(1)
Medical Sciences Division Northern Ontario School of Medicine West Campus, Lakehead University, Thunder Bay, Ontario, Canada
Key Topics
Molecular pathology of melanoma
Circulating melanoma cell-free nucleic acid biomarkers
Circulating melanoma epigenetic biomarkers
Circulating melanoma genetic biomarkers
Circulating melanoma protein biomarkers
Circulating melanoma cells
Key Points
The molecular pathology of melanoma is well established to include promoter methylations, mutations, and loss of heterozygosity (LOH) of key genes (e.g., BRAF) that alter signaling pathways including the MAPK, PI3K, and cell cycle control. The ability to detect and measure these molecular changes in circulating tumor DNA provides real-time noninvasive means of patient management.
A number of proteins show differential circulating levels in melanoma patients. Although not specific to melanoma, their utility in disease management, such as prognostic and treatment predictions, is clinically informative.
Melanoma is a highly metastatic cancer that spreads mostly via lymphatics to sentinel lymph nodes (cutaneous) and through the blood stream (uveal). Advanced stage disease is associated with the shedding of cancer cells into the circulation, the detection of which offers prognostic and treatment prediction applications in disease management.
1.1 Introduction
Melanoma accounts for about 4 % of all skin cancers, but is the most aggressive form, being responsible for >70 % of deaths from cutaneous tumors. The global incidence is rising more rapidly than any other forms of cancer, with an estimated doubling in incidence every couple of decades. The 2012 global incidence, mortality, and 5-year prevalence rates are 232,130, 55,489, and 869,754, respectively. The 2016 estimated incidence and mortality for the US stand at 76,380 and 10,130, respectively. The high prevalence is partly attributed to increased patient survival due to early detection and improved management, especially in the more developed world where >80 % of all cases are diagnosed.
There are multiple predisposing risk factors of melanomagenesis. Sun exposure (UV skin damage), especially early in life, is an established risk factor. People with fair skin, blond or red hair, blue eyes, and who are unable to tan properly, or who sunburn or freckle easily have elevated risk above the general population. Other risk factors include the presence of precursor lesions such as atypical moles (dysplastic nevi) and congenital melanocytic nevus. About 10 % of people who develop melanoma have family history of the disease. Globally, the highest rates are among Australians and New Zealanders.
Early stage lesions are curable by surgery. Hence, early detection increases the cure rate, because stage I disease is associated with 90 % 5-year survival, but fatality rate increases with stage progression. The 5-year survival rate is 60 % for stage II, 10 % for stage III, and very dismal for stage IV. Late diagnosis, early distant organ invasion, and lack of efficacious therapeutic approaches for metastatic disease are some of the reasons responsible for the increased mortality from melanoma.
Clinical features of melanoma are quite distinct, making early detection relatively easy, if screening were established. While diagnosis may easily be established with skin examination, complemented by the use of modern diagnostic techniques such as whole-body photography, dermoscopy, and in vivo confocal microscopy, disease biology can be unpredictable, thus necessitating the need for evidence-based objective biomarkers for clinical management of melanoma. Advances made in the study of melanoma molecular genetics, coupled with current available targeted biotherapeutic agents, should improve the future outlook for patients with this disease. Biomarkers that can be assayed serially and noninvasively for assessing disease progression, prognosis, therapy selection, and monitoring, as well as for early detection of recurrences, should improve clinical management of melanoma.
Melanoma is a known solid tumor with the highest metastatic potential. Vertical-phase melanoma spreads and metastasizes via the circulation (lymphatic and blood) to sentinel and regional lymph nodes, as well as visceral organs including the lungs, liver, brain, bone, and the gastrointestinal tract. Because of the high propensity to spread, melanoma biomarkers are abundant in the circulation and are usually elevated at a much higher level in advanced than early stage disease. These biomarkers are measurable for clinical applications.
1.2 Screening Recommendations for Melanoma
Based on insufficient evidence of benefit vs. harm, the US preventive services task force does not recommend routine skin examination for the early detection of melanoma in the general adult population. However, skin examination is necessary as a surveillance for people with a family history of melanoma in two or more blood relatives, as well as individuals with multiple atypical moles (dysplastic nevi) or actinic keratosis. In this high-risk population, whole-body photography is periodically performed to screen for suspicious lesions.
1.3 Molecular Pathology of Melanoma
The majority (~90 %) of melanomas are cutaneous, with the remaining being uveal (ocular), mucosal, or leptomeningeal. Cutaneous melanomas originate from neural crest-derived melanocytes in the epidermis or very occasionally in the dermis. The molecular pathology of melanoma is fairly well characterized. Alterations in specific molecules that control oncogenic signaling pathways are implicated in this disease, and hence targeted therapies are at various stages of clinical trials. Established are alterations in the MAPK and PI3K pathways, as well as RB/TP53 and cell cycle control in melanoma progression. In fact, a number of targeted agents, specifically vemurafenib and dabrafenib (BRAF targets) and trametinib (MEK target), are clinically available. This session provides a synopsis of genes altered in melanoma that influence the designated signaling pathways.
