Fig. 8.1
Localization of p53 gene on chromosome 17p13.2
The origins of p53 gene’s ancestors are initially seen in descendants of single cell choanoflagellates and early metazoan sea anemone. The ancestral p53 gene is much related to both p63 and p73 genes. Interestingly, this p63/p73 like genes are responsible for guarding the DNA of germ-line gametes against damage in sea anemone which is also conserved in insects, worms, clams, and vertebrates. It was proposed that p53 gene is as a result of gene duplication of these ancestral genes to keep on their critical responsibility in both somatic and progenitor cells. The vertebral p63 and p73 genes are as consequences of second duplication which has been occurred in the ancestral genes. The functions of p63/p73 genes have been complicated during the development of vertebrates and were extended to regulation of transcription within the skin and other organ cells (Yang et al. 2002; Yang and McKeon 2000; Hernandez-Acosta et al. 2011; Mills et al. 1999).
Transcription of p53 expression is regulated through several transcription factor binding motifs which are distributed within the promoter sequence of p53 gene and are evolutionary conserved (Neduva and Russell 2005; Neduva and Russell 2006) The Myc/Max, USF, YY1, NF1, AP-1 and NFκB were described to activate the expression of p53 gene whereas binding the PAX2, PAX5, PAX8, and BCL6 transcription factors to their specific motifs negatively regulate the transcription of p53 (Reisman et al. 1993; Kirch et al. 1999; Ronen et al. 1991; Roy et al. 1994). Moreover, HOXA5 was found as another activators of p53 gene owing to the up-regulation of p53 following to its over-expression which was associated with apoptosis of breast cancer cells (Raman et al. 2000). Both the human and mouse have two interferon stimulated response element (ISRE) within their p53 promoters that are induced by a complex including Stat1, Stat2 and IRF-9. These ISRE could be targeted for IFNα/β therapy in cancer in order to activate immune system against tumor cells (Takaoka et al. 2003; Pfeffer et al. 1998). There is an important regulatory element between the BCL6 binding motif and CpG Island of p53 promoter which is called as CTCF motif that activates the expression of p53 through blocking the negative effects of transcription silencers (Soto-Reyes and Recillas-Targa 2010; Su et al. 2009). Moreover, protein kinase C δ (PKC δ) is another transcription factor that by accompanying Btf, plays pivotal roles in induction of apoptosis in response to DNA damage through binding to its core promoter element (CPE) on p53 promoter (Liu et al. 2007).
8.1.2 p53 Protein
In human, the full length of p53 protein is composed of 393 amino acids. It has a close homology in structure and function with its two famous family members including p63 and p73 (Fig. 8.2). Although it remains at low level in the cell by degradation effect of 26S proteasome, DNA damage or every other stress signals immediately stabilize and activate it. The first 42 amino acids build up the NH3 or amino terminal (N-terminal) of the p53 protein which its 22 and 23th residues forms the transactivation domain (Teufel et al. 2007). MDM-2, the major negative regulator of p53, exerts its inhibitory effects on transcription and ubiquitination of lysines residing in carboxyl terminal (C-terminal) domains (Rippin et al. 2002). The amino acids between the residues 43–63 construct the second transactivation domain of p53 protein probably designed to induce the transcription of other genes which are under the control of p53. Immediately after these transactivation domains, there is another domain spanning within the 61–94 residues which is proline rich and plays pivotal roles in apoptosis and protein-protein interactions through SH-3 signals (Walker and Levine 1996; Neduva et al. 2005a, b). DNA binding domain (DBD) and oligomerization domain (OD) are the other domains of p53 protein which are composed of the 101–306 and 356–393 residues, respectively. Of note, there are three nuclear localization signals (NLS) in OD domain. The last domain at the C-terminal has regulatory function and is spanned within the 356–393 amino acids. Both C and N-terminals of p53 protein are actively involved in post-translational modifications including phosphorylation, ubiquitination, acetylation and methylation (Bode and Dong 2004).
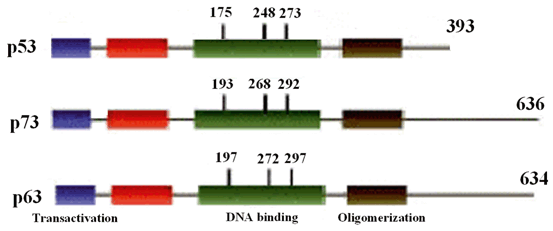
Fig. 8.2
Schematic structure of p53 and its two homologous proteins.
8.1.2.1 p53 Protein Methylation
Protein methylation is almost defined as transferring a methyl group to amino acids especially lysines, arginines and histidines that is important in modulation of gene expression, RNA metabolism and also influences the function of protein (Chen et al. 1999; Shen et al. 1998). The p53 protein is one of the few proteins which its stabilization and transcription factor activity is regulated through methylation and demethylation of its residues.
