FIGURE 2.1 G1 pathways that can trigger cell cycle arrest, senescence, or apoptosis. A variety of threats to genomic integrity lead to activation of pathways that result in cell cycle arrest. Signaling via ATM/CHK2 and ATR/CHK1 leads to p53 activation, among other effects. One of the principle downstream effects of p53 is activation of p21 expression, with resultant cyclin E(A)/CDK2 inhibition and cell cycle arrest. Senescence, which results in a more sustained cell cycle exit, also involves the up-regulation of p14ARF and p16INK4A. Both proteins ultimately lead to retinoblastoma protein (RB) activation and G1 arrest via cyclin/CDK inhibition. In response to DNA damage during S phase, activation of PP2A can lead to dephosphorylation of RB and inhibition of DNA synthesis.
Oncogene-induced senescence has been shown to occur in vivo, limiting tumor progression in models of lung and prostate cancer, melanoma, and lymphoma.33 Whereas inactivation of the PTEN tumor suppressor and resultant activation of the AKT signaling pathway in prostate epithelial cells appears to promote senescence through p14ARF, inappropriate activation of RAS signaling in other tissues results in p16INK4A-mediated senescence. Nonetheless, targeted activation of oncogenic K–RAS alleles in somatic tissues in mice predisposes to a wide variety of tumor types, including early-onset lung cancer, suggesting that this barrier may be readily overcome or that the consequence of RAS expression may vary depending on the context.38,39 Moreover, expression of endogenous levels of oncogenic K-RAS can promote proliferation, and it has been demonstrated that oncogene-induced senescence due to RAS activation can be dose-dependent.40–42
RAS-induced senescence in lymphocytes depends on heterochromatin formation via the Suv39h1 histone methyltransferase, the disruption of which facilitates lymphoma development in response to RAS activation.33 Furthermore, disruption of Suv39h1 by itself has been shown to disrupt heterochromatin formation and to promote genetic instability, contributing to lymphomagenesis.43 This may occur at least in part through cell division failure and the generation of unstable tetraploid cells (see later discussion). These findings confirm the in vitro observations that changes in chromatin structure contribute to cell cycle exit by senescence. In addition, they suggest that emerging epigenetic therapies such as histone deacetylase inhibitors and DNA methyltransferase inhibitors that interfere with chromatin silencing may disrupt this senescence barrier, a potential caveat to their use.
MUTATIONS IN CANCER
Despite multiple levels of protection against the development of genomic instability, with age, cells can develop genetic alterations that escape detection.4 One path to genetic instability is inactivation of checkpoint proteins such as those previously described. The subsequent deregulation of the cell cycle and impairment of the response to genomic injury allows the progressive accumulation of lesions that can drive oncogenesis. Additionally, mutations can also occur in genes encoding the proteins that repair DNA damage and protect against the development of chromosome abnormalities. In this setting, accelerated mutation rates and chromosomal instability destabilize the genome and facilitate progression through the steps of oncogenic transformation. The basic types of genetic alterations observed in tumors, and the mechanisms by which genome destabilization can occur, are outlined in the next sections. Cancer predisposition syndromes that result from inherited defects in genome maintenance are highlighted in Table 2.1.
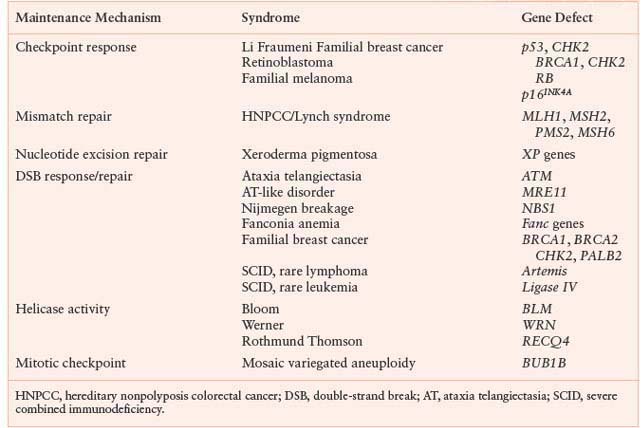
TABLE 2.1
INHERITED GENOME MAINTENANCE DEFECTS WITH CANCER PREDISPOSITION
Point Mutations
Changes in the nucleotide sequence can arise from spontaneous mutation, exposure to endogenous or exogenous mutagens, or defects in the ability to detect and/or repair simple sequence errors.4,21 The spontaneous mutation rate per nucleotide per cell division has been estimated to be on the order of 10−9 in somatic cells and 10−11 in stem cells.44 Despite the remarkable fidelity of DNA polymerase and its inherent proofreading capacity, a variety of endogenous and exogenous chemical and radiation exposures can introduce additional DNA lesions, requiring the presence of multiple repair pathways for further protection of genomic integrity. Mutations arise when such errors are not detected and repaired by this machinery, which can occur when repair pathways are overwhelmed or defective. As a result, point mutations are frequently detected via sequencing of both oncogenes and tumor suppressor genes in multiple cancers. Notable examples include activating mutations in oncogenic kinases such as K-RAS in colorectal, pancreatic, and lung cancer, B-RAF in melanoma, and JAK2 in myeloproliferative disorders. In some instances, specific mutations have been linked to epidemiologic features such as tobacco exposure, with oncogenic K-RAS mutations in non–small cell lung cancer (NSCLC) occurring more frequently in smokers and epidermal growth factor receptor (EGFR) mutations in nonsmokers.45
Next-generation sequencing technology has ushered in a new era in cancer genomics.46,47 Massively parallel DNA sequencing platforms now allow for the routine sequencing of billions of bases of DNA per week and the identification of point mutations, insertions and deletions, copy number changes, and genomic rearrangements on a genome-wide basis. Large-scale sequencing of coding sequences from a panel of colorectal and breast tumors, for example, revealed that these cancers harbor approximately 100 mutant genes, with computational methods predicting that 14 to 20 of these mutations will be bona fide tumor suppressor genes or oncogenes.48 In this unbiased effort, both known and unknown mutations were identified, with each tumor possessing a relatively unique cancer gene mutational signature. In another study, sequencing of coding regions of protein kinases in a large number of cancers identified “driver” mutations in approximately 120 genes across all samples.49 Although these studies identified a greater number of mutational events associated with oncogenesis than previously thought (the “state” of genome integrity), they do not necessarily imply a high “rate” of mutation, which remains low in most mature tumors.21
In two early studies, targeted gene resequencing was also applied to lung adenocarcinoma and glioblastoma multiforme (GBM) tumor samples.50,51 Both studies integrated the somatic mutation data with other genome-wide characterizations and clinical data. In the lung adenocarcinoma samples, for example, the sequencing data were used to identify multiple pathways, including MAPK signaling, p53 signaling, and the mTOR pathway, that are targeted by a combination of point mutations, copy number amplifications and deletions, and loss of heterozygosity (LOH). In a different study of GBM tumor samples, the sequencing of approximately 20,000 protein coding genes led to the discovery of a variety of genes that were not known to be altered in GBM, including the enzyme isocitrate dehydrogenase (IDH1).52 The cancer-associated IDH1 mutations result in the novel ability of the enzyme to catalyze the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG), which has been shown to lead to an elevated risk of malignant brain tumors.53 Subsequently, mutations in IDH1 have also been identified in acute myeloid leukemia (AML) genomes.54,55
More recently, there has been a rapid progression from targeted gene sequencing to targeted whole-genome and whole-transcriptome sequencing. The first sequencing of a whole cancer genome was reported for AML.56 Acquired mutations in coding sequences of annotated genes were identified in ten genes in the AML genome by comparing the genomic DNA of leukemia cells with normal skin cells obtained from a patient with FAB M1 AML. Two of the identified mutations were in genes previously described to have a role in leukemogenesis, FLT3 and NPM1. The other eight mutations, however, were in genes that were not previously implicated in the pathogenesis of AML. Four of the affected genes (PTPRT, CDH24, PCLKC, and SLC15A1), though, are in gene families that are strongly implicated in cancer pathogenesis. Intriguingly, the remaining genes (KNDC1, GPR123, EBI2, and GRINL1B) are involved in metabolic pathways.