1.3.1 BRAF Mutations in Melanoma
The BRAF proto-oncogene belongs to the RAF family of serine/threonine kinases. Other members are ARAF and CRAF (RAF1). All activate the MAPK pathway, but their phosphorylation targets and effects are different. BRAF is mutated in ~90 % of human cancers. It is commonly mutated in melanoma and papillary thyroid carcinoma (at ~69 % frequency). Somatic missense mutations that affect the kinase domain are found in ~66 % of malignant melanomas. Specifically, the T to A substitution in exon 11 at codon 600 (V600E) is the most frequent mutation occurring in over 80 % of melanomas. The V600E mutation confers over tenfold kinase activity than the wild-type allele. BRAF mutations are early events in melanoma development, being observed in ~82 % of benign nevi. However, BRAF mutations alone are insufficient to initiate tumor formation. Body fluid analysis for BRAF mutations as diagnostic, prognostic, and therapy monitoring for targeted therapy (e.g., with PLX4032 and GSK2118436) has been explored with promising clinical applications.
1.3.2 RAS Mutations in Melanoma
The RAS gene family includes KRAS, HRAS, and NRAS. KRAS is most commonly mutated in many cancers, but NRAS mutations are mostly associated with melanoma. Activating mutations, commonly at codons 12, 13, and 61, are found in 15–22 % of cutaneous melanomas. These mutations lead to constitutive activation of RAS. While not sufficient to initiate melanoma development, RAS mutations are important in maintaining the malignant phenotype, because loss of activated RAS causes tumor regression. NRAS and BRAF mutations are mutually exclusive in melanomas, possibly due to functional redundancy. In very rare occasions, the NRAS Q61R mutation is found with BRAF V600E in the same melanoma sample. Similarly, HRAS and KRAS mutations are found in ~1 % and ~2 % of melanomas, respectively.
1.3.3 Alterations in Cell Cycle Control Genes in Melanoma
Loss of cell cycle control is a common feature of almost all malignancies. The cell cycle has positive and negative regulators that function to restrain unwanted cell growth. The cyclin-dependent kinase (CDK) inhibitors (e.g., p16 and p21) are negative regulators, while CDKs drive cell cycle progression by interacting with specific cyclins. Cyclin D1/CDK4, cyclin D1/CDK6, and cyclin E/CDK2 complexes control the G1-S transition phase, where DNA synthesis occurs and the cell is committed to progress through the cycle. The RB protein is the gatekeeper of this transition phase. In non-proliferating cells, RB sequesters E2F, preventing its translocation into the nucleus and induction of genes needed for transition from G1 to S phase. This control is made possible because the cyclin/CDK complexes are inhibited when they bind to the 16 kDa inhibitor of CDK4A (p16INK4A, also known as cyclin-dependent kinase inhibitor 2A (CDKN2A) or major tumor suppressor). In proliferating cells such as the cancer cell, this control is abolished, and hence, cyclin/CDK complexes phosphorylate and inactivate RB leading to the release of E2F transcription factor to induce expression of genes that mediate cell cycle progression.
In familial atypical multiple mole melanoma where there is elevated risk of melanoma and pancreatic cancer, germline CDKN2A/INK4A mutations are found in as many as 40 % of cases. In sporadic melanomas, several alterations in cell cycle genes are also reported. CDKN2A/INK4A is silenced, probably through promoter hypermethylation in as many as 75 % of melanomas. CCND1 amplifications are observed, so are amplifications in CDK4 that are commonly found in acral and mucosal melanomas. CDK4 amplification is however dispensable (not observed) in melanomas with loss of both CDKN2A/INK4A alleles. Activating CDK4 mutation (R24C) interferes with p16 binding, but not cyclin D1, leading to constitutive cyclin D/CDK4 activity and cell cycle progression. CDK6 overexpression has also been reported in melanomas.
1.3.4 TP53 Mutations in Melanoma
As the guardian of the genome, TP53 tumor suppressor gene maintains genomic integrity by responding to cellular stress such as exposure of the skin to UV radiation. Being a transcription factor, p53 responds to cellular stress by initiating expression of a set of genes involved in DNA repair, cell cycle arrest, and apoptosis. There are multiple regulatory circuitries for TP53; however, a well-characterized regulator of p53 is MDM2. In cancerous cells, MDM2 binds to p53 and targets it for ubiquitin-mediated proteasomal degradation. Another safeguard for the cell is stabilization of p53 by p14/ARF, an alternate spliced translated product from the CDKN2A locus on chromosome 9p21. The p14/ARF complexes with MDM2 and hence frees p53. TP53 and CDKN2A/ARF mutations are rare in melanoma. Mutations in CDKN2A/ARF are found in ~2 % of melanomas.
1.3.5 KIT Mutations in Melanoma
KIT is a 109.865 kDa receptor tyrosine kinase encoded by KIT (CD117) located on chromosome 4q12. Following receptor ligand (stem cell factor of kit ligand) interaction, a number of signaling pathways are modulated. These include the MAPK, PI3K, and phospholipase C pathways. Additionally KIT targets the induction of genes such as MITF and Src. KIT is involved in melanocyte survival, proliferation, differentiation, and migration and melanin production. Mutations lead to lack of melanoblast migration and loss of KIT-negative melanoblasts. C-KIT is activated in many malignancies including melanoma. Work by Curtin and colleagues revealed that C-KIT mutations or increase in copy number occurs in 39 % of mucosal, 36 % of acral, and 20 % of chronically sun-damaged skin melanomas but not in non-chronically sun-damaged skin melanomas [1]. Mutations in KIT include K642E (common in gastrointestinal stromal tumors) and N566D. The genetic alterations in KIT are associated with increased expression and constitutive tyrosine kinase receptor activation.
