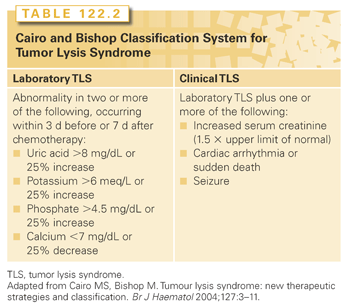
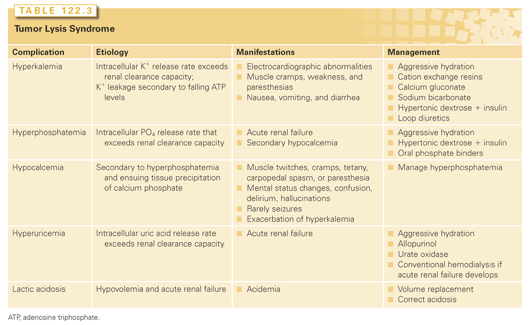
The highest incidence of TLS occurs in patients with rapidly growing, chemosensitive myelo-lymphoproliferative malignancies with high white blood cell counts (acute leukemias) or large bulky adenopathy (high-grade non-Hodgkin lymphomas [NHL], especially Burkitt lymphoma).3–6 In high-grade NHL, an incidence of TLS as high as 42% has been reported, clinically significant in only 6% of patients.7 The latter agrees with a Pan-European retrospective chart review that identified TLS in 3.4%, 5.2%, and 6.1%, of patients with acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), and NHL, respectively. In an era antedating modern interventions, this review found an overall mortality of 0.9% for all patients and 17.5% for patients who developed TLS.8 By comparison, TLS occurs infrequently in solid tumors, most likely due to the lower growth fractions, and slower response to treatment—occurring with tumors that are highly or moderately sensitive to chemotherapy, or if bulky, metastatic disease is present.9 TLS has also been reported following ionizing radiation including total-body irradiation in the transplant setting, embolization, radiofrequency ablation, monoclonal antibody therapy, glucocorticoids, interferon, and in the setting of hematopoietic stem cell transplantation. As new therapies emerge, TLS may occur in unanticipated settings, as seen in indolent chronic lymphocytic leukemia treated with flavopiridol.10 Table 122.2 summarizes the Cairo and Bishop Classification System for TLS. Table 122.3 summarizes recent guidance from a tumor lysis expert panel.11
Hyperuricemia can occur in TLS as noted previously, or more frequently as an isolated finding in patients with cancer. For example, in the Pan-European retrospective chart review previously noted, among 755 patients with ALL, AML, or NHL hyperuricemia without TLS was identified in 13.6% of cases, and with TLS in an additional 5.3%.8
Pathogenesis
Cell lysis with the release of contents at a rate exceeding the kidney’s clearance capacity is the most important etiologic factor in TLS (see Table 122.3 and Fig. 122.1). Rapidly dividing cells have high nucleic acid turnover, and some cancer cells, particularly lymphoid cells, contain higher levels of phosphate than their normal counterparts.
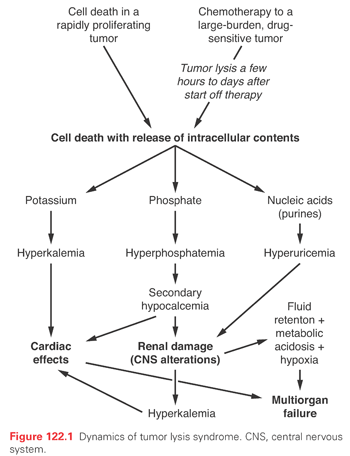
Hyperkalemia poses the greatest immediate threat. Although release of potassium from dying cells is the principal cause, falling adenosine triphosphate (ATP) levels before cell lysis may lead to potassium leakage, explaining why a rise in serum potassium is often the first sign of TLS.
Hyperphosphatemia, like hyperkalemia, follows cell lysis. The initial adaptation involves increased urinary excretion and decreased tubular reabsorption of phosphate. However, as transport becomes saturated, phosphorus levels rise, the calcium phosphorus multiple exceeds 70, and calcium phosphate precipitates in tissues resulting in hypocalcemia. Hypocalcemia leads to increased levels of parathyroid hormone, with decreased proximal tubule phosphate reabsorption, accentuating hyperphosphaturia and the risk of calcium phosphate crystals in renal tubules (nephrocalcinosis) with tubular obstruction.