Methylation of p53 protein is fulfilled through transferring methyl group to either lysines residues residing in the basic C-terminal domain or argenine amino acids within the DBD domain performing by histone lysine methyltransferases and arginine methyltransferases, respectively.
The histone lysine methyltransferases include KMT5 (Set9), KMT3C (Smyd2), and KMT5A (Set8) (Allis et al. 2007). KMT5 methylates the K372 and thereby enhances the stability and nuclear localization of p53 protein to be enough strong to activate the expression of its target genes as well as p21 and BAX and induce apoptosis through arresting cell cycle at G2/M checkpoint (Ivanov et al. 2007). The activity of KMT5 protein was shown to be increased in response to DNA damage without increasing its gene expression, possibly via post-translational modification with acetylation and phosphorylation. It was demonstrated that K372 has been methylated by KMT5 followed by acetylation of K373/382 when p53 induced the expression of p21. It is indicating that first methylation and then acetylation of p53 are required for inducing the expression of target genes. Interestingly, methylation of p53 by the two other methytransferases, KMT3C and KMT5A, leads to reducing the capability of p53 to induce the expression of p21, PUMA and MDM2 (Brown et al. 2006; Shi et al. 2007). Although, expression of KMT3C remains constant and is not changed by detecting the stress signals, but, methylation of K372 by KMT5 prevents methyltransferase activity of KMT3C which leads to p53 methylation (Huang et al. 2006).
Argenine methylation of p53 protein is performed by two protein groups belonged to class I of protein arginine methyltransferases (PRMTs) including PRMT1 and CARM1 through methylating the argenines within the transactivation and basic C-terminal domains, respectively. In contrast to lysine methyltransfrases, methylation of p53 by both PRMTs is associated with increase in its activity. They also regulate the p53 function through modulation of the methylation status of histone proteins wrapping around its target genes. p53 is able to induce the expression of GADD45 in response to UV radiation when the CARM1 and PRMT have methylated the respective H3 and H4 histones which are enhanced via their prior acetylation by KAT3B (An et al. 2004).
Demethylation of lysine residues of p53 protein is carried out by lysine-specific demethylase KDM1 which has shown to pay critical roles in immediate p53 induction in response to cellular stresses. Knocking down the expression of KDM1 led to decrease in expression of p21 and MDM2 while its over-expression has changed neither the cellular proliferation rate nor the ability of p53 to activate its target genes (Scoumanne and Chen 2007). KDM1 can only demethylates p53 protein when it is dimethylated at K370 and K372 and thereby prevails the binding of it to its major activator, 53 binding protein 1 (53BP1) (Huang et al. 2007). In general, it is assumed that demethylase agents act as the repressors of p53 protein when the PRMTs increase its transactivation of target genes. Further studies are warranted to clarify the role of other demethylase enzymes in demethylation of p53 protein.
8.2 P53 Functions
8.2.1 Cell Cycle Control
There are four main cell cycle checkpoints ensuring that all parts of cell division have been perfectly occurred. When the cell encounters to a stress resulting in DNA damage, the division cycle should be stopped until the damage will be repaired or it immediately being deviated to be mortal. It was shown that p53 is involved in arresting the cell cycle at two major G1/S and G2/M checkpoints (Giono and Manfredi 2006). This function of p53 has been strongly investigated and various molecules have been determined in different pathways acting as upstream and downstream of it. The p21 is the most important downstream molecule of p53 which leads to G1/S arrest when it being activated. Given that inactivation of p21 was not as harmful as p53 loss, indicates that there are some other molecules that do this function as well (Brugarolas et al. 1995). The p53 induces G1/M arrest through repression of cyclin B1/cdc2 complex forming the maturation-promoting factor (MPF). After DNA damage, cell cycle arrest is carried out through two major mechanisms including inactivation of a phosphatase involved in promoting of mitosis known as cdc25c and, activation of 14-3-3σ (Hermeking et al. 1997; Clair St and Manfredi 2006). Increase in expression of 14-3-3σ is associated with interference with nuclear localization of cyclin B1/cdc2 complex following DNA damage. It was demonstrated that 14-3-3σ depletion in HCT116 cell line caused cell death after DNA damage (Chan et al. 1999).There are two final fates for cell cycle arrest: (1) Permanent arrest known as cell senescence; (2) Cell death (Fig. 8.3).