The genomes of a small cell lung cancer, melanoma, and breast tumor have also been described.57–59 Interestingly, the mutations in both the lung cancer and melanoma genomes were not distributed evenly throughout the genome—many were present outside the gene-coding regions, suggesting that cells had repaired damaged DNA in those key regions. Sequencing of the melanoma genome, for example, revealed multiple levels of selective DNA repair, including the preferential targeting of repair to transcribed regions compared with nontranscribed regions, to exons compared with introns, to transcribed DNA strands compared with nontranscribed strands, and to the 5’ end of genes compared with the 3’ end.
Next-generation sequencing has also been applied to RNA (“RNA-Seq”) extracted from tumor cells for complete transcriptome characterization.60,61 This approach is more sensitive than microarrays and also provides data that can be used to evaluate for allele-specific expression, structural and copy-number alterations, alternative splice isoforms, fusion transcripts, and single nucleotide mutations.60 RNA-Seq, for example, was applied to four granulosa-cell tumors and identified missense point mutations in the FOXL2 gene, which encodes a transcription factor known to be crucial in granulosa cell development.62 In a different study, RNA-Seq was used to identify both known and novel fusion transcripts in prostate cancer samples.63 RNA-Seq has also been used to study the role of microRNAs in the regulation of gene expression in both normal and cancerous cells.
The number of sequenced cancer genomes is likely to expand substantially in coming years. The Cancer Genome Atlas Program of the U.S. National Cancer Institute, for example, initially focused its large-scale genomic analysis on only three tumor types, GBM, ovarian serous cystadenocarcinoma, and lung squamous carcinoma. The scope of the Cancer Genome Atlas Research Network, however, has now expanded to include more than 20 tumor types and thousands of samples. The clinical and translational implications of routine cancer genome sequencing are profound and include the identification of new drug targets, as well as the generation of new insights into the genetic patterns of disease phenotype, prognosis, and therapeutic response. One shortcoming of direct sequencing, though, is its failure to detect epigenetic changes, such as DNA methylation, which may alter gene expression indirectly.
Although the role of 2HG in cancer development remains unclear, the identification of this unexpected class of mutations validates the high-throughput cancer genome sequencing approach. Also, because the mutations in IDH1 result in a gain of function, there is much excitement about developing small molecule inhibitors of mutant IDH1. Although major clinical impact of large-scale sequencing projects is yet to be realized, the discovery of IDH1 illustrates the potential of this approach.
Translocations
Unlike point mutations, small insertions, or deletions of nucleotides, larger chromosomal changes such as translocations, amplifications, and deletions may be observed using cytogenetic analysis4,21 (Fig. 2.2). Chromosome translocation involves juxtaposition of two different chromosome segments, resulting in fusion of two different genes or placement of a gene next to an inappropriate regulatory element. Examples include t(9;22) in chronic myelogenous leukemia, resulting in expression of the growth promoting BCR-ABL gene product, and t(14;18) in follicular lymphoma, resulting in overexpression of the antiapoptotic protein BCL2 as a result of its fusion with the immunoglobulin heavy chain promoter. Recent work has also identified a translocation between the immunoglobulin heavy chain and the cytokine receptor CRLF2 in a subset of precursor B-cell acute lymphoblastic leukemia associated with a poor outcome and activating JAK mutations.64,65

FIGURE 2.2 Common cytogenetic abnormalities. Metaphase spread derived from a mouse tumor model (combined telomerase and p53 deficiency). Individual chromosomes are highlighted by spectral karyotyping (SKY) using fluorescent chromosome probes (right panel). Normal mouse cells contain a diploid complement of 40 chromosomes. In this sample, more than 260 chromosomes are observed, with multiple dicentric chromosomes and nonreciprocal translocations.
One exciting recent development is that translocations not only create chimeric genes or alter promoter sequences, but can also affect the expression of microRNAs.66,67 MicroRNAs are short regulatory RNAs that control mRNA stability and/or translation, and changes in microRNA expression have been linked to prognostic factors and progression in diseases such as chronic lymphocytic leukemia.68
Although translocations and gene fusions are a hallmark of cancer, the mechanisms underlying their genesis are unclear. Recent work, however, has begun to elucidate the mechanisms of some tissue-specific translocations, such as TMPRSS2 to ERG and ETV1 in prostate cancer.69,70 A clever bioinformatics approach, termed cancer outlier profile analysis, was used to first identify this recurrent translocation in prostate cancer.71 This approach was used to successfully identify recurrent gene fusions of TMPRSS2 to ERG or ETV1 in prostate cancer. Further mechanistic work has shown that this translocation requires two roles of the androgen receptor.69 First, ligand-dependent binding of the androgen receptor to intronic binding sites near the tumor translocation sites creates specific intra- and interchromosomal interactions that result in the spatial proximity of tumor translocation partners. Second, the intron-bound androgen receptor alters local chromatin architecture and recruits the ligand and genotoxic stress-induced enzymes, including the activation-induced cytidine deaminase and LINE-1 repeat-encoded ORF2 endonuclease, to these regions, which results in the generation of DNA double-stranded breaks (DSB). The DSB are subsequently ligated by the nonhomologous end-joining (NHEJ) machinery to create translocations. Further elucidation of the mechanisms leading to gene- and tissue-specific translocations will advance the understanding of basic mechanisms of cancer, as well as possibly facilitating the development of new therapeutic strategies.
Amplifications and Deletions
Amplifications can be detected cytogenetically as double-minute chromosomes or regions of excess signal intensity using fluorescence in situ hybridization. Such “amplicons” may range in size from 0.5 to 10 megabases of DNA, resulting in multiple copies of both oncogenes and their neighboring sequences. Conversely, deletions result in loss of chromosomal regions, and can involve small interstitial segments or entire chromosome arms. Genetic alteration of tumor suppressor genes frequently involves mutation in one allele and deletion of the second allele as part of a larger chromosomal segment, resulting in regions of uniform sequence with LOH.
A recent study reported the high-resolution analysis of somatic copy-number alterations (SCNAs) from 3,131 cancer specimens belonging to 26 histologic types.72 The most prevalent SCNAs were either very short (focal) or almost the length of a chromosome arm or whole chromosome (arm level). The focal SCNAs occurred at a frequency inversely related to their lengths, with a median length of 1.8 megabases. Arm-level SCNAs occurred approximately 30 times more frequently than expected by the inverse-length distribution associated with focal SCNAs. This observation was seen across all cancer types and applied to both copy gains and losses. The study also identified 158 regions of focal SCNAs that were altered at significant frequency across several cancer types, of which 122 could not be explained by the presence of an oncogene located within the region. Several gene families were enriched among the regions of focal SCNA, including the BCL2 family of apoptosis regulators and the NF-kB pathway. Interestingly, the finding that most of the SCNAs were found in multiple cancer types suggests that the diversity across cancer genomes may reflect the combinations of a limited number of functionally relevant events.
Whole-Chromosome Loss/Gain
Nearly all solid tumor types exhibit whole-chromosome loss or gain, resulting in alterations in chromosome number or aneuploidy.21,73 As will be described later, such defects are generally the result of chromosomal mis-segregation during mitosis. Glioblastomas, for example, frequently exhibit loss of chromosome 10, inactivating the tumor suppressor PTEN, while melanomas often show gain of chromosome 7, from which B-RAF is expressed. Monosomy 7 and trisomy 8 are associated with myelodysplasia and AML. Whole-chromosome loss may be underestimated by karyotypic analysis, as loss of one parental chromosome may be accompanied by duplication of the other parental chromosome, resulting in an abnormal “allelotype” with accompanying LOH, or copy neutral loss of heterozygosity (CN-LOH).74
CN-LOH, also referred to as uniparental disomy, is common in cancer and has been described in AML, breast cancer, multiple myeloma, basal cell carcinoma, childhood acute lymphoblastic leukemia, chronic lymphocytic lymphoma, myelodysplastic syndrome, and glioblastoma.75 Furthermore, CN-LOH has been shown to have prognostic significance in a number of these cancer types, including primary and secondary AML.76 The specific role of CN-LOH in tumorigenesis remains undefined, but possible mechanisms include the duplication of oncogenes, loss of tumor suppressors, or acquisition of improper epigenetic patterns.77 In mice, it has been demonstrated that Bub1 insufficiency can drive tumor formation through tumor suppressor gene LOH.78 Specifically, Bub1 insufficiency predisposed p53+/− mice to thymic lymphomas and ApcMin/+ mice to colonic tumors. These tumors demonstrated CN-LOH and lacked the nonmutated tumor suppressor allele, but had gained a copy of the mutant allele.