1.3.6 MITF Mutations in Melanoma
MITF is a basic helix-loop-helix leucine zipper transcription factor on chromosome 3p14.2-p14.1 that belongs to the MiT family of transcription factors. The members form homodimers and heterodimers. MITF-M isoform regulates melanomagenesis because of the presence of a melanoma-restricted promoter in this isoform. MITF-M usually induces the transcription of genes including tyrosinase (TYR), tyrosinase-related protein (TYRP1), and dopachrome tautomerase (DCT) required for melanin production. Signaling pathways that target MITF include the C-KIT/MAPK, WNT/β-catenin, and α-MSH/cAMP and gp130 pathways. MITF amplifications (up to 100-fold in some cases) and overexpression are found in ~10 % of primary and ~20 % of metastatic melanomas. Increased transformation and anchorage independence and poor survival are features of MITF overexpression. MITF G1075A (E318K) mutation causes impaired sumoylation and aberrant regulation of MITF targets leading to increased cellular proliferation, migration, and invasion. This mutation is found in both familial and sporadic melanomas [2].
1.3.7 PI3K Pathway Alterations in Melanoma
The PI3K pathway that controls important cellular processes such as survival, proliferation, invasion, and glucose metabolism is deregulated in several tumors. The AKT3 isoform is overexpressed in melanoma, and phospho-AKT is detected in 54 % of nevi and at a frequency of 71 % in primary and metastatic melanomas [3]. Loss of chromosome 10 (PTEN locus) occurs in 30–60 % of sporadic melanomas [4]. Loss of PTEN expression is observed in 30–50 % of melanoma cell lines and in 5–20 % of primary melanomas [5]. PTEN somatic mutations occur in association with BRAF mutations (but not NRAS). PTEN epigenetic silencing is also demonstrated in melanomas [6].
1.3.8 Molecular Subtypes of Melanoma
Using comparative genomic hybridization (CGH), DNA sequencing, and immunohistochemistry (IHC), Curtin et al. provided a genetic classification of melanoma [7]. Based on skin sites involved and UV exposure, four groups of cutaneous melanoma with defined genetic alterations have been identified. These groups are melanomas associated with chronic sun-induced damage (CSD), without CSD (non-CSD), as well as mucosal and acral melanomas. Chronic sun-induced damage melanomas rarely harbor BRAF and NRAS mutations. The predominant findings in these tumors are CCND1 copy number gains. Non-CSD melanomas are associated with BRAF (59 %) and NRAS (22 %) mutations. These tumors also harbor chromosome 10 loss (PTEN locus) and amplifications in CDK4 and CCND1. Landi et al. provided evidence that MCIR variant alleles confer melanoma risk in non-CSD skin, and these melanomas have BRAF mutations [8]. Deletions in CDKN2A characterize mucosal melanomas, while acral melanomas have increased incidence of CDK4 amplification, but melanoma cells with homozygous CDKN2A deletions lack CDK4 amplification.
1.3.9 Molecular Progression Model of Melanoma
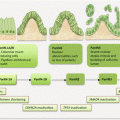
Melanoma develops in a very defined progressive model proposed by Clark and coworkers in 1984 (Fig. 1.1) [9]. The identified five defined steps include acquired or congenital nevi with normal melanocytes, dysplastic nevi with atypical melanocytes, radial growth phase primary melanoma without metastatic activity, vertical growth phase primary melanoma with metastatic competence, and finally metastatic melanoma. However, this model accounts for ~35 % of melanomas, indicating the presence of alternative pathways of melanoma development. Various epigenetic and genetic alterations drive melanoma progression. The classic Clark pathway involves early alterations in BRAF and NRAS as observed in nevi, followed by CDKN2A and PTEN mutations that drive the development of melanoma but in the radial growth phase. The subsequent evolution into vertical growth phase melanoma with invasive propensity requires additional alterations in CDK2, CCND1, and PI3K pathway, among several others. Finally, the loss of normal homeostatic interactions between melanoma cells and the extracellular stromal matrix, keratinocytes, endothelial cells, fibroblasts, and immune cells propel these cells into invasive state. Thus, loss of cell-cell and cell-microenvironmental communications mediate progression. Cellular adhesions play an important role in this process. Alterations in the major groups of adhesion molecule receptors, cadherins, integrins, and cellular adhesion molecules of the immunoglobulin superfamily mediate metastasis.
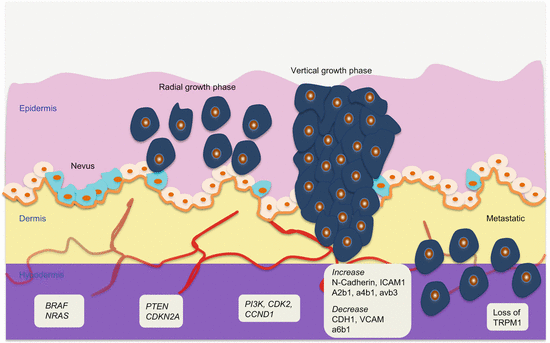
Fig. 1.1
Molecular pathology of multistep melanoma progression model
The molecular pathways of uveal melanoma are not well understood. However, emerging work indicates the involvement of GNAQ, a gene that encodes the q class of G protein α-subunit, involved in signaling between G protein-coupled receptors and their downstream components. Mutations in GNAQ, especially at codon 209, are common (~46 %) in uveal melanomas, while activating BRAF and inactivating CDKN2A mutations are mostly absent. Understandably, these GNAQ activating mutations are capable of engaging the MAPK pathway (without the need for BRAF and NRAS mutations) [10].