While hyperuricemia alone may not be an acute threat, it is the most common finding and contributes to renal failure, a complication that in the setting of tumor lysis is usually multifactorial. Within the cell, there is continuous turnover and salvage of bases (Fig. 122.2). In humans, the end product of purine metabolism is uric acid. Adenine is catabolized to hypoxanthine, and this is converted by xanthine oxidase to xanthine and in turn to uric acid. Guanine forms xanthine that is converted to uric acid by xanthine oxidase. In most mammals, urate oxidase catalyzes the oxidation of uric acid to allantoin, a more soluble catabolite. However, a nonsense mutation in the coding region acquired during evolution precludes urate oxidase expression in humans, making uric acid, not allantoin, the final purine metabolite.12
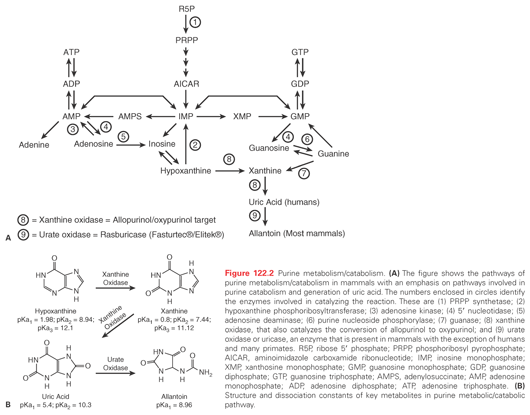
Hyperuricemia occurs primarily as a result of accelerated cellular breakdown. Acute uric acid nephropathy occurs when urate and uric acid crystals obstruct renal tubules. Urate is filtered at high concentrations from the plasma and is further concentrated along the course of the tubular system. As the pH becomes more acidic, uric acid can precipitate, obstructing tubules, collecting ducts, and even pelves and ureters. Crystal deposition increases tubular and intrarenal pressure with extrinsic compression of the vasa recta, an increase in renal vascular resistance, and a fall in renal blood flow. The elevated tubular pressure and decreased renal blood flow reduce glomerular filtration and can lead to acute renal failure.
Therapy
In the modern era, laboratory abnormalities alert physicians and guide preventive management (see Tables 122.1 and 122.3). In adults, preventive measures include foremost hydration, allopurinol, and oral phosphate binders, beginning preferably 24 hours before chemotherapy administration.
Aggressive hydration, the most important intervention, should begin immediately, administering at least 3,000 mL/m2 per day, when possible delaying tumor therapy so hydration can be administered. Urine alkalinization remains controversial as it favors precipitation of calcium/phosphate complexes in renal tubules—calcium phosphate, unlike uric acid, becomes less soluble at an alkaline pH. Furthermore, metabolic alkalemia can worsen the neurologic manifestations of hypocalcemia. Administration of 100 mEq sodium bicarbonate will maintain urine pH above 7.5, a pH that is needed not because of uric acid, which has a pKa of 5.4 (so that above a pH of 6.4 >90% is sodium urate), but because of xanthine which as a pKa of about 7.4.
Hyperkalemia should be treated aggressively. Cation exchange resins should be used, recognizing their value will be delayed. Calcium gluconate antagonizes cardiac effects of hyperkalemia and can be especially helpful if there is concomitant hypocalcemia. Sodium bicarbonate corrects acidemia and shifts potassium back into cells; administering hypertonic dextrose and insulin can augment this. Loop diuretics can eliminate excess potassium in patients without renal failure; hemodialysis is indicated in renal impairment.
Hyperphosphatemia and its resultant hypocalcemia require oral phosphate binders except to manage hyperkalemia avoid calcium administration, because it can promote metastatic calcifications.