Epigenetics
Significant evidence now indicates that epigenetic modifications, or heritable changes in gene expression that are not caused by changes in DNA sequence, are critical factors in the pathogenesis of cancer.79 Although beyond the scope of this chapter, epigenetic mechanisms controlling the transcription of genes involved in cell differentiation, proliferation, and survival are often targets for deregulation in the development of cancer. These epigenetic alterations include DNA methylation, covalent modifications of histones, and noncovalent changes in nucleosome position. The role of epigenetic modifications in cancer pathogenesis is illustrated by the tumor suppressor SNF5, which regulates the epigenome as a member of the SWI/SNF chromatin remodeling complex.80 Biallelic inactivation of SNF5 is found in the majority of malignant rhabdoid tumors. Most human SNF5-deficient cancers are diploid, lack genomic amplifications/deletions, and are genomically stable.81 Furthermore, the epigenetically based changes in transcription that occur following loss of SNF5 correlate with the tumor phenotype.
Recent work with an NSCLC cell line, PC9, has also identified a novel role for epigenetics in acquired drug resistance.82 Treatment of PC9 cells, which have an activating mutation in the EGFR, with the drug erlotinib results in the death of nearly all of the parental cells. A small percentage of the PC9 cells, however, demonstrate significantly reduced drug sensitivity and remain viable through activation of IGF-1 receptor signaling and an altered chromatin state that requires the histone demethylase RBP2. This drug-tolerant phenotype is transiently acquired at low frequency by individual cells within the population. The drug-tolerant subpopulation can be selectively ablated by treatment with IGF-1 receptor inhibitors or chromatin-modifying agents, such as HDAC inhibitors. There has been much discussion and controversy about the existence of drug-resistant cancer stem cells; the epigenetic effects described in this work provide a potential mechanism for the generation of some “cancer stem cells.” Furthermore, this research suggests that the potentially reversible nature of epigenetic changes, unlike genetic mutations, may provide a unique therapeutic avenue in the treatment of cancer.
MECHANISMS OF GENOME DESTABILIZATION IN HUMAN TUMORS
Microsatellite Instability
One of the earliest insights into the contribution of genome destabilization to carcinogenesis came from the study of the familial cancer syndrome HNPCC.4,11,83 It had been recognized that a subset of sporadic colon cancers and a majority of cancers derived from patients with HNPCC exhibited frequent mutations, particularly in regions of simple repeat sequences known as microsatellites. This type of genetic instability, termed microsatellite instability (MIN), had been described in bacteria and yeast mutants defective in mismatch repair. Linkage analysis in kindreds with HNPCC revealed germ line mutations in hMSH2 and hMLH1, which are human homologues of the mutL and mutS mismatch repair genes in Escherichia coli. It is now known that mutations in other components of the human mismatch repair process, hPMS2, and hMSH6, are also observed in families with HNPCC.
Mismatch repair corrects mispaired bases that can result from errors during DNA replication, as well as mismatched bases occurring in recombination intermediates or occurring as a result of some types of chemical damage to DNA.84 Mismatched bases are recognized by a complex of MSH2 and MSH6, recruiting MLH1 and PMS2 to the site to initiate the subsequent steps of repair, including excision, DNA synthesis, and ligation (Fig. 2.3). Larger insertion/deletion mispairs due to slippage of the replication machinery in repetitive sequences or recombination errors form a loop structure that is alternatively recognized by a complex of MSH2 and MSH3, with recruitment of an MLH1/MLH3 complex promoting subsequent repair. Cancer cells that exhibit MIN from defects in these components have a nucleotide mutation rate that has been estimated at two to three orders greater than that of normal cells. MLH1 and MSH2 have also been shown to have functions outside mismatch repair, as defects in these proteins have been associated with an impaired G2/M cell cycle checkpoint in response to alkylating agents as well as abnormalities in meiotic recombination in mouse knockout models.9
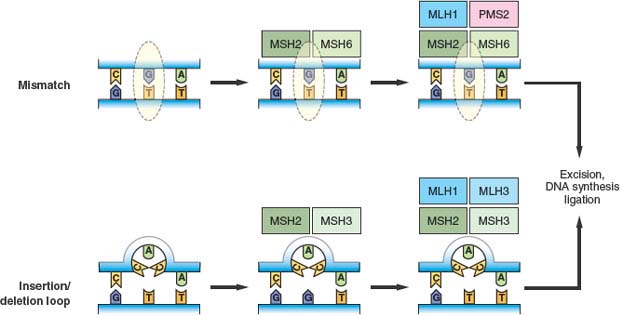
FIGURE 2.3 Mismatch repair pathways. Mispaired bases due to errors in DNA replication or other causes are recognized by the mismatch repair machinery. The initial step involves recognition of simple mismatches by MSH2 and MSH6 (upper panel), or recognition of insertion/deletion loops by MSH2 and MSH3 (lower panel). Subsequent steps involve recruitment of MLH1 and PMS2 to mismatch sites, or MLH1 and MLH3 to insertion/deletion loop sites. This is followed by excision of the respective lesions, DNA synthesis, and ligation to complete the repair.
HNPCC is associated with a 60% to 80% lifetime risk of developing colorectal cancer and is responsible for 2% to 5% of all cases of colorectal cancer.85 Nearly 85% of HNPCC patients have mutations in MLH1 or MSH2, with median age at colon cancer diagnosis being significantly lower in MLH1 mutation carriers and in males, and with more frequent extracolonic tumors observed in MSH2 carriers. The MIN phenotype is also observed in 15% of sporadic colon cancers, often due to epigenetic silencing of mismatch repair genes such as MLH1. Colorectal cancers exhibiting MIN are typically diploid, in contrast to the remaining 85% of cases, which are associated with chromosomal instability (CIN).86 Experimental evidence supports the idea that MIN occurs very early in sporadic colorectal cancer formation, prior to anaphase-promoting complex (APC) inactivation, increasing genomic instability and thus obviating the selection pressure to develop another mechanism of genomic instability, CIN. In both sporadic cases and HNPCC, MIN has been associated with more favorable prognosis, lack of p53 mutation, and a potential resistance to 5-fluorouracil chemotherapy. These observations are in accordance with the hypothesis that the accelerated mutation rate facilitates cancer evolution, but at the same time compromises fitness of cells, presumably by the accumulation of deleterious mutations.
Nucleotide Excision Repair/Base Excision Repair Defects
Whereas the mismatch repair pathway functions primarily in the recognition of and repair of replication errors, the nucleotide excision repair (NER) and base excision repair (BER) pathways respond principally to lesions created by exogenous or endogenous DNA damaging agents.4,9,84,87 It is becoming apparent that defects in components of these repair pathways have impacts on both cancer pathogenesis and the efficacy of cancer therapy.88 Ultraviolet radiation or exogenous chemicals such as polycyclic aromatic hydrocarbons and platinum chemotherapeutic drugs can result in bulky, helix-distorting lesions that are recognized by the NER machinery. Components of the NER pathway were in part discovered by mutation in the genetic syndromes xeroderma pigmentosa (XPA-XPG) and Cockayne syndrome (CSA and CSB).89 Mutation of XP genes can also be seen in the related disorder trichothiodystrophy. Whereas all three disorders exhibit dramatic sun sensitivity, only xeroderma pigmentosa is associated with a marked incidence of sun-induced skin cancer.90 Deletion of CSB has been shown to impair tumor formation in cancer-prone mice, and it has been hypothesized that the lack of cancer in Cockayne syndrome may be related to a particular sensitivity of cells to apoptosis or impairment of transcription.91
Two separate NER pathways have been identified, one that involves scanning the entire genome for lesions (global genome NER) and another that detects lesions that interfere with elongating RNA polymerases (transcription coupled repair or TCR)9,87 (Fig. 2.4). XP genes are involved in the recognition and repair of lesions in global genome NER, while CS genes play a specific role in transcription coupled repair. Subsequent stages of NER are similar and involve ERCC1, an endonuclease involved in excision of the lesion, followed by DNA replication to complete the repair process. Notably, mutant mice defective in NER also accumulate DNA damage, with a more pronounced cancer phenotype and evidence of premature aging. Furthermore, reduced expression of ERCC1 in NSCLC has been associated with response to cisplatin-based adjuvant chemotherapy.92 A subgroup analysis of the International Adjuvant Lung Cancer Trial (IALT) demonstrated that ERCC1 deficiency was observed in tumor samples from 56% of patients and correlated with a significant improvement in survival following cisplatin-based chemotherapy, whereas no benefit was seen in tumors in which normal ERCC1 expression was maintained. Thus, while reduced ERCC1 expression may promote genetic instability and facilitate NSCLC development in a significant fraction of patients, the resulting cancers are “stuck” with the deleterious effects of ERCC1 deficiency and become sensitive to certain therapies. Potentially, this provides a general model for targeted therapy based on fixation of mutations that compromise genomic integrity during tumorigenesis.