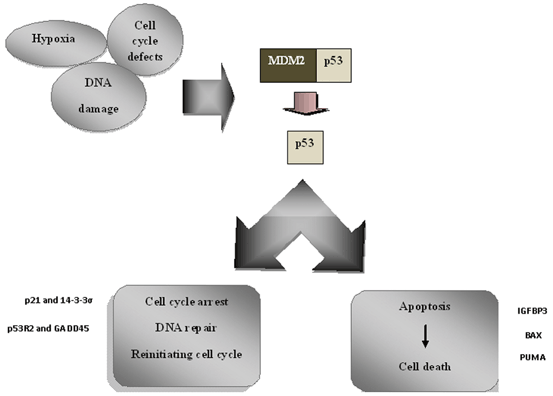
Fig. 8.3
p53 central role in modulation of DNA damage
8.2.2 Cell Senescence
Cellular senescence was initially proposed for normal fibroblast with human origin (Hayflick 1965). According to suggested hypothesis, cellular senescence can be harmful or beneficial to the involved cells. Based on the former hypothesis, cellular senescence could move the cell toward aging which is critical especially when the potential of regeneration is necessary. The later hypothesis reflects that cellular senescence would be an inhibitory mechanism against tumor’s cells proliferation (Campisi and d’Adda di Fagagna 2007).
In the senescent cell, there is a remarkable but regular change in gene expression profile which is similar to the process of wound healing (Krizhanovsky et al. 2008). Although, there is no special marker to be assigned to a senescent cell, senescence associated β-galactosidase, increased in p16INK4a and p53INK4b expressions have been detected when the cell goes through senescence (Campisi and d’Adda di Fagagna 2007).
The diversity of cellular senescence are usually face some stresses including DNA damage, inactive telomere, abnormal chromatin structure and extra mitogenic signaling generated by oncogenes. It was demonstrated that viral oncoproteins to invade from senescence through inactivation of p53 (Stewart and Weinberg 2006). Many chemotherapeutic drugs are designed to induce senescence in tumor cells which can be inhibited in cells carrying mutant and inactive p53 (Roninson 2003). Based on type of the involved cell and trigger molecule, both the p53-p21 and p16-RB pathways can drive the cellular senescence. p14ARF functions at the center of both mentioned pathways and links them together. The senescence signal is, firstly, received by p14ARF and then activates p53 followed by p21 which in turn suppresses the cyclin-dependent kinases upstream of RB (Martín-Caballero et al. 2001; Matheu et al. 2008). RB itself, induces subsequent activation of p14ARF and p53 by inhibition of E2F1 and its target genes through formation of senescence associated heterochromatin foci (SAHFs) around them (Narita et al. 2006). It was proposed that p53 exerts its tumor suppressive activity in induction of senescence through overexpression of extracellular matrix (ECM) degrading enzymes and inflammatory cytokines to target and kill the tumor cells by dynamic action of innate immune system (Xu 2008).
8.2.3 Apoptosis
The role of p53 in apoptosis was initially identified in mouse thymocytes after radiation (Clarke et al. 1993). There are many known targets for p53 gene function in apoptosis indicating that such role of p53 is more indicative than its other functions. The most important targets of p53 include p53 upregulated modulator of apoptosis (PUMA) and Noxa which are the only proteins that have BH3 repeats, Bax, PIG3, CD95 or Fas, Killer/DR5, p53AIP1 and Perp (Riley et al. 2008). It seems that active involvement in apoptosis is shared by other family members of p53 gene which is modulated with various external and internal signals and is also dependent on cell type (Fridman and Lowe 2003).
Induction of apoptosis is fulfilled through activation of cellular p53 depending on apoptosis pathway in mitochondria. Some apoptotic specific signals as well as radiation pull the p53 to be localized in mitochondria to induce secretion of proapoptotic factors from the intermediate space of mitochondria through increasing the permeability of outer membrane of mitochondria. The major effect of p53 in induction of apoptosis through mitochondria pathway could be observed through activation or repression of inhibitors of Bcl2 family including Bcl-XL, Bak and Bax (Chipuk et al. 2005). When the p53 becomes activated in response to any stress, requiring cell to be undergone apoptosis, then it will immediately increase the level of PUMA to make the p53 free through binding with Bcl-XL.The TP53 then will be able to activate the Bax gene. Among all of the target genes of p53 in apoptosis, PUMA is as critical as p53 which its loss has shown the same defects as in p53 loss (Jeffers et al. 2003).
8.2.4 Response to Stress
The p53 protein protects cell against any type of stress including DNA damage, any temperature shock (heat or cold), spindle toxins, hypoxia, telomere shortening and even activation of oncogenes or inactivation of other tumor suppressor genes. Activation of p53 in each of the mentioned cellular stresses is modulated through induction of its activators as well as HAUSP or inactivation of its negative regulators which include MDM-2, MDM-4, WIP-1 phosphatase (Perry 2010; Lu 2010; Meek and Anderson 2009). Some of these regulatory molecules, similar to WIP1- and MDM-2, are responsible for dimorphic pattern between two sexual identities of human. MDM-2 contains sequence recognized by estrogen receptor (ER) to be combined with the alleles of a SNP within the intron 1 which was associated with higher risk of breast cancer in ER positive premenopausal women (Hu et al. 2007). Of note, the cancer predisposition of this SNP and the age of onset of cancer incidence is different between male and females making sexual dimorphism (Grochola et al. 2010). WIP-1 encodes a phosphatase and was found to be one of the estrogen regulated genes (Han et al. 2009).