1.4 Circulating Melanoma Biomarkers
Noninvasive biomarkers for the management of melanoma patients will greatly improve patient outcomes. Thus, the genetic alterations in melanoma tissues are being actively pursued in circulation. Specifically, ctDNA and the associated epigenetic and genetic alterations, as well as altered protein levels have shown promise. Attention has also been given to the clinical importance of circulating melanoma cells.
1.4.1 Circulating Melanoma Cell-Free Nucleic Acid Biomarkers
Studies on circulating cell-free DNA (ccfDNA) in melanoma have primarily focused on qualitative tumor-specific alterations such as analysis of methylation and mutations in genes including RASSF1A and BRAF. Work by Orlando’s group on ccfDNA in melanoma patients had initially indicated that DNA integrity index (DII), defined as the ratio of 180 bp to 67 bp PCR fragments, was a reliable discriminatory biomarker for identifying patients with melanoma [11]. The explanation for this finding is that melanoma and other cancer patients have mostly larger fragmented DNA of sizes 181–307 bp compared to healthy control individuals that harbor mostly shorter apoptotic fragments (67–180 bp). In a follow-up study, this group included measurement of total ccfDNA concentration, and detection of BRAF V600E mutation and RASSF1A promoter methylation in the analyses. Whereas ccfDNA concentration may not be accurate enough for the detection of many cancers, this study found that the best performance in detection of melanoma was total ccfDNA concentration with area under the receiver operating characteristic curve (AUROCC) of 0.85, followed by DNA integrity index (AUROCC of 0.79), with the cancer-specific biomarkers performing poorly (RASSF1A; AUROCC of 0.69, and BRAF V600E ; AUROCC of 0.64). The explanation could be that not all tumors will express a specific genetic marker (e.g., not all melanomas harbor BRAF mutations). The combination of the three biomarkers (DNA concentration, mutations, and methylation), however, increased the accuracy of melanoma detection (AUROCC of 0.95) [12].
1.4.2 Circulating Melanoma Epigenetic Biomarkers
In melanoma, a CpG island methylator phenotype (CIMP) has been described. There are over 50 genes with promoter hypermethylation in melanoma. An epigenetic control of DNA methylation is exemplified by the regulatory functions of miR-29c. miR-29c controls the expression DNMT3A and DNMT3B, which are upregulated in melanoma progression. This may have effects on the observed hypermethylation of multiple genes, including WIF1, RASSF1A, TFP12, and SOCS1 in melanoma progression. The tumor suppressor RASSF1A, for example, is methylated in ~57 % of melanomas. This gene silencing, and loss of tumor suppressor function, strongly correlates with melanoma progression and prognosis. Similarly, methylations of methylated-in-tumor 17 and methylated-in-tumor 31 (MINT17 and MINT31) are associated with specific tumor gene methylation patterns. Another miRNA, miR-532-5, which is upregulated in metastatic melanoma, controls RUNX3 expression, and RUNX3 promoter methylation occurs in a subset of primary and metastatic melanomas.
Genes frequently methylated in melanoma have been examined in serum as biomarkers of disease progression, prognosis, and/or therapy response. Work primarily from Hoon’s laboratory has associated methylation of RASSF1A, RARβ2, MGMT, and ERα with melanoma prognosis and response to biochemotherapy. Gene promoter methylation in sera as predictive biomarkers in patients on concurrent biochemotherapy has been assessed. Methylation of RASSF1A, RARβ2, and MGMT was assayed in pretreatment sera and related to response to treatment. Responders had significantly less methylated RASSF1A (13 %) than nonresponders (42 %). The presence of at least one methylated gene conferred worse survival outcome in patients than those without methylated genes. RASSF1A methylation was the only gene alteration significantly associated with overall survival and biochemotherapy response [13]. In another study, promoter hypermethylation of ERα appeared to be a biomarker of melanoma progression, being more frequent in advanced metastatic disease than in localized melanoma. The clinical relevance of serum ERα methylation in patients on biochemotherapy and tamoxifen was also explored. Consistent with tissue analysis, methylation in serum was associated with advanced stage disease and was the only predictive factor of progression-free survival (PFS) and overall survival (OS) in patients on biochemotherapy [14]. Koyanagi et al. conducted a study on the predictive value of methylation markers and CTCs in peripheral blood from melanoma patients [15]. CTCs were targeted by amplification of MART-1, GalNACT, and MAGEA3 transcripts. CTCs were detected in 86 % of patients with methylated RASSSF1A and RARβ2 DNA compared to 37 % of those without gene methylation. The CTC markers significantly correlated with methylation, and both biomarkers correlated with biochemotherapy outcome. The presence of both methylated DNA and CTCs significantly predicted worse response to biochemotherapy treatment and shorter time to PFS and OS. Methylation status of LINE and absent in melanoma 1 (AIM1) were assessed in tissue and sera from melanoma patients. LINE hypomethylation was observed more frequently in sera from patients with primary melanoma, while AIM1 promoter hypermethylation was associated with metastatic disease. In multivariate analysis, methylation of AIM1 was a significant predictor of OS. Consistent with tissue alterations, in serum analysis, LINE hypomethylation was more frequent in stage I/II disease than in healthy controls, and AIM1 promoter hypermethylation was a predictor of OS in stage IV patients [16].