BER is primarily involved in the response to damage caused by small chemical alterations and x-rays, as well as spontaneous reactions such as base loss from hydrolysis of glycosyl DNA bonds, which has been estimated to occur on the order of 104 times per day per cell.9,24 Damaged bases are removed by DNA glycosylases, and abasic sites are recognized by a complex that includes the APEX1 endonuclease, poly(ADP-ribose) polymerase (PARP), DNA polymerase and ligase, and XRCC1, a scaffolding protein that interacts with most of the core components (Fig. 2.4). Disruption of any of these genes in the BER pathway results in the cellular accumulation of oxidative DNA damage.
Despite being integrally important to the maintenance of genome stability, human disorders or inherited cancer susceptibility syndromes due to mutation of components of the BER machinery have yet to be described. This may be because of partial redundancy of DNA glycosylases, the fact that mutation of core BER components (in mice) results in embryonic lethality, or simply to the need for additional investigation. As described later, pharmacologic inhibition of PARP may selectively sensitize cancer cells with a pre-existing defect in another repair pathway to death by DNA damage.93,94
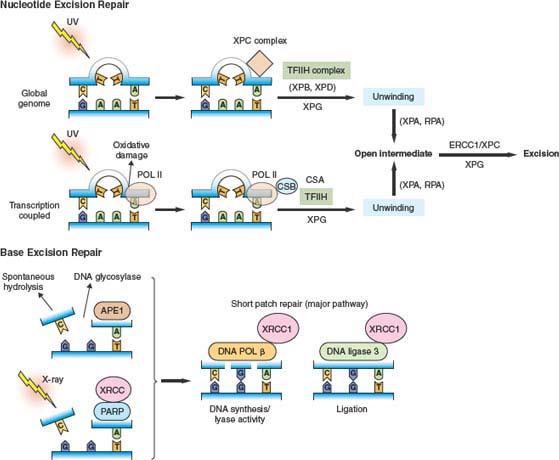
FIGURE 2.4 Nucleotide excision repair and base excision repair. Nucleotide excision repair (NER) is activated in response to bulky lesions that are generated, for example, by UV irradiation (upper panels). Global genome (GG) repair involves proteins identified by complementation groups in patients with xeroderma pigmentosa (XP proteins). Initial recognition of lesions occurs by a complex containing xeroderma pigmentosa C (XPC). Transcription coupled repair (TCR) also involves proteins identified by mutation in Cockayne syndrome (CS proteins), and occurs when RNA polymerase II stalls at the site of lesions. Stalled RNA polymerase II recruits Cockayne syndrome B (CSB) to the site of damage. Subsequently, DNA is locally unwound around the injured site by a TFIIH complex containing XPB and XPD. This process also involves XPG, CSA, and other proteins for TCR. Once unwound, XPA and replication protein A (RPA) contribute to stabilization of an open intermediate and recruitment of the ERCC1 and XPF endonucleases that excise the lesion. Subsequent steps involve DNA synthesis and ligation to complete the repair. In base excision repair (BER) (lower panels), abasic sites generated by spontaneous hydrolysis, action of DNA glycosylases, or x-ray–induced single-strand breaks are recognized by the APE1 endonuclease, as well as PARP and XRCC1. Subsequent repair is influenced by PARP-mediated ADP ribosylation of histones and other proteins, while XRCC1 serves as a scaffold for recruitment of DNA polymerase β and DNA ligase 3. These latter enzymes catalyze nucleotide reinsertion and ligation into the injured strand as part of the short patch repair pathway (major BER pathway).
DNA Damage Response to Double-Strand Breaks
DNA DSBs represent a significant threat to genomic integrity.4,9 They can occur during DNA replication at sites of stalled replication forks, or after ionizing radiation or oxidative damage.24 In addition, single-strand nicks can be converted into DSBs by DNA replication. Even a single DSB in budding yeast can trigger a DNA damage response checkpoint, a finding that is not surprising as DSBs can promote major cytogenetic abnormalities such as chromosome translocations, amplifications, and deletions. DSBs are detected and repaired by an intricate cascade of proteins, ultimately involving the processes of homologous recombination (HR) or NHEJ. Defects in multiple components of this process have been linked to genomic instability and cancer predisposition.
The initial detection and activation of signal transduction pathways mediating repair of DNA DSBs involves the PI(3)K-like kinases, ATM and ATR.10,11,24,95 DSBs induced by DNA damage activate ATM, while regions of single-stranded DNA at stalled replication forks recruit and activate ATR. Once activated, ATM and ATR kinase activity results in phosphorylation of multiple targets, including histone H2AX, resulting in the local alteration of chromatin structure. Key downstream targets of ATM and ATR include the checkpoint mediator proteins (CHK), with ATM principally activating CHK2 and ATR activating CHK1. As previously described, ATM/CHK2 as well as ATR/CHK1 can phosphorylate and activate p53, mediating downstream checkpoint activation. ATM/CHK2 and ATR/CHK1 have also been shown to slow progression through S phase by down-regulating CDC25A.28
ATM was identified by virtue of its association with the neurodegenerative disorder ataxia telangiectasia, in which patients are also predisposed to malignancies such as acute lymphoblastic leukemia and lymphoma.9,10 Inactivating mutations in ATR (AT and Rad3-related) result in embryonic lethality in mice and are not observed in familial human cancer syndromes, presumably reflecting the key role of this kinase in normal DNA replication. However, hypomorphic alleles of ATR that result in low levels of expression have been associated with the Seckel syndrome, which results in dwarfism, microcephaly, and chromosome instability in cells treated with mitomycin C.96 Patients with Seckel syndrome are not significantly predisposed to cancer development, perhaps reflecting the general balancing of fitness effects and oncogenic potential; in Seckel syndrome the disadvantages of compromised ATR for cell viability may outweigh the potential for an increase in cancer-causing mutations. As previously described, the important caretaker function of CHK2 is evidenced by its mutation in a subset of patients with Li Fraumeni syndrome. CHK2 mutation has also been observed in familial breast cancer, and in multiple sporadic tumor types.97 Mutation of CHK1 is observed less frequently in tumors, which, similar to ATR, could reflect strongly disadvantageous effects of CHK1 loss on viability.
The initiation of the repair process itself involves recruitment of multiple other proteins to sites of DSBs in conjunction with ATM and ATR.4,9 DSBs are recognized by the MRN complex, consisting of MRE11 (meiotic recombination 11)/RAD50/NBS1 (Nimjen breakage syndrome 1).98 Mutations in MRE11 result in an ataxia telangiectasia-like disorder, while NBS1 is mutated in the Nijmegen breakage syndrome, and both diseases are characterized by immunodeficiency, cellular sensitivity to ionizing radiation, chromosomal instability, and a high frequency of malignancies. In addition to their role in DSB repair by HR, the MRN proteins have important roles in telomere maintenance, and, at least in yeast, in NHEJ. ATR/CHK1 signaling is linked with activation of the Fanconi anemia pathway and BRCA2.
