter is hypermethylated in various cancers (Merlo et al. 1995). Alterations of SWI/SNF remodeling complex activity in various mammalian cells and organs have been implicated in transcriptional silencing through site-specific genomic methylation (Banine et al. 2005; Dennis et al. 2001; Gibbons et al. 2000). hSNF5, a core subunit of SWI/SNF induces demethylation of p16 INK4A to promote transcriptional activation. DNMT3b is involved in de-novo methylation during development. Induction of hSNF5 causes removal of DNMT3b from the p16 INK4A promoter that can explain alterations of methylation in this region (Kia et al. 2008). Higher expression of DNMT3b has shown in human oesophageal squamous cell carcinoma in correlation with low p16 INK4A expression (Simao Tde et al. 2006). Disruption of DNMT1 and DNMT3b resulted in demethylation of the p16 INK4A in other human cancer cells (Rhee et al. 2002).
EZH2 a Key for Differentiation, Aging and Cancer
EZH2 is the catalytic subunit of PRC2, which is a highly conserved histone methyltransferase that targets lysine-27 of histone H3. This methylated H3K27 chromatin mark is commonly associated with silencing of differentiation genes in organisms ranging from flies to human. EZH2 is frequently overexpressed in wide variety of cancerous tissue types including prostate, breast, lymphoma, myeloma, bladder, colon, skin, liver, endometrial, lung, gastric (Simon and Lange 2008). EZH2 and EED co-immuneprecipitate with all three human DNMTs and silencing of certain target genes requires both EZH2 and DNMTs (Vire et al. 2006). PRC2 is required for the expression of proliferative genes. EZH2 and EED are targets of the pRB-E2F pathway, and deregulation of the pathway, as is frequently observed in human cancer, would result in higher levels of EZH2 and EED. The EZH2 and EED promoters are direct targets of the E2F transcription factors in vivo. Overexpression of EZH2, like MYC shortens the G1 phase of the cell cycle, results in accumulation of cells in the S phase of the cell cycle, and confers a proliferative advantage by suppression of Macrophage Stimulating 1 ( MST1) (Kuser-Abali et al. 2014). The role of E2F and pRB in the control of embryonic development is likely related to their ability to regulate the abundance of the PRC2 complex (Bracken et al. 2003). EZH2 is accumulated in undifferentiated progenitor cell population, such as hematopoitic cells (Su et al. 2003). Genome wide mapping of PRC2 target genes in different cells in human, mouse and Drosophila by ChIP-on-ChIP method have been performed by different groups. These studies revealed that PRC2 target genes are highly enriched for transcription factors and signalling components that control cell differentiation (Simon and Lange 2008). Correspondence between silenced genes bound by Oct4, Sox2 and Nanog, which promote expression of proliferation genes and silencing of differentiation genes, with PRC2 target genes suggests that PRC2 is a key co-repressor in ES cells (Lee et al. 2006; Pasini et al. 2007). It has been shown that epidermal basal cells like ES cells are rich in Ezh2 and other PcG proteins, but as they differentiate, Ezh2 expression is turned off and the cells exit the cell cycle concomitant with induction of INK4b/a. How PcG genes themselves are regulated still remains unresolved. It is not mechanistically clear how PcG proteins repress their targets, PcG and histone marks may silence target genes through the looping process. It has been shown that PcG-occupied region can form chromatin loops and physically interact in cis around a single gene in mammalian cells. Ezh2 knock down was shown to affect this long-range interaction (Tiwari et al. 2008a). Analysis of the spatial organization of the INK4/ARF locus in vitro by 3C (Chromosome Conformation Capture) technology revealed that at least in Mon cells, Neonatal fibroblasts and Megacaryocyte-Erythrocyte progenitor cells, there is a physical and spatial interaction between p15INK4B and p16INK4A but not p14ARF (Dekker et al. 2002). Importantly, p15INK4B loses its physical interaction with p16INK4A upon induction of hSNF5, aging and differentiation. It is shown that inhibition of EZH2 in atypical rhabdoid teratoid tumor cells suppresses the self-renewal (Alimova et al. 2013). There is strong evidence that PcGs play a role in generating the repressive loop at the INK4/ARF locus (Dekker et al. 2002).
Related posts:
 Reduced Risk of Cancer in Schizophrenia, a Bridge Toward Etio-Pathology and Therapy of Both Diseases
Biodynamic Phenotypic and Epigenetics Changes of Circulating Tumor Cells: Their Application in Cancer Prognosis and Treatment
An Introduction to Impact of Bio-Resonance Technology in Genetics and Epigenetics
Sentinel Gene Within Cell Territory and Molecular Platforms in Cancer: Methylation Diversity of p53 Gene in Brain Tumors
Exploring ATM and Methylation in Cancer: Emphasizing on Brain Tumors
Methylation in Colorectal Cancer
Reduced Risk of Cancer in Schizophrenia, a Bridge Toward Etio-Pathology and Therapy of Both Diseases
Biodynamic Phenotypic and Epigenetics Changes of Circulating Tumor Cells: Their Application in Cancer Prognosis and Treatment
An Introduction to Impact of Bio-Resonance Technology in Genetics and Epigenetics
Sentinel Gene Within Cell Territory and Molecular Platforms in Cancer: Methylation Diversity of p53 Gene in Brain Tumors
Exploring ATM and Methylation in Cancer: Emphasizing on Brain Tumors
Methylation in Colorectal Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