Methylation of selected genes has shown potential for melanoma detection. Methylation of five genes (SOCS1, SOCS2, RASSF1A, CDKN, and MGMT) was assayed in sera from patients with melanoma, nevi, other skin tumors, metastatic non-cutaneous cancers (breast and colon cancers), chronic inflammatory diseases, and healthy controls. Methylation of all these genes was detected in melanoma patient samples, with frequencies ranging from 43 to 75 %. Eighty three percent (83 %) of the samples had methylation in at least one gene, and as many as 20 % harbored methylation of all five genes, and these methylation profiles were distinct from those in the other tumors examined. In primary melanoma tissue samples, SOCS2, CDKN, and RASSF1A levels were downregulated, while MGMT was upregulated 12-fold [17].
1.4.3 Circulating Melanoma Genetic Biomarkers
The mutations in genes as well as microsatellite alterations with disease progression are detectable and measurable in circulating fluids from patients.
1.4.3.1 Mutations as Circulating Melanoma Biomarkers
Specific gene mutations in melanoma have been assayed in circulating blood as noninvasive biomarkers for disease management. Expectedly, BRAF mutations dominate the various studies, which primarily have come from David Hoon’s group. One study by this group indicates that BRAF mutations in serum can be used to predict OS (i.e., mutation is associated with poor outcome) and response to biochemotherapy (responders are often mutation negative). In this initial study, 37 % and 39 % of stage I/II and III/IV disease patients, respectively, were positive for BRAF mutations in serum samples, and this was significantly associated with worse OS. Before treatment, BRAF mutations were detectable at a frequency of 42 % each in responders and nonresponders. However, following biochemotherapy, the detection rates of BRAF mutation were 10 % and 70 % in responders and nonresponders [18]. In another study, the detection rate of BRAF V600E mutation in plasma/serum was obtained in 80 % of patient but not in any control samples [19]. The positive samples were mostly from patients with advanced stage (IV) disease, suggestive of the usefulness of circulating BRAF V600E mutation detection in monitoring advanced stage disease patients.
BRAF mutation analysis in serum DNA may be useful for patient selection for therapy as well. The clinical relevance of BRAF mutation analysis in ccfDNA was examined in advanced stage melanoma patients in phase II clinical trial with MEK1/2 inhibitor [20]. BRAF mutations were detected in 47.9 % and 26.2 % of tissue and serum samples, respectively. However, the frequency of detection was 55.6 % (higher) in those with tissue mutations. The potential noninvasive use to select patients for treatment is evident from this study [20]. In BRAF V600E/K mutation-positive stage IV patients in phase II trial with BRAF inhibitors, dabrafenib, clinical outcome prediction was evaluated by comparing BRAF mutation status in ctDNA and tissue samples, as well as ccfDNA levels. Baseline ccfDNA levels predicted PFS in patients with BRAF V600E mutations in this trial. This finding shows potential for monitoring treatment response [21]. A sensitive detection method (able to detect 3:100 mutant alleles) was used to quantify BRAF V600E mutation in plasma from patients with melanoma and non-melanoma skin cancer. The performance of this assay using absolute concentrations of BRAF mutations was diagnostic at a sensitivity of 97 % and a specificity of 83 %. BRAF mutations in plasma were concordant at 80 % with tissue mutations [22]. This assay should augment the translational potential of BRAF mutation detection in circulation.
Mutation analyses in circulation have also been performed in patients with uveal melanoma. Circulating tumor cells and gene mutations in ccfDNA were assessed in samples from patients with metastatic uveal melanoma. GNAQ c.626 A > T, c.626 A > C, and GNA11 c.626 A > T copy numbers were quantified in plasma, and CTCs were assayed using CELLSEARCH® technology. CTCs were detected at a rate of 30 % compared to ccfDNA using gene mutations, which were found in 84 % of cases. Circulating tumor DNA concentration and CTCs were associated with hepatic spread, metastatic volume, as well as PFS and OS. In multivariate analysis, ctDNA was superior to CTCs as a prognostic biomarker [23]. Activating mutations in exon 4 (R183) and exon 5 (Q209) of GNAQ and GNA11 are almost exclusive to uveal melanoma. As a proof of principle, ultra-deep sequencing was used to detect ctDNA targeting these mutations. The Q209 mutation (present in 2–38 % mutant reads) was detected in either GNAQ or GNA11 in 40.9 % of plasma from patients with metastasis. The potential for early detection of micrometastasis needs pursuance [24].