DNA Repair of Insterstrand DNA Crosslinks
Fanconi anemia is another autosomal recessive human disease characterized by bone marrow failure, congenital abnormalities, cellular hypersensitivity to DNA cross-linking agents such as mitomycin C and cisplatin, and predisposition to malignancies such as acute leukemia and squamous cell carcinomas.99,100 There are 13 known FA genes, and the encoded FA proteins cooperate in a common DNA repair pathway. Eight of the FA proteins are assembled in a multisubunit ubiquitin ligase complex. In response to DNA damage, or during normal S phase progression, this ligase monoubiquitinates two additional FA proteins, FANCD2 and FANCI. Monoubiquitination is activated by the ATR/CHK1 pathway.101 Monoubiquitinated FANCD2 and FANCI are required for normal homologous recombination repair, and these proteins further interact with the downstream FA proteins, FANCD1, FANCJ, and FANCN/PALB2.102,103 Interestingly, FANCD1 is identical to the BRCA2 gene.104
The three downstream FA genes are breast/ovarian cancer susceptibility genes, and heterozygote carriers, with mutations in a single copy of FANCJ, FANCD1, or FANCN have an increased cancer risk. Biochemical functions of monoubiquitinated FANCD2/FANCI were recently elucidated. In a cell-free system, this complex was required for nucleolytic incisions near an interstrand cross-link and for translesion DNA synthesis past the cross-link. Thus, the FA pathway was shown to be required for the generation of a new, partially processed DNA substrate that can be further repaired by the downstream homologous recombination machinery.105
Homologous Recombination
In repair by HR, sequences from a homologous DNA duplex are used to provide a template for reconstruction of the damaged DNA segment.4,9 The template for repair can either be the identical sister chromatid (the preferred substrate in mitotic cells) or the homologous chromosome (the preferred substrate during meiosis). The classic HR pathway involves the following basic steps (Fig. 2.5). DSBs are recognized by the MRN complex and by checkpoint proteins as previously described. A 5’-3 exonuclease generates 3’ overhangs, which are then coated with replication protein A (RPA). “Mediator” proteins such as BRCA2 or Rad52 then facilitate the recruitment of Rad51-related proteins, which form filaments on the single-stranded DNA, replacing RPA. A homology search ensues, followed by strand invasion and DNA synthesis. The links between DNA strands (double Holliday junctions) can be resolved to produce exchange between chromosomes (crossovers) or no exchange (non-crossovers). Enzymes such as the RecQ helicase BLM, in conjunction with topoisomerase IIIα can resolve these double Holliday junctions.106
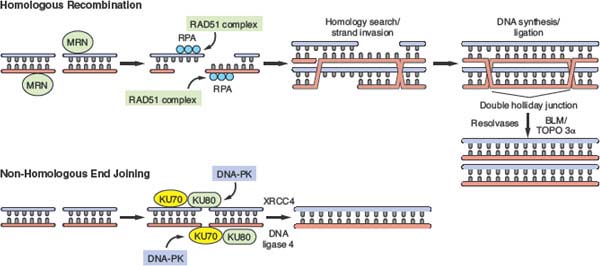
FIGURE 2.5 Double-strand break (DSB) repair by homologous recombination and nonhomologous end-joining (NHEJ). In homologous recombination (HR), DSBs are recognized by the MRN complex, among other proteins. 5’-3’ exonuclease activity results in the generation of single-strand overhangs that are coated with RPA. Mediator proteins such as BRCA2 and RAD52 stimulate assembly of a RAD51 nucleoprotein filament complex that guides subsequent homology search and strand invasion into the homologous strand (e.g., the identical sister chromatid in late S/G2 phase and mitosis). Subsequent DNA synthesis and ligation results in the formation of recombination intermediates that contain double Holliday junctions. These are resolved by resolving enzymes such as the RecQ helicase BLM, in conjunction with topoisomerase 3α. The process of NHEJ involves recognition of DSB ends by the Ku70-Ku80 heterodimer, with subsequent recruitment of DNA-dependent protein kinase. DNA ends are then ligated following recruitment of XRCC4 and DNA ligase 4.
Several other HR pathways also exist. A potentially important mechanism for cancer development is break-induced replication (BIR).107 During BIR a broken chromosome end invades a homologous site and replication proceeds to copy the entire sequence of the template chromosome. This is relevant to cancer because it can result in large-scale LOH. Furthermore, BIR is an important mechanism for healing breaks at chromosome ends resulting from telomere attrition.108 Finally, in yeast, aging is accompanied by a switchlike increase in mutagenesis and BIR after a certain number of generations.109
Disruption of recombination pathways produces complex effects that pose the danger of chromosomal rearrangement due to the accumulation of recombination intermediates.4,9 These are then channeled into alternate, often suboptimal, repair pathways, increasing the potential for errors and rearrangements. Cancer-causing mutations have been associated with multiple steps in HR. Given that RecQ helicases are particularly important resolving enzymes in this process, their inactivation results in widespread accumulation of recombination intermediates.106,110,111 Mutation of BLM results in Bloom syndrome, a disease characterized by immunodeficiency, male sterility, dwarfism, skin disorders, and a high incidence of both leukemia and solid tumors. In addition, the WRN helicase was identified by virtue of its mutation in Werner syndrome, a disorder characterized by premature aging, with early atherosclerosis, type 2 diabetes, osteoporosis, and age-associated malignancies. Rothmund-Thomson syndrome is associated with mutation in the related RecQ helicase RECQ4, and affected patients exhibit characteristic photosensitivity with poikilodermatous skin changes, early alopecia and hair graying, juvenile cataracts, growth deficiency, and an elevated frequency of malignancies such as osteogenic sarcomas. RecQ helicases have also been shown to facilitate NHEJ, interact with components of the MMR and BER machinery, and play an important role in telomere maintenance. The combination of aging and cancer predisposition phenotypes associated with RecQ lesions provides yet another example of the balance between the deleterious and growth-promoting effects of genomic instability on developing cancer cells.
BRCA1 and BRCA2 are perhaps the most extensively studied cancer susceptibility genes required for HR. They are mutated in familial breast and ovarian cancer syndromes and represent key components of the response to DSBs and subsequent repair by HR.112–114 BRCA1 forms a heterodimer with the structurally related protein BARD1, and as previously described, forms a complex together with BRCA2, RAD51, and other proteins involved in the regulation of repair by HR. Another large, multiprotein complex termed BASC (BRCA1-associated genome surveillance complex) has been identified, containing tumor suppressors, mismatch repair proteins, ATM, MRN, and BLM, with a presumed role in the global sensing and coordinated response to DNA damage. BRCA1 has also been implicated in S phase and G2/M checkpoint control, and in the organization of heterochromatin.
Mutations in BRCA1 and BRCA2 are associated with a 60% to 85% lifetime risk of developing breast cancer and a 15% to 40% lifetime risk of developing ovarian cancer, although only 2% to 3% of all breast cancer cases are associated with mutation in one of these genes.113 They account for approximately 40% of cases of familial breast cancer, with mutations in CHK2 and p53 responsible for an additional 5% and 1% of cases, respectively. Recently, mutation in PALB2, which encodes a BRCA2-binding partner, has also been described in familial breast cancer, and may contribute to inherited prostate cancer as well.115 PALB2 is also identical to the Fanconi anemia gene FANCN.116 Germ line biallelic mutations in PALB2 or BRCA2 (FANCD1) result in Fanconi anemia.117
Although it is clear that mutations in BRCA1 and BRCA2 can destabilize the genome and promote cancer susceptibility, it remains poorly understood which roles of these proteins are specifically involved in tumor suppression and why patients with germ line defects primarily develop breast and ovarian cancer. Somatic inactivation of BRCA1 and BRCA2 has been reported in other tumor types such as colorectal cancer, as have defects in other components of DSB repair such as ATM and FANC genes. A subset of sporadic breast cancer, defined by lack of expression of the estrogen and progesterone receptors and absence of HER2 amplification (termed triple-negative breast cancer) and a “basal-like” phenotype shares strong similarities with the tumors that develop in patients with germ line BRCA1 and BRCA2 mutations. These sporadic basal-like cancers also exhibit defects in X chromosome inactivation,118 and co-cluster with BRCA10-deficient breast cancers on transcriptional arrays. Although these sporadic triple-negative breast cancers appear to have normal BRCA1, these similarities have led to the suggestion that they may be defective at another point within the BRCA1 pathway. Another possibility is that breast tissue selectively accumulates genotoxins that induce a heightened requirement for BRCA1 and BRCA2. Further study is needed to elucidate the mechanism behind the tissue-specific nature of BRCA1 and BRCA2 mutant cancer.