Strikingly, p53 differently respond to stress in various types of cells even when the genetic and environmental conditions of the cells are the same (Lahav et al. 2004). It remained many questions to be answered especially about the roles of p53 regulators in these different patterns of response.
8.2.4.1 Metabolic Stress
The main factor that a cell may encounters to it, is metabolic stress. p53 plays critical roles in switching the metabolic pathways in cancers in order to disable the malignant cells to survive and proliferate under stress conditions (Gottlieb and Vousden 2010). Cell proliferation is induced with glucose through activation of AMP-activated protein kinase (AMPK) which phosphorylates and activates p53 (Hardie 2004). In metabolic stress, AMPK also induces the tuberous sclerosis complex 2 (TSC2) protein which in turn inhibits GTP-binding protein, Rheb, and then mammalian target of rapamycin1 (mTORC1) are noatable. In nutrient restriction, mTORC1 dephophorylates p53 on Ser15 (Inoki et al. 2003; Feng et al. 2006; Imamura et al. 2001) (Jones et al. 2005). The p53 targets which are activated one by one in mTORC1 and IGF-1/AKT pathways modulate their pathways in opposite directions. For instance, Sestrin 1 and 2 are induced by p53 in IGF-1/AKT pathway play role in response to oxidative stress through two critical functions: (1) Indirect activation of TSC1/2 by phosphorylation and activation of AMPK through interaction with its α-catalytic subunits which leads to suppression of m-TORC1 pathway; (2) Reducing the cellular level of reactive oxygen species (ROS) through deoxidizing the ROS products by oxidized peroxiredoxins (Budanov et al. 2002; Budanov et al. 2004; Velasco-Miguel et al. 1999).
In general, p53 is actively involved in maintaining the baseline metabolic status of cell and genomic integrity in response to stresses threatening them. These stresses are created through three major mechanisms which will be discussed in detail in the following sections including regulating the glycolysis, oxidative phosphorylation and metabolism of fatty acids.
8.2.4.2 Regulating Glycolysis
Glycolysis is a non-oxidative pathway of energy production mainly used in neoplasms including the malignant cells to supply their energy (Warburg 1956). In glycolysis pathway, there is a protein called TP53-induced glycolysis regulator (TIGAR) which under the induction of p53 shifts glycolysis toward pentose phosphate pathway (PPP) through fructose-2, 6-bisphosphate. TIGAR, actually by this switching, helps to keep the metabolic status of cell stable through reducing the generation of ROS and resistance against it through production of nicotinamide adenine dinucleotide phosphate (NADPH) in PPP pathway (Bensaad et al. 2006). Another attempt of p53 in stablizing the cell in metabolic stress is fulfilled through inhibition of phosphoglycerate mutase (PGM). Although, both the TIGAR and PGM are considered as intracellular antioxidants, in contrast to TIGAR , PGM increases the glycolysis to decrease the ROS generation by mitochondrial respiration (Brand and Hermfisse 1997). Nonetheless, enhancing of the glycolysis pathway in cancer cells would be dangerous, and PGM is suppressed by p53. Mutant p53 acts in reverse direction in cancerous cells in order to increase the glycolysis pathway through induction of PGM and hexokinase 2 (HK2) which is another driver of glycolysis oxidative pathway (Bustamante and Pedersen 1977; Mathupala et al. 1997).
8.2.4.3 Role of p53 in Oxidative Phosphorylation
Oxidative phosphorylation is the preferred and more proficient energy producing pathway in normal cells which is carried out in mithochondria. This pathway is aimed to oxidate the pyruvate, as the end product of glycolysis pathway, and generates ATP in a more efficient manner. P53 enhances the mithochondrial respiration and the expression of cytochrome c oxidase (SCO2) which is essential for formation of cytochrome c oxidase (COX) complex (Bensaad et al. 2006). Cancer cell with abnormal or missing p53 function had shown the pathway switching from oxidative phosphorylation to glycolysis (Ma et al. 2007). The p53 increases the mitochondrial respiration and ATP generation through glutaminase 2 (GLS2) induction which is required for converting the glutamine to glutamate and production of α-ketoglutarate (Hu et al. 2010).
8.2.4.4 How Does p53 Play Role in Metabolism of Fatty Acids
The p53 protein controls the metabolic status of the cell through regulating the functions of two important molecules in fatty acids metabolism pathways including guanidinoacetate methyltransferase (GAMT) and carnitine palmitoyltransferase (CPT1).