1.4.3.2 Circulating MSA as Melanoma Biomarkers
In addition to BRAF mutations, microsatellite alterations (MSA) are detectable in circulation of melanoma patients. Once again, the Hoon’s group questioned whether tumor-specific genetic changes could be detected in plasma from melanoma patients, and whether this had any clinical relevance. First, ten microsatellite markers on six chromosomes commonly harboring LOH in melanoma were used to assess alterations in tissue and matched plasma samples from patients with early and advanced stage melanoma. Forty matched tissue and plasma samples were significantly concordant for LOH, which tended to be more frequent in advanced stage disease. Clinical disease progression was significantly associated with plasma LOH at D3S1293, as well as the combination of LOH at D9S157/D3S1293, D9S157/D1S228, and D11S925/D3S1293 [25]. A follow-up study assessed the prognostic value of MSA in preoperative blood samples from melanoma patients. LOH on the six chromosomes was used in this study of patients undergoing surgical resection of melanoma. LOH was present in 56 % of patients and was associated with advanced stage disease. LOH in plasma samples was an independent preoperative indicator of increased risk of death. The microsatellite alterations at D1S228 significantly correlated with poor survival after surgery [26]. Microsatellite analysis as a measure of allelic instability as well as mutations in specific genes has been explored in the circulation as prognostic biomarkers of melanoma. Nine microsatellite markers were used for analyses of sera from stage IV melanoma patients. The findings indicated that allelic instability at these sites was associated with a much lower response to therapy and independently predicted disease progression [27]. Because allelic instability at the APAF-1 locus (12q22–23) is more frequently found in metastatic melanoma, serum as a noninvasive sample was assayed for possible use in prognostic and therapy response monitoring. Instability at this locus is significantly higher in patients who do not respond well to treatment and is also associated with poor survival [28]. Takagi et al. targeted detection of recurrence of mucosal melanoma after radiotherapy by analyzing LOH in plasma samples [29]. Pretreatment samples were interrogated using 4 (D1S243, D6S311, D9S161, D19S246) out of 20 known markers of mucosal melanoma. All markers demonstrated LOH, but the highest frequency was at D9S161 (65 % of plasma samples). LOH in plasma DNA at one or more loci was associated with recurrence and metastasis, and LOH at two loci tended to relate to larger mean tumor volume. Tagagi’s group performed a follow-up study using the same markers. Using triplicated whole genome amplification and triplicated PCR, alterations were detected on at least one locus in 70.6 % of samples, and this was associated with metastasis and recurrence. Importantly, LOH in plasma using these markers (D1S243, D6S311, D9S161, and D19S246) strongly correlated with melanoma, and hence could serve as noninvasive screening biomarkers of mucosal melanoma [30].
1.4.4 Circulating Melanoma Coding RNA Biomarkers
Altered gene transcripts, primarily tyrosinase (TYR) mRNA, have been targeted in circulation of melanoma patients. As a proof of principle that extracellular mRNA integrity is stable and can be amplified, serum from six patients with malignant melanoma and 20 controls were subjected to tyrosinase mRNA analysis. Four cancer patient samples and none of the 20 control samples were positive for this marker. Even sera frozen for several years could still be assayed for mRNA detection [31]. CTCs are commonly detected in melanoma patients by targeting tyrosinase mRNA. Because CTCs are rare, and that melanoma cells are transient in circulation, molecular CTC assay data have been inconsistent depending on whether isolated cells or whole blood is used for analysis. To address this problem, Hasselmann et al. targeted melanoma-specific mRNA in ccfRNA in serum/plasma samples in comparison to data using blood cell samples from stage IV melanoma patients [32]. In the small samples analyzed, tyrosinase mRNA was detected in 60 % of serum ccfRNA samples compared to 100 % of the blood cell samples [32]. Thus, ccfRNA only samples released TYR mRNA from other sources, with possible exclusion of CTCs.
1.4.5 Circulating Melanoma Noncoding RNA Biomarkers
MicroRNA deregulation was initially reported in three primary melanoma samples and two melanoma cell lines included in the analysis by Lu et al. [33]. Zhang et al. included 45 primary cultured melanoma cells in their analysis of miRNA copy number variations [34]. In this series, 85.9 % of the genomic loci harboring the 283 miRNAs examined had copy number abnormalities in melanoma samples. In the NCI-60 panel of cell lines including melanoma cell lines, 15 miRNAs were significantly differentially expressed between melanoma and normal, as well as other non-cutaneous cancer cell lines [35]. However, the first well-designed study of miRNA alterations in melanoma was by Mueller et al. [36]. They profiled the miRNAome of melanoma, normal human melanocytes, and melanoma cell lines from primary and metastatic melanoma. MicroRNA deregulation could be associated with disease progression from early to metastatic stages. The data was also validated by RT-PCR and in primary melanoma tissue samples.
The targets and mode of regulation of some deregulated miRNA in melanoma are provided in some studies. Several relevant tumor-associated genes (NRAS, MITF, C-KIT, TFAP2, CDKN1B, RUNX3, ITGB3, CCND1, MET, MNT, PLZF, and FOXO3) are controlled by miRNAs in melanoma progression. Similarly, some tumor-associated genes act by targeting and regulating miRNA in melanoma. For example, MITF regulates a number of miRNAs including let-7 family, miR-29, miR-125b, miR-146a, miR-148b, as well as miR-221/222, miR-17-92, and miR-106-363 clusters. RAS oncogene is a target of let-7 in many cancers. In melanoma, let-7b directly and indirectly targets genes involved in cell cycle control [37]. Let-7b interacts with the 3′ UTR of CCND1, interfering with cell cycle progression. Consistently, some members of let-7, including let-7a, are significantly downregulated in melanoma. Let-7a controls later stages of melanoma progression by regulating integrin β3 expression through interaction with its 3′ UTR. This repression is lost with increasing integrin expression associated with melanoma invasion and metastasis. Additionally, let-7a controls RAS oncogene expression in melanoma [38].