Nonhomologous End-Joining
Given that HR uses an identical sister chromatid as template to guide repair, in principle it should be error-free.4,95,119,120 In yeast, where the genome lacks extensive repetitive sequences, HR is indeed mostly error-free. However, in humans, where the genome contains extensive repetitive sequences, HR poses the danger of repeat sequence recombination resulting in gross chromosomal rearrangements. One factor that prevents this type of error is that repair by HR is limited to late S and G2 phases of the cell cycle, after DNA replication. Thus, HR predominantly occurs when a homologous template sequence is held in close physical proximity to the break by the cohesion between sister chromatids. In humans and other higher eukaryotes it appears that NHEJ is relatively more important than in other organisms such as fungi. It seems that, at least in G1 cells, the small-scale errors generated by NHEJ are less detrimental than the potential large-scale errors (deletions or translocations) that could arise from HR. NHEJ involves direct end-joining of the broken double-strand DNA ends, without a template for repair. Thus, NHEJ can occur during any phase of the cell cycle, although it primarily occurs during G1 phase.
In the process of NHEJ, broken DNA ends are recognized by a heterodimer of Ku70/Ku80, which recruits the catalytic subunit of DNA-dependent protein kinase (DNA-PK) and the Artemis nuclease120–122 (Fig. 2.5). DNA-PK–mediated phosphorylation of Artemis facilitates its activation and results in the processing of DNA ends in a subset of DSBs, contributing to the error-prone nature of NHEJ. DNA ligation is subsequently mediated by a complex that contains XRCC4 and DNA ligase IV. NHEJ is integrally involved in the V(D)J recombination and class switching that occurs during normal lymphocyte maturation. During V(D)J recombination and certain other physiologic settings, cleavage induced by the RAG nuclease may generate regions of microhomology that allow for relatively precise end-joining.
While mutations in these core NHEJ components in mice cause severe immunologic defects, apoptosis, and premature senescence of cultured fibroblasts, concomitant p53 inactivation results in a high frequency of lymphomas with recurrent, clonal rearrangements.123 Certain chromosome regions, such as the c-myc and the immunoglobulin heavy chain (IgH) loci, are targeted in a recurrent fashion in these lymphomas, similar to common translocations observed human lymphomas. These regions may contain specific sites that are recognized by the RAG nuclease, and the initial cleavage combined with defective NHEJ may be responsible for these aberrant chromosome fusions. Alternatively, NHEJ deficiency has been linked to impaired telomere capping and end-to-end fusions, with so-called breakage-fusion-bridge cycles (see later discussion) promoting translocations and gene amplifications. It is also possible that fragile sites within these chromosomal loci and elsewhere throughout the genome may account for the particular susceptibility of certain chromosome regions to breakage and rearrangement.
Although there is abundant evidence that NHEJ defects can promote tumorigenesis in mouse models—at least in the setting of concomitant p53 deficiency—there are few reports that implicate NHEJ deficiency in human cancer. The reasons for this are unclear. Loss of NHEJ may be cell-lethal in humans. Consistent with this idea, Artemis deficiency, which results in a very restricted NHEJ defect, is observed in rare lymphoma-prone patients. Similarly, there is a report of a ligase IV mutation in a leukemia patient.124 However, it may also be the case that more human tumors need to be carefully characterized for subtle mutations, haploinsufficiency, and epigenetic silencing.
Telomere Maintenance
Telomeres, the structures at the ends of chromosomes composed of repetitive sequences and a 3’ G-strand overhang, are key mediators of genomic instability.125 In humans, telomerase is expressed at low to undetectable levels in somatic tissues, leading to an age-dependent compromise of telomere integrity and resultant telomere dysfunction. By contrast, telomerase is expressed in many tumors, resulting in stabilization of telomere length and restoration of capping function. The activation of telomerase expression during cancer progression supports the idea that telomere dysfunction contributes to chromosome instability during a specific window in oncogenesis.126,127 For example, telomerase activity is low in small and intermediate-sized colon polyps, and high in late adenomas and colorectal carcinomas, consistent with the observation that CIN arises early during colorectal tumorigenesis, at a time of short telomere length. Furthermore, telomere shortening as a consequence of cell turnover in the setting of chronic inflammatory states such as hepatocellular cirrhosis may contribute to chromosome instability and carcinogenesis in these settings. Conversely, the lack of complex karyotypes in many lymphomas may be related to early activation of telomerase in the setting of the frequent dysregulation of Myc, a positive regulator of telomerase expression.128
Studies of mice deficient in telomerase activity have yielded powerful evidence that telomere dysfunction can drive chromosomal instability and epithelial carcinogenesis. Strongly reminiscent of the shift in tumor spectrum to epithelial malignancies on aging humans, aging mice deficient in telomerase and heterozygous for mutant p53 exhibit carcinomas of the breast, colon, and skin.128 Such tumors are exceedingly rare in wild type mice, which primarily develop sarcomas and hematopoietic malignancies. Moreover, the presence of cytogenetic profiles similar to human carcinomas supports a role for telomere dysfunction in epithelial carcinogenesis. A breakage-fusion-bridge cycle is believed to be responsible for chromosomal fragmentation and the nonreciprocal translocations observed in such tumors (Fig. 2.6). With progressive erosion of telomeres, unprotected chromatid ends can undergo end-to-end fusion, with the formation of a dicentric chromosome. During mitosis, the fused chromosome ends form anaphase bridges as sister centromeres are pulled to opposite centrosomes, resulting in chromosome breakage. The further generation of atelomeric chromosomes by this process can result in propagation of breakage-fusion-bridge mechanisms and continued chromosome instability. In addition to generating translocations, this form of genetic instability can also result in amplifications and deletions.129,130 The observation that mouse epithelial tumors in this model exhibit amplified and deleted regions syntenic to those seen in human carcinomas lends further support to the notion of chromosomal fragile sites that may be conserved between species.
More recently, key regulatory elements of telomere structure have been implicated in genomic instability and tumorigenesis.131,132 TRF2, an important regulator of telomere protection and telomere length, is overexpressed in a variety of epithelial malignancies, including lung, skin, and breast cancer, and has been shown to interact with a number of DNA repair proteins, such as the MRN complex, the WRN and BLM helicases, DNA-PK, PARP, and ERCC1/XPF. Moreover, mice develop an XP-like syndrome when TRF2 is expressed in the skin at high levels, with UV-induced skin cancer, severe telomere shortening, and chromosomal instability.133 Concomitant telomerase inactivation in these mice dramatically accelerates carcinogenesis, with TRF2 promoting recombination at telomeres and de-repression of pathways that lead to alternative lengthening of telomeres (ALT).131 ALT involves recombination between telomeres as an alternative means of telomere extension and is operative in a small minority of tumors that are telomerase-negative. Taken together, these studies identify a fundamental role for telomeres and their regulatory proteins in the genesis of chromosome abnormalities and epithelial malignancies.
Telomerase may also play important roles beyond telomere maintenance in the regulation of genome stability.134 It is expressed at low levels during S phase in normal cells, and targeted disruption of hTERT, the catalytic subunit of telomerase, has been shown to impair heterochromatin formation on a global level and to disrupt the response to DNA damage in normal human fibroblasts.134,135 These results are consistent with the emerging ties between regulators of telomere maintenance, heterochromatin structure, and components of DNA damage response.136–138 In addition, they lend further support to the idea that stem cells, which express higher levels of telomerase than their differentiated progeny, are protected from genomic injury. It is important to note, however, that telomerase-deficient mice are viable and do not exhibit significant phenotypic defects until later generations. The specific role of telomerase in chromatin maintenance and the DNA damage response remains to be elucidated, and the reason it is down-regulated in association with cell differentiation remains to be determined.