GAMT is involved in creatine pathway and changes the guanidineacetate to creatine to be used in ATP generation pathways (Ide et al. 2009). GAMT is related to fatty acid synthase (FAS) which has demonstrated high expression in various types of cancer cells to drive glucose independent energy generating pathways (Alo et al. 1996; Swinnen et al. 1997)
Another strategy of p53 in keeping the baseline status of cellular metabolism in a calm manner, is fulfilled by induction of β-oxidation of fatty acids through activation of CPT1 which attaches the carnitine to fatty acids to be imported in to the mitochondria (Buzzai et al. 2005; Vousden and Lu 2002).
8.2.5 Tumor Suppression
The successfulness of p53 in suppression of tumor is determined by three major factors including perfect detection of oncogenic signals, differentiating between healthy and cancerous signaling and efficient conquering on neoplastic cells (Tyner et al. 2002). Tumor suppressive activity of p53 can be overcome through five main mechanisms; (1) Oncogenic signaling below the threshold of p53 activation which is in line with first model described above. In these conditions, the cell containing damaged DNA can go through carcinogenesis prior to be repaired by ARF or p53 induced DNA repair pathways (Zilfou and Lowe 2009). Complementary studies on subordinating the threshold of p53 signal detection have shown that over-activity in tumor suppression role of p53 also increases its regenerative capacity which would be associated with premature aging (Cao et al. 2003; Lanni et al. 2008; Maier et al. 2004; Medrano et al. 2009; Campisi 2003). (2) Inactivating mutations in the upstream molecules of p53 which can be influenced by several factors including the type of mutation and gene, the extent of degeneracy of affected pathway and its contribution to tumor suppression. (3) Mutations in other contributory molecules rather than upstream which can abrogate the major functions of p53 in either apoptosis or cell senescence. Overexpression of BCL-2 or BCL-XL, decreases the expression of apoptosis effectors as well as PUMA or NOXA and loss of INK4A and CDKN1A involved in cell senescence, are some examples. (4) Tumor may arise, when, the overall outcome of p53 activation is switching from cell senescence or apoptosis to temporary arrest or incomplete repair of damaged DNA in favor of cancer evolution. This mechanism makes the growth of tumor more plausible through unbalancing between statue of cell loss and cell gain. (5) Genetic or epigenetic mutations in p53 gene which directly affect its function or expression and sometimes changes its tumor suppressor character to a new functionally oncogene discussed in detail in next section (Strano et al. 2007; Xu 2008; Lozano 2007).
8.3 Role of p53 Gene Mutations
P53 is the key gene which is altered in more than 50 % of different types of cancers. Li Fraumeni’s syndrome (LFS) which is characterized with multiple generations susceptible to some types of cancers was the first evidence of the role of germline p53 mutations in risk of cancer (Li and Fraumeni 1969b; Li and Fraumeni 1969a). LFS is inherited with autosomal dominance pattern and affected families are prone to get breast cancer, sarcoma and different types of neoplasms (Malkin et al. 1990)
Single nucleotide non-synonymous mutations are the most common type of p53 alteration usually associated with replacement of amino acid with another one with new function. Mutations almost change the half life of p53 protein in order to be prolonged and this apparently gain of function led to assume that it is an oncoprotein. This special characteristics of p53 mutations, makes it unique among all the tumor suppressor proteins those alterations almost lead to insufficient expression with normal function (Levine et al. 1995). The second dominant aspect of p53 alteration could be described through this evidence that loss of heterozygosity (LOH) of an allele of p53 gene is subsequently occurred after the episode of mutation in another allele (Brosh and Rotter 2009).
Similar to other genes, mutations can be seen in every nucleotides of p53 protein encoding gene, and some hot spots have been determined in this protein. DBD is the most common target of mutation in p53 protein wherein many various and famous alterations have been identified (Petitjean et al. 2007; Olivier et al. 2004).
8.3.1 p53 Gene Defects in Different Cancers
Involvement of the p53 mutations in different cancers can open the promising windows toward improvements in treatment schedules and more precise determination of prognosis and survival for cancer patients (An et al. 2004). Interestingly, more than 80 % of the p53 gene mutations are missense alterations which usually ends in absolute intact and functional protein in contrast of non- or dysfunctional mutations in other tumor suppressor genes (Soussi and Beroud 2001). Loss of function of p53 gene has shown controversial clinical effects on the outcome of chemotherapy which are usually cell specific (Soussi and Beroud 2003). Although p53 gene mutations are the most frequent genetic alterations in various cancers, it seems that it may have no or scant role in some of cancers as well as testis cancer and melanoma. However, it was demonstrated that genetic mutations of Apaf gene, as one of the downstream molecules of p53, had influenced the success rate of chemotherapy in melanoma patients (Soussi and Beroud 2003). Furthermore, although the frequency of p53 gene mutations was reported to be low in inflammatory breast cancer and neuroblastoma, high amounts of normal p53 protein accumulated in cytoplasm of tumor cells may interfere with its immediate action after DNA damage (Zaika et al. 1999).