Using database search for miRNAs located on chromosome 1p22 that is characterized by melanoma susceptibility genes, Bemis et al. identified miR-137 to be of importance in melanoma [39]. Further computational work showed that MITF was a target of this miRNA and that it directly binds and suppresses MITF expression. Amplification of variable number tandem repeat (VNTR) located on the 5′ UTR of miR-137 primary transcript interferes with the processing of miR-137. VNTR amplification is observed in some MITF-expressing melanoma cells. Another miRNA that controls MITF is miR-182 [40]. MicroR-182 is overexpressed in melanoma and is associated with invasive and metastatic propensities of melanoma cells through suppression of MITF and FOXO3.
MicroRNA-221 and miR-222 are transcribed as a bicistron precursor from the X-chromosome. They are overexpressed in a variety of cancers and target genes including CDKN1B and C-KIT receptor. In melanoma, C-KIT downregulation is associated with disease progression, and p27Kip1/CDKN1B suppression causes increased proliferation via cell cycle deregulation. In normal melanocytes, miR-221/miR-222 cluster is repressed by promonocytic leukemia zinc finger (PLZF) that is a transcriptional repressor of the miR-221/miR-222 promoter. Thus, PLZF is lost in melanomas overexpressing these miRNAs [41, 42]. It is even suggested that C-KIT may be regulated primarily by miRNAs [43]. Promoter methylation and decreased expression of miR-34a are observed in two thirds of melanomas and are a tumor suppressor in uveal melanoma [44]. MicroRNA-34b, miR-34c, and miR-199a* downregulate MET oncogene in melanocytes leading to impaired MET-mediated motility. A number of miRNA biomarkers with emerging functional importance in melanomagenesis are found in Tables 1.1 and 1.2.
Table 1.1
Some important melanoma oncomirs and their targets
Oncomirs | Targets |
|---|---|
let-7 | CDK4, 6 |
MiR-15b | BCL2 |
MiR-17-92 cluster | MYC |
MiR-21 | PTEN, STAT3, PDCD4, TIMP3 |
MiR-27a | PSMA1 |
MiR-30b, d | GALNT7 |
MiR-33a | PIM1, CDK6, CCND1 |
MiR-100 | PLK1 |
MiR-137 | MITF, EZH2, MET, YB1 |
MiR-145 | TP53, MYC |
MiR-149 | GSK3α |
MiR-150 | NOTCH3, EGFR2 |
MiR-182 | MITF, FOXO3 |
MiR-195 | WEE1 |
MiR-199a-3p | APOE, DNAJA4 |
MiR-199a-5p | SWI/SNF, APOE, DNAJA4 |
MiR-210 | HIF1α, ATM, FAS, TNFR1 |
MiR-214 | ITGA3, MET, TFAP2C |
MiR-221/222 | P27, CCND1 |
MiR-340 | RAS, RAF, MAPK |
MiR-424 | HIF1α, HIF2α |
MiR-506-514 | HOXB7, PBX |
MiR-532-5p | RUNX3 |
MiR-1908 | APOE, DNAJA4 |
Table 1.2
Some important melanoma tumor suppressormirs and their targets
Tumor suppressormirs | Targets |
|---|---|
Let-7a | NRAS |
MiR-9 | NK-jB, SNAI1 |
MiR-29 | DNMTS |
MiR-31 | DNMTS |
MiR-126 | ADAM9, MMP7 |
MiR-155 | SKI |
MiR-193b | CCND1 |
MiR-148 | MITF |
MiR-203 | E2F3 |
MiR-205 | E2F1, E2F5 |
MiR-211 | MITF, AP1S2, SOX11, KFBP5 |
MiR-455 | PAX6, NEDD9 |
MiR-573 | NCAM |
The clinical relevance of circulating miRNA in melanoma patients has been addressed. Serum levels of miR-221 are significantly elevated in patients with malignant melanoma compared to healthy controls. Levels were much higher in stages I–IV disease patients than those with in situ melanoma and correlated with tumor thickness. The levels decreased following surgical tumor removal and returned to elevated levels following recurrence [45]. Shiiyama et al. evaluated the utility of circulating miRNA in detection of metastatic melanoma [46]. Six miRNAs (miR-9, miR-145, miR-150, miR-155, miR-203, and miR-205) were examined in patients with and without metastatic disease. A panel of all the miRNAs except miR-203 was sensitive enough to discriminate patients with metastasis from those without. Another study focused on circulating miRNA in sera from healthy controls and those with metastatic melanoma. Loss of normal serum miR-29c and miR-324-3p was predictive of metastasis, and this was even accurate at differentiating melanoma metastasis from colon and renal cancer metastasis, indicating a high level of specificity for melanoma spread [47]. It will be informative to know whether the other remaining altered miRNAs in melanoma tissue samples are measurable in circulation, as well as their clinical relevance.
1.4.6 Circulating Melanoma Protein Biomarkers
Melanoma serum biomarkers have been explored for multiple applications in patient management. Among them, they are used to complement clinical staging (e.g., serum lactate dehydrogenase—LDH), for prognostication (survival predictions of newly diagnosed patients), for monitoring response to various therapeutic interventions, for monitoring stage progression, and for predicting lymph node status. Detailed in this section are established serum biomarkers and the emerging circulating melanoma proteome and peptidome.