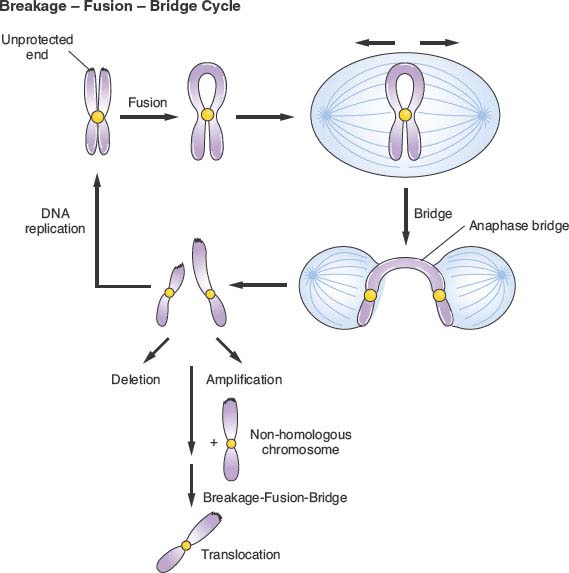
FIGURE 2.6 Breakage-fusion-bridge cycle. In the setting of telomere dysfunction and uncapping of chromosome ends, telomeric fusions may occur between identical sister chromatids or between different chromosomes (dicentrics). During anaphase, as sister chromatids are pulled to opposite poles, the fused chromosome ends are placed under tension and form anaphase bridges. These pulling forces result in chromosome breaks that contribute to deletions, amplifications, and translocations. In addition, because of the further generation of unprotected chromosome ends, the cycle may be repeated.
The role of telomere dysfunction in promoting cancer is further supported by findings in a rare genetic disorder, dyskeratosis congenita (DC). DC is a progressive bone marrow failure syndrome that is classically associated with the clinical triad of abnormal skin pigmentation, leukoplakia, and nail dystrophy. X-linked DC is caused by mutations in dyskerin, which leads to reduced levels of TERC and telomerase activity. Autosomal dominant DC can be caused by mutations in TERT, TERC, or the telomere binding protein TRF1-interacting nuclear factor 2. Autosomal recessive DC can be caused by mutations in the dyskerin-associated proteins NHP2 and NOP10. In all cases, patients have very short germ line telomeres. In addition to bone marrow failure, patients with DC are also prone to develop myelodysplastic syndrome, leukemia, and solid tumors. Thus, telomere-shortening syndromes in humans recapitulate the impaired maintenance of proliferative tissues and the tumor predisposition seen in telomerase-knockout mice.
WHAT CAUSES CHROMOSOMAL INSTABILITY AND WHOLE-CHROMOSOME ANEUPLOIDY?
Chromosomal instability (CIN), defined as a persistently high rate of loss and gain of whole chromosomes, is a common characteristic of many cancers. CIN, or the “rate” of karyotypic change, should be distinguished from aneuploidy, or the “state” of the karyotype. Although CIN leads to aneuploidy, not all aneuploid tumors exhibit CIN; some tumors are aneuploid with a uniform, stable karyotype. The first breakthrough in understanding the mechanisms of CIN came from studying different colon cancer cell lines that exhibited either a CIN or MIN phenotype.139 In a single-cell cloning assay, the colon cancer cell lines with MIN maintained a stable chromosome content, but the aneuploid colon carcinoma cells exhibited deviations from the modal chromosome number, indicating the presence of CIN. The fusion of MIN and CIN cells resulted in hybrid cells that retained the CIN phenotype, suggesting that the mechanisms that cause CIN act in dominant fashion. Subsequent work demonstrated that a compromised spindle assembly checkpoint could result in CIN.140
Chromosome Cohesion Defects
Accurate chromosome segregation is achieved through carefully orchestrated interactions between the mitotic spindle, kinetochores, and cohesin.11,29 Replicated chromosomes attach to the spindle microtubules via the kinetochore, an organelle that is assembled onto centromeric chromatin. Prior to anaphase, replicated sister chromatids are held together by cohesin, a protein ring that physically links the sisters.141,142 The detailed molecular mechanism of how cohesin holds sisters together is a topic of much current research.143 Most cohesin is lost from chromosome arms prior to metaphase in a manner that requires the Polo and Aurora B kinase. Centromere cohesion is then lost after anaphase onset, a direct consequence of the protease separase cleaving a cohesin subunit (Fig. 2.7). Recently, mutations in genes involved in sister chromatid cohesion, including subunits of the cohesion complex, were identified in colorectal tumors through the sequencing of human homologues of genes known to cause CIN in budding yeast.144 The frequency and functional consequences of these mutations has not yet been tested experimentally. Additional evidence suggesting a role for the disruption of cohesion in the development of CIN comes from data demonstrating that some breast cancer tumors, as well as osteosarcoma and prostate tumors, express high levels of separase.145–147 Although, in theory, excessive separase could cause premature chromatid disjunction and subsequent chromosome mis-segregation events, the significance of this mechanism in generation of CIN has not been definitively established.
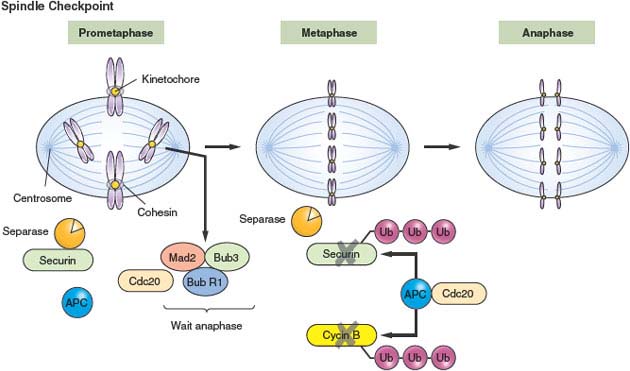
FIGURE 2.7 Spindle checkpoint. During metaphase, paired sister chromatids attach to the bipolar mitotic spindle apparatus at kinetochores, organelles that are assembled onto centromeric chromatin. Sister chromatids are held together by a cohesin, a protein ring that physically links them together. Kinetochores that remain unattached to the spindle catalyze the formation of an active MAD2 complex (“wait” anaphase signal) that binds and inhibits CDC20. Once the final kinetochore is occupied by the spindle, the wait anaphase signal is lost, and CDC20 activates the anaphase-promoting complex to ubiquitinate substrates such as cyclin B and securin. The resultant proteasomal degradation of securin releases the enzyme separase to cleave cohesin and allow for sister chromatid separation under the tension of the mitotic spindle.
Spindle Assembly Checkpoint Defects
Attachment of kinetochores to microtubules is monitored by the spindle checkpoint (Fig. 2.7).29,148,149 A number of spindle checkpoint proteins have been identified, initially via screens in budding yeast, and include MAD1, MAD2, BUB1, BUBR1, BUB3, and a BUB3-related protein RAE1. Although mutations in these spindle assembly checkpoint proteins do occur in human tumors it appears to be a relatively rare event, as discussed in the next section.150 To prevent chromosome mis-segregation, the spindle checkpoint proteins bind to kinetochores that are improperly attached to the spindle and form a “stop” or “wait” anaphase signal. Ultimately the wait anaphase signal prevents cleavage of cohesin by separase. This involves a cascade of events that include activation of spindle checkpoint proteins at the kinetochore, diffusion of the activated signal throughout the cell, and binding of spindle checkpoint proteins to CDC20, which is the key activator of the E3 ubiquitin ligase complex, the APC. Once all kinetochores are attached to the spindle, the wait anaphase signal is lost and CDC20 is released. Activated APC then triggers mitotic cyclin degradation, separase activation, and cohesin cleavage. How this checkpoint signal is rapidly reversed is poorly understood.