It is worth to note that more than 70 % of the p53 mutations take place in the sequences that are not hot spot and around 4.4 % of them have been found once and their importance in the cancer progression merited to be determined (Soussi and Beroud 2003). Since most of the p53 mutation screening studies have focused on hot spot DNA binding domain; it has been famous to have the most frequent of alterations in p53 mutation database (Soussi and Beroud 2001). The most well-known hot spot regions of p53 gene include the codons 175, 248 and 273, those alterations comprise 19 % of all the p53 mutations. Based on the effects of these alterations on structure and function of p53 protein, these could be classified into two major classes (Cao et al. 2006; Joerger et al. 2004). So mutations that change the amino acids which are involved in interaction of p53 with DNA sequence are categorized in the class I. These types of p53 mutations as well as alterations in codon 248 comprise 7.6 % of all the p53 mutations recorded in p53 gene mutations database (http://p53.free.fr). However, these mutations have no effect on overall p53 protein conformation determined by spatial monoclonal antibodies and low affinity of mutant p53 protein to be bound with chaperon molecules (Ory et al. 1994; Hinds et al. 1990; Vakifahmetoglu-Norberg et al. 2013). In contract, class II of mutations make mutant protein to be strongly recognized by the major chaperon, hsp70, due to their conformational changes (Ory et al. 1994). Alterations of codon 175 are the best examples of this type of p53 mutations. Class II mutations include 4.9 % of the p53 mutations, however, most but not all of them have irreparable effect on protein conformation (Selivanova et al. 1997).
Mutations in hot spot regions have different frequency in various cancers and sometimes they have no crucial impact on the structure and function of p53 protein. An explanation for different frequency in various cancers may be due to the inactivation of p53 protein in special and diverse mutagenic pathways in each cancer (Soussi and Beroud 2003). For instance, G > T transversion is predominantly seen in lung cancer and head and neck cancer which have shown strong correlation with tobacco smoking. The G > A transversion is considered to be the most frequent mutation of codon 175 found in breast and colon cancers and, include the 5 % of the total pool of p53 mutations. This variant leads to substitution of an Arginine with one Histidine amino acid that accompanies excessive defects in normal biological and biochemical activities of p53 protein. It was shown that R175H mutation leads to increased cell proliferation and transformation in heterozygous mice carrying this alteration (Lang et al. 2004). Moreover, it was associated with a gain of function in p53 protein which was resistance to chemotherapeutic agents in cultured cells and breast cancer patients (Blandino et al. 1999; Aas et al. 1996). It was shown that p53 protein carrying R175H mutation is able to decrease the expression of Fas pro-apoptotic gene through binding with the sequence which is different from the site recognized by normal p53 protein to induce its expression (Zalcenstein et al. 2003). Although, it was demonstrated that the residues 62–69 of p53 protein play crucial roles in its apoptosis function, deletion of this region would be associated with decrease in induction of BAX and PIG3 pro-apoptotic genes (Garcia and Attardi 2014; Venot et al. 1998). In lung cancer its importance had been attenuated by the alterations of codons 157 and 158 which are the hot spots of nucleotide change in exposing to benzo (α)pyrene adducts found in tobacco (Denissenko et al. 1996; Caron de Fromentel and Soussi 1992).
The GC > AT transition occurs in frequency of less than 5 % in each of codons 175, 248 and 273 while comprise 51 % of all the point mutations and 59 % of all the CpG dinucleotide transitions within the p53 gene. This high rate of transversion is due to the deamination of methylcytosine to thymidine nucleotide which could not be efficiently recognized by repair system and makes replacing of CpG with AT in the following rounds of replication (Denissenko et al. 1997). However, the position of CpG in the codons arrangement can affect the fate of transition. When the CpG lies in the CGN pattern of codon which is called the type I of CpG dinucleotide, it will always be associated with amino acid replacement. In the second and third types (NCG, or NNC, GNN), since there is degeneracy in genetic code including G nucleotide, mutation can only occurrs in the transition of modified C nucleotide. Given that the GC > AT transition usually provide no selectable growth advantage for infected cell, most of them are harmless and doesn’t lead the cell to become cancerous (Smela et al. 2001; Soussi and Beroud 2003).