1.4.6.1 Serum S100β as Melanoma Biomarker
The S100 family of acidic calcium-binding proteins comprises 21 members encoded by different genes. They are expressed by diverse cell types and have multiple intracellular and extracellular functions. They control intracellular signal transduction processes through inhibition of protein phosphorylation. They also control cellular morphologic and structural changes through modulation of cytoskeletal dynamics, and are involved in cell-cell communication, cell growth, and energy metabolism. Their extracellular functions include macrophage activation, leukocyte chemoattraction, and control of cell proliferation, apoptosis, and p53 functions.
The expression of S100β was initially identified by Gaynor et al. in cultured human melanoma cells, and later confirmed by Nakajima et al. using immunohistochemistry as being present in human malignant melanoma and pigmented nevus [48, 49]. Fagnart et al. were first to report the elevated levels of S100β in sera from cancer patients, among whom 81.8 % were patients with metastatic melanoma [50]. Subsequently, Guo et al. revealed the clinical value of serum S100β measurements in malignant melanoma as it mirrored clinical stages [51]. Using a cutoff value of 0.15 ug/L, sensitivity was 1.3 % for early stage I/II disease, 8.7 % for stage III, but as high as 73.9 % for stage IV disease. In advanced stage disease, a rise or decline in serum S100β was predictive of disease progression or decline, respectively. Since these initial findings, the clinical relevance of serum S100β levels as a biomarker for clinical staging, prognostic, and possible predictive potential for malignant melanoma has been confirmed by several other studies. However, the levels of S100β are elevated in other neoplastic diseases, as well as in liver and renal injury, inflammatory conditions, infections, and also liver metastasis from any primary, which compromises diagnostic specificity.
Prognostic Utility of Serum S100β in Melanoma
The survival rate of patients with malignant melanoma strongly correlates with S100β serum levels. The observed-to-expected death ratio was significantly increased with elevated serum S100β. At levels >0.6 μg/L, a fivefold increase in relative hazard was observed, and prognostication was independent of clinical stage [52]. In other studies, increasing levels of S100β were associated with clinical stage, disease progression, and lack of response to therapy [53, 54].
Abraha et al. measured serum levels in patients and controls [55]. Median concentrations were 0.11 μg/L, 0.24 μg/L, and 0.39 μg/L in patients with stage I/II, III, and IV, respectively, and these levels were significantly higher than in control individuals (0.1 μg/L). Sensitivity and specificity at a cutoff value of 0.2 μg/L were 82 % and 91 %, respectively, for detection of advanced stage disease. The levels also correlated with Breslow tumor depth. The use of serum S100β levels at a cutoff value of 0.22 μg/L and Breslow thickness >4 mm increased the sensitivity and specificity to 91 % and 95 %, respectively, for detection of secondary spread. Indeed, a large study conducted by Martenson et al. demonstrated the prognostic utility of circulating S100β [56]. S100β was measured by luminescent immunoassay (LIA) method in a large cohort of >1000 patients with stage I–III cutaneous melanoma. Clinical disease stage was significantly related to serum levels, with the lowest levels found in stage I and the highest in stage III disease. In multivariate analysis, serum S100β was the strongest predictor of disease-specific survival in stages II and III disease but not in stage I [56]. A meta-analytical review of 22 studies involving 3393 patients was performed to assess the prognostic value of serum S100β in patients with malignant melanoma. Serum levels were significantly associated with poor survival, with a hazard ratio (HR) of 2.23 (p < 0.0001). The studies were homogenous in stage I/II disease (1594 patients). In this subgroup, S100β levels still remained a strong prognostic factor (HR 2.28, p < 0.0001). Studies using multivariate analysis confirmed serum S100β has prognostic value independent of TNM staging system [57].
As initially reported by Abraha et al. [55], others have confirmed the fact that serum levels increase with disease progression. The mean S100β concentration was determined to be 0.075 μg/L in stage I–III compared to 0.441 μg/L in stage IV disease patients. Median survival was 256 days for patients with levels >0.150 μg/L compared with 561 days for those with normal values [58]. A multiple biomarker (S100β, LDH, MIA, YKL40) study of advanced melanoma (stages IIIB/C and stage IV) patients revealed that S100β, MIA, and LDH levels were significantly much higher in cancer patients than disease-free controls. S100β and MIA had the best diagnostic sensitivity. MIA was a prognostic factor for OS. However, patients with simultaneous increase in serum S100β and MIA had significantly shorter survival period than those with lower levels [59]. Preoperative S100β levels predicted nodal tumor load, and increased levels were associated with shorter disease-free survival (DFS) in stage III melanoma patients [60]. A retrospective prognostic study of the value of S100β and LDH in patients with metastatic melanoma found in multivariate analysis that S100β (but not LDH) and brain metastasis were independent predictors of OS [61]. S100β is much superior to LDH in prognostication of stage IIIB/C malignant melanoma. Preoperative and postoperative days 1 and 2 S100β levels were all associated with DFS. In multivariate analysis, preoperative day 2 levels were the strongest predictor of DFS (HR 2.55). Preoperative S100β levels are strongest independent predictor of disease-specific survival (HR 2.81). LDH was not prognostic in these series [62].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