Given its central role in acute renal failure, hyperuricemia should be managed aggressively. Allopurinol, an analog of the purine base hypoxanthine, lowers uric acid by inhibiting xanthine oxidase, the enzyme responsible for converting hypoxanthine to xanthine and xanthine to uric acid (see Fig. 122.2). Its active metabolite, oxypurinol, also inhibits xanthine oxidase. Because both allopurinol and oxypurinol inhibit uric acid synthesis, but have no effect on preexisting uric acid, uric acid levels usually do not fall until after 48 to 72 hours of treatment. Furthermore, inhibition of xanthine oxidase leads to an increase in plasma hypoxanthine and xanthine levels, with increased renal excretion of both metabolic products. Like uric acid, xanthine (pKa = 7.4) but not hypoxanthine (pKa = 1.98) may precipitate contributing to acute renal failure. Oral allopurinol has a bioavailability of 50%; alternately, intravenous allopurinol (Aloprim, Bioniche Pharma, Belleville, Ontario, Canada) may be administered. Allopurinol should be discontinued if allergic reactions such as skin rashes and urticaria occur (incidence increased in patients receiving amoxicillin, ampicillin, or thiazides). Finally, the doses of allopurinol should be adjusted for creatinine clearance as follows:

An alternate approach to the treatment of hyperuricemia involves the use of rasburicase (Fasturtec, Sanofi Synth, Paris, France; Elitek, Sanofi US, Bridgewater, NJ), a recombinant urate oxidase developed to replace uricozyme, a nonrecombinant urate oxidase. Because urate oxidase degrades uric acid rather than prevent its synthesis, as does allopurinol, rapid reduction in uric acid occurs following its administration without the accumulation of precursors. Studies in adults and children have shown uric acid levels fall to 0.5 to 1 mg/dL within 4 hours of rasburicase injection with low levels maintained throughout treatment. Despite normalization of uric acid levels with rasburicase, a small percentage have required dialysis—a not unexpected observation, given the multifactorial nature of renal failure in TLS and the fact rasburicase affects only one component, albeit an important one.
Table 122.4 contrasts allopurinol and rasburicase, and notes that rasburicase is more expensive.13 The manufacturer recommends a rasburicase dose of 0.2 mg/kg for up to 5 days (http://products.sanofi.us/elitek/elitek.html). However, except in rare patients with very high serum levels of uric acid, much less is sufficient. A 5-day course of rasburicase is about 15,000 times more expensive than a 5-day course of allopurinol and 15 to 30 times more than intravenous allopurinol, so cost must be factored into decision making. With a 2013 price of ≈$521/mg (Bluebook Average Wholesale Price is $2,345 for three vials containing 1.5 mg each) the 5-day tab for a 70-kg patient receiving the manufacturer’s recommended dose of 0.2 mg/kg for 5 days exceeds $36,470. At this price, rasburicase can be justified only if it will prevent acute renal failure, and the existing data is not compelling. While some studies suggest rasburicase can reduce the need for dialysis, no differences or only clinically insignificant differences in serum creatinine have been reported in other studies, and at the present time the data do not definitively proof that less dialysis is required in patients treated with rasburicase.14–16 It appears rasburicase therapy is beneficial in children with hematologic malignancies; however, randomized controlled trials in adults have been less convincing. Nevertheless, rasburicase is approved by the US Food and Drug Administration for use in adults based on a randomized phase 3 trial.14 Because outcomes other than laboratory parameters were not different, the authors could only conclude “the incidence of laboratory TLS, which can be regarded as an indicator of the risk of clinical TLS, was significantly lower with rasburicase than with allopurinol” and “the rapid reduction in uric acid to less than 1.0 mg/dL with rasburicase simplifies uric acid control by reducing the need for urinary alkalinization in patients who are often critically ill with compromised organ function.”14 An earlier Pan-European multicenter economic evaluation of rasburicase in prevention and treatment of hyperuricemia and TLS in patients with hematologic malignancies concluded rasburicase is economically attractive in the treatment of hyperuricemia both in adults and children while in the prevention of hyperuricemia, it “has an attractive economic profile in children and is cost-effective in adults with ALL and NHL . . . whereas in adult AML patients, the short average life expectancy and the lower baseline risk of hyperuricemia and TLS limits the cost-effectiveness of rasburicase.”8