8.3.2 p53 Polymorphisms and Cancer
P72R is the first single nucleotide polymorphism identified in p53 gene which has demonstrated heterogeneity in frequency of proline proline (PP) genotype among different populations from Scandinavia (16 %) to Africa (63 %) (Harris et al. 1986; Li and Fraumeni 1969b). The role of this polymorphism has been detected in open angle glaucoma, endometriosis, recurrent pregnancy loss, glioma, breast, cervix, and esophageal cancers (Hou et al. 2013; Jia et al. 2012; Zhao et al. 2013; Tang et al. 2011; Zhou et al. 2012; Shi et al. 2012; Guo et al. 2012). Codon 72 is located in the proline reach domain (PRD) of protein which is the major regulatory domain of p53 protein during apoptosis (Venot et al. 1998). It was shown that p53 protein containing proline has less potential proapoptotic activity relative to variant p53 proteins with Argenine that is may be due to stronger interaction of variant p53 protein with nuclear-export protein chromosomal region maintenance 1 (CRM1) (Bergamaschi et al. 2006). This would be associated with increased nuclear export of p53 protein leading to more entry of it into the mitochondria (Dumont et al. 2003). In addition, it was found that mutant R72 p53 protein provides a new malfunction which is greater affinity to p73 protein to inhibit its apoptotic activities. The later side effects of R72 mutant p53 would be a convincing explanation for poor prognosis and response to chemotherapy in head and neck cancer patients (Bergamaschi et al. 2003).
P47S is the second main exonic p53 polymorphism that is associated with substitution of proline to serine in codon 47 and had demonstrated different frequency among various studies (Felley-Bosco et al. 1993; Kashima et al. 2007). Although the clinical importance of this variant remains elusive, Li and his coworkers have demonstrated that this amino acid conversion may interfere with the proper phosphorylation of adjacent S46 codon leading to defective apoptotic function of p53 protein (Li et al. 2005).
8.3.3 Other Genes Variants and Mutations Affecting p53 Signaling
Mutations and variants in other genes interacting with p53 can have some pivotal influences on its crucial functions that will describe the most important of them in two following sub-sections. Although it is established that the response element (RE) of p53 protein has a highly degenerate sequence, several studies have found some of SNPs in RE that can influence the p53 signaling. SNP309 in the MDM2 gene is an example that include conversion of T to G nucleotide in intron 1 which is close to the p53 RE sequence. It causes the MDM2 mRNA and protein would be increased in response to enhanced recognition of DNA binding site with specifity protein 1 (Sp1) (Bond et al. 2004).
High level of MDM2 expression will be associated with decreased p53 expression and its defective apoptotic response to DNA damage (Bond et al. 2004).The GG genotype of T309G has a frequency of around 1 % and the G allele was found to be associated with earlier onset of cancer in family members of Li-Fraumeni syndrome. Interestingly, since the promoter of MDM2 gene is recognized by hormonal signals, it was shown that penetrance of this variant was more in women (Bond et al. 2006). Another variant (SNP354) was found in the exon 12 of MDM2 gene which has demonstrated strong association with risk of breast cancer, even though its biological effects remains undetermined (Boersma et al. 2006). AKT gene is negatively regulated by p53 through activation of Phosphatase and tensin homolog (PTEN) phosphatase and has a suppressive function on p53 activity by phosphorylating and stabilizing MDM2 (Zhou et al. 2001). Several SNPs have been identified in AKT gene that have shown association with overexpression of it and poor apoptotic response to irradiation (Harris et al. 2005). Fms-like tyrosine kinase 1 (FLT-1) is another gene with a polymorphism in its p53 response element which can affect the strength of FLT-1 gene expression induced by p53. FLT-1 is indirectly involved in angiogenesis through encoding a receptor which coupled with VEGF (Menendez et al. 2006).
Among the genes which their mutations could affect the p53 activity, BRCA1 seems to be the most important. Coexistence of BRCA1 and p53 mutations is a relatively common genetic alteration especially in breast cancer families. It was shown that p53 mutations have risen in a mutated BRCA1 background in these families almost with different pattern more residing in non hot spot p53 gene codons (Crook et al. 1997). Although they were susceptible to get cancer, rescue of mice carrying Brca1Δ11 (deletion of carboxyl terminal of BRCA1 protein) in p53+/− background is relying on that p53 mutations act as the passenger alterations (Cao et al. 2006). However, co-selection of BRCA1 and p53 mutations has shown to be associated with normal p53 protein functions. It may be due to that the lack of impact of these mutations on DNA repair activities of p53 owing to its close cooperation with BRCA1 or may alter some of the unknown p53 functions (Bourdon et al. 2005; Mihara et al. 2003).
Interestingly, the spectrum of p53 has shown to be low in colorectal cancers with positive status of microsatellite instability (MSI+ ). The tendency of MSI+ tumors to driving mutations in genes including BAX, TGFβR and IGFR which contain repeated polynucleotide tract provides the optimal condition for tumor development (Konishi et al. 1996; Young et al. 2001).
In general, further studies merited to define other genetic variations in genes interacting with p53 protein and affect the strength of its crucial activities.
8.4 Methylation Defects in Cancers
The primary evidences relying on the effect of promoter hypermethylation on p53 expression were reported in vitro studies on rat and human cell lines (Schroeder and Mass 1997; Pogribny et al. 2000). However, there are a few reports indicating the role of promoter methylation in regulation of its mRNA and protein expression which will be described separately in each cancer based on the available publications.
8.4.1 Leukemia
Molecular assessment of bone marrow samples of patients affected with acute lymphoblastic leukemia (ALL) demonstrated that p53 promoter was methylated in 31 % of the patients. p53 promoter methylation was compatible with decreased expression of p53 protein versus normal high expression in unmethylated samples (Agirre et al. 2003). Of note, p53 mutations have very low frequency (2–3 %) in ALL patients and therefore promoter methylation may plays more pivotal role in suppression of p53 functions in ALL and different types of leukemia and myeloma (Hurt et al. 2006).
8.4.2 Breast Cancer
Almost low frequency of p53 mutations in breast cancer families and patients (20–25 %) encouraged methylation analysis of p53 promoter in two separate investigations (Pharoah et al. 1999). In the first one, 4/16 CpG dinucleotides sites were found to be methylated in 19 % of the patients (Kang et al. 2001). The second study was extended to analyze the promoter methylation of TP53 gene in four groups including BRCA1/BRCA2 mutation carriers with or without breast cancer, sporadic breast cancer patients and healthy controls. Interestingly, no one of the studied group had shown methylated p53 promoter (Kontorovich et al. 2009).
8.4.3 Other Cancers
Methylation study of TP53 promoter gene was failed to find methylated promoter in 10 patients affected with sporadic adrenocortical carcinoma (ACC) and 5 healthy controls (Sidhu et al. 2005). Methylation analysis in Li-Fraumeni’s and Li-Fraumeni like syndromes patients has revealed strong association with splice site mutations and exonic methylation of p53 gene (Kouidou et al. 2009).
8.4.4 Brain Tumors
Although there are more frequent studies on the methylation of brain tumors than other human cancers, literature of the methylation status of p53 gene promoter is relatively poor and is restricted to almost cell line studies and a few pathological classification of brain tumors. In the primary trial carried out on 67 astrocytic gliomas patients, p53 promoter methylation was found in only 2 % of glioblastomas and 8 % of astrocytoma patients (Gonzalez-Gomez et al. 2003a; Gonzalez-Gomez et al. 2003b). In the following study on 41 oligodendroglial patients, none of the analyzed patients had methylated p53 promoter (Alonso et al. 2003). The methylation status of p53 promoter was then examined through methylation specific polymerase chain reaction (MSP-PCR) using the same primer pairs employed in the two previous mentioned studies in three U87MG, LNT-229, T98G glioma cell lines. It was revealed that the p53 promoter methylation was in direct correlation with mRNA and protein expression in U87MG and T98G cell lines. They then analyzed the p53 promoter methylation in various types of low grade glioma’s primary tissues. P53 promoter was methylated in 60 % of low-grade astrocytoma and oligoastrocytoma and, 74 % of all the oligodendroglioma tumors (Amatya et al. 2005). Huang and his coworkers have found the same correlation in their study on human GBM cell line and verified it using 5-Aza-2’-deoxycytidine (Aza) as a major DNA methyltransferase inhibitor (Huang et al. 2009). However, in spite of restoring the p53 expression in T98G cell line in both studies, in contrast to the results contributed by Amatya and his co-worker, p53 promoter was not methylated in the study performed by Soto-Reyes et al (Soto-Reyes and Recillas-Targa 2010).
Related posts:
 Reduced Risk of Cancer in Schizophrenia, a Bridge Toward Etio-Pathology and Therapy of Both Diseases
Biodynamic Phenotypic and Epigenetics Changes of Circulating Tumor Cells: Their Application in Cancer Prognosis and Treatment
An Introduction to Impact of Bio-Resonance Technology in Genetics and Epigenetics
Malignant Rhabdoid Tumor: Epigenetic Mechanism of Tumorogenesis
Exploring ATM and Methylation in Cancer: Emphasizing on Brain Tumors
Methylation in Colorectal Cancer
Reduced Risk of Cancer in Schizophrenia, a Bridge Toward Etio-Pathology and Therapy of Both Diseases
Biodynamic Phenotypic and Epigenetics Changes of Circulating Tumor Cells: Their Application in Cancer Prognosis and Treatment
An Introduction to Impact of Bio-Resonance Technology in Genetics and Epigenetics
Malignant Rhabdoid Tumor: Epigenetic Mechanism of Tumorogenesis
Exploring ATM and Methylation in Cancer: Emphasizing on Brain Tumors
Methylation in Colorectal Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