In both children and adults, it appears the manufacturer’s recommended dose of 0.2 mg/kg/day for up to 5 days is excessive.17–20 Evidence indicates a single 3-mg dose is effective in the prophylaxis and treatment of hyperuricemia in adults with uric acid levels up to 12 mg/dL, with uric acid levels continuing to decline beyond 24 hours in most patients without additional treatment.19 For a 70-kg patient, a 3-mg dose—approximately one-fifth the recommended dose of 0.2 mg/kg—is convenient as vials contain 1.5 mg rasburicase, making it more affordable. Note that as the half-life of rasburicase in serum is approximately 18 to 21 hours, about one-half of the administered dose is still present 24 hours after administration. Given this, a reasonable strategy to consider might be the following: (1) continue to rely on the triad of aggressive hydration, management of electrolyte disturbances, and institution of allopurinol beginning immediately upon presentation; (2) evaluate patients for their TLS risk (see Table 122.1) and administer a single 3-mg dose of rasburicase at presentation to patients with “high risk disease”; and (3) follow the levels of uric acid and administer additional rasburicase doses only to patients with uric acid levels above an arbitrary number such as 5 mg/dL 24 hours after a rasburicase dose. Remember a few additional uric acid measurements are far less expensive than a dose of rasburicase. (Important: Because serum samples contain rasburicase, immediately cool samples obtained for uric acid to 0°C to 4°C, so as to prevent ex vivo enzymatic uric acid degradation that will falsely lower levels. Label test tubes as requiring chilling to avoid removal from ice upon arrival in the laboratory.)
Finally, if despite aggressive management acute renal failure develops, immediate hemodialysis is recommended. Indeed, in patients presenting with TLS, hemodialysis should be started before cytotoxic therapy is administered. Conventional hemodialysis is more effective in eliminating uric acid and phosphate than peritoneal dialysis.
General
Water and sodium homeostasis is frequently disordered in patients with cancer. Optimal homeostasis requires a delicate balance between renal free water clearance and renal sodium metabolism. Hyponatremia, commonly defined as serum sodium <130 mEq/L, occurs with a reported incidence of 3.7% in patients with cancer.21 Clinical manifestations of hyponatremia range from none to impaired consciousness progressing to coma and generalized hypotonia, or seizure activity all secondary to cerebral edema. Less dramatic symptoms, including anorexia, nausea, and asthenia, are often difficult to ascribe to a single cause in patients with cancer.
The differential of hyponatremia is extensive and includes many conditions patients with cancer experience including inappropriate secretion of arginine vasopressin (AVP = antidiurectic hormone = ADH)22 and sodium depletion secondary to reduced intake and gastrointestinal or renal losses each accounting for about one-third of cases (Table 122.5).23 Other etiologies more likely to occur in patients with cancer than the general patient population include ectopic atrial natriuretic peptide (ANP) production, hyponatremia associated with third spacing of fluids (ascites), and “pseudohyponatremia” as might occur in a patient with multiple myeloma and hyperproteinemia—although one can argue the latter is in fact true hyponatremia with the reduced sodium an adaptation at electrical neutrality imposed by the increase in positively charged myeloma proteins.24

Pathogenesis
Only cell membrane impermeable solutes that are “relatively” restricted to either the extracellular or intracellular compartments can effectively function as solutes and create osmotic gradients across cell membranes. Sodium and its accompanying anions are the major cell membrane impermeable plasma solutes, so that hyponatremia is in effect a surrogate for hypoosmolality and these terms are usually synonymous. Under normal circumstances, plasma osmolality is closely regulated in the range of 280 to 295 mOsm/kg through (1) thirst perception, (2) control of free water clearance in the kidney by ADH also known as AVP, and (3) renal sodium excretion regulated by ANP and the renin angiotensin system. Understanding this helps one understand the causes of hyponatremia.
In patients with cancer, hyponatremia secondary to inappropriate secretion of AVP (or ADH; syndrome of inappropriate antidiuretic hormone [SIADH] or syndrome of inappropriate antidiuresis) occurs as a paraneoplastic syndrome, or as a complication of therapy.22 SIADH is defined as a hypo-osmolar or dilutional hyponatremia with excessive natriuresis. Hyponatremia occurs because AVP secretion continues even after plasma osmolality falls below the threshold for AVP release. Key and ancillary features are summarized in Table 122.6.22,23
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