It is important to note that although some CIN cell lines have a defective spindle assembly checkpoint (SAC), several live-cell imaging studies have demonstrated that most CIN cells actually have an intact and functional checkpoint.151–153 Likewise, in another study, both diploid and CIN cells underwent mitotic arrest in response to spindle poisons with equal efficiency.151
Kinetochore-Microtubule Attachment Defects
Attachment of kinetochores to microtubules is necessary but not sufficient for proper chromosome segregation. The spindle checkpoint is also activated if kinetochores are attached but not under proper tension, an indication of successful biorientation.154 Improper kinetochore-microtubule attachments that are not under normal tension are disassembled by a mechanism involving phosphorylation of kinetochore proteins by the Aurora B kinase.155,156 Recent work has shown that phosphorylation of an Aurora B substrate at the kinetochore depends on its distance from the kinase at the inner centromere. Repositioning Aurora B closer to the kinetochore prevents stabilization of bioriented attachments and activates the spindle checkpoint. Thus, centromere tension can be sensed by increased spatial separation of Aurora B from kinetochore substrates, which reduces phosphorylation and stabilizes kinetochore microtubules.157
The efficient correction of microtubule-kinetochore attachment errors requires the release of incorrectly attached microtubules. Thus, interactions that inappropriately stabilize microtubule attachments might be expected to increase chromosome mis-segregation errors and generate CIN. In fact, recent work demonstrated that kinetochore-microtubule attachments were more stable in cancer cells with CIN than in a noncancerous, diploid cell line.158 Furthermore, increasing the stability of kinetochore-microtubule attachments in the diploid cell line was sufficient to cause chromosome segregation defects at levels comparable to those in cancer cells with CIN. Conversely, overexpression of proteins that cause increased kinetochore-microtubule dynamics was sufficient to restore stability to chromosomally unstable tumor cell lines.159 This work demonstrates that the temporal control of microtubule attachments to chromosomes during mitosis is central to genome stability in human cells.
Supernumerary Centrosomes
CIN is also known to be correlated with extra centrosomes. Although this relationship was long-standing, it was not until recently that a mechanism was proposed to link these two common characteristics of cancer cells. Early theories hypothesized that extra centrosomes generate CIN by promoting a multipolar anaphase, which results in three or more aneuploid daughter cells. Recent long-term, live-cell imaging experiments, however, revealed that cells with extra chromosomes typically cluster the extra chromosomes during mitosis to ensure that anaphase occurs with a bipolar spindle.160–164 The imaging also demonstrated, though, that these cells with extra centrosomes had a significantly increased frequency of lagging chromosomes during anaphase. Further analysis revealed that these segregation errors were a result of the cells with supernumerary centrosomes passing through a transient “multipolar spindle intermediate” in which merotelic kinetochore-microtubule attachment errors accumulated before centrosome clustering and anaphase (Fig. 2.8). Merotely, a conformation where a single kinetochore is attached to microtubules arising from opposite spindle poles, was previously known to generate lagging chromosomes during anaphase and cause chromosome segregation errors.165,166 The results of these live-cell imaging experiments, consequently, provide a direct mechanistic link between extra centrosomes and CIN, two common characteristics of solid tumors. Thus, for chromosome segregation, “CIN geometry” may be as important as “CIN genes.”
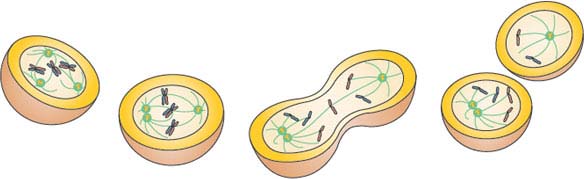
FIGURE 2.8 A mechanism linking extra centrosomes to chromosomal instability. The first cell illustrates a cancer cell with extra centrosomes undergoing a multipolar metaphase. Note that the geometry predisposes to “merotelic” attachments (see chromosome in the middle) where a single chromatid forms attachments to different spindle poles. The second cell illustrates the clustering of centrosomes, which is required for cancer cell survival. Note that the merotelic attachment persists and does not activate the spindle assembly checkpoint. Cell three has undergone anaphase and the merotelic attachment results in a lagging chromosome. Lastly, daughter cells are illustrated with resulting aneuploidy. (Figure provided by L. Kelley and J. DiGianni.)
DOES WHOLE-CHROMOSOME ANEUPLOIDY CAUSE CANCER?
Whole chromosome losses or gains are very common in human cancer.73 Although the specific gains or losses vary from tumor to tumor, some changes are recurrent in a given tumor type. For example, the gain of chromosome 3 or 3q is reportedly as common in cervical cancer as the Philadelphia chromosome is in chronic myelogenous leukemia.167 Likewise, glioblastomas frequently exhibit loss of chromosome 10 and melanomas often show gain of chromosome 7. Despite the fact that whole chromosome loss or gain is frequently observed in both solid and hematologic malignancies, a causal role for aneuploidy in cancer progression has been controversial.168,169
The identification of germ line mutations in the gene encoding BUBR1 in the disease mosaic variegated aneuploidy has provided the strongest evidence for a causal link between aneuploidy and cancer in humans because patients with this disorder exhibit growth retardation, microcephaly, and multiple childhood malignancies. Premature chromatid separation is frequently seen in more than 50% of lymphocytes from patients with mosaic variegated aneuploidy and many tissues exhibit more than 25% aneuploid cells. Mutations in other components of the spindle assembly checkpoint, including MAD1 and MAD2, have also been described in sporadic cancers and cell lines. Deregulated expression of spindle checkpoint proteins is potentially more common in tumors, but it is always difficult to know which cells to compare with the tumor cells.
Mouse models support the notion that spindle checkpoint misregulation can contribute to tumorigenesis.150,170–172 However, they also illustrate that there is no simple one-to-one correspondence between checkpoint defects and cancer. Homozygous null mutations in spindle checkpoint genes are early embryonic-lethal. Heterozygous wild type/null animals are viable, but display increased aneuploidy. This illustrates an important point that partial loss of spindle checkpoint function is biologically significant. Although heterozygous checkpoint mouse models do not display a dramatic increase in spontaneous tumors, many models display an increase in tumors after carcinogen exposure. Overexpression of MAD2, which is commonly observed in sporadic tumors and can result from RB inactivation, causes extensive chromosomal abnormalities associated with a wide variety of tumors in mice.173,174 Over 50% of mice engineered to overexpress MAD2 exhibit tumors including hepatocellular carcinomas, lung adenomas, fibrosarcomas, and lymphomas. Furthermore, MAD2 is not required for tumor maintenance in this setting, as restoration of normal MAD2 levels had no effect on subsequent tumor progression. This observation is consistent with the idea that MAD2 overexpression triggers genetic instability during an early phase of tumor development, promoting subsequent self-sufficiency in tumor growth.
However, at least in theory, spindle checkpoint genes, including MAD2, may have additional functions outside mitosis, making it difficult to isolate a primary mitotic defect as the cause of tumor formation in these mice. This issue has been addressed by targeted deletion of CENP-E, a kinetochore-associated motor protein that is specifically expressed during mitosis. Heterozygous loss of CENP-E results in an age-dependent increase in aneuploidy in mice, with associated formation of splenic lymphomas and benign lung tumors.175 Nonetheless, CENP-E heterozygosity also inhibits tumor formation in the setting of p19ARF loss, suggesting that aneuploidy can act both oncogenically and as a tumor suppressor depending on the genetic context. The tumor-suppressing effect of aneuploidy likely reflects the fitness cost from gene expression imbalance.176,177 Consistent with this idea, mice with significantly reduced BUBR1 display an array of early aging phenotypes: reduced lifespan, cachectic dwarfism, muscle atrophy, cataracts, and infertility.177
An additional, facile way to accumulate whole-chromosome aneuploidy is via a tetraploid intermediate.178–180 Interestingly, many studies have linked tetraploidy to tumorigenesis. A number of cell-division defects can lead to cytokinesis failure and tetraploidy: errors in DNA replication or repair, or errors in spindle function and chromosome segregation. Mitotic defects lead to spindle checkpoint activation, but cells eventually recover and fail cytokinesis in the face of persistent chromosome segregation errors. This phenomenon is known as mitotic slippage. Recent work has also identified a pathway linking telomere damage to tetrapoloidy.181 Persistent telomere dysfunction in p53-deficient cells activates an ATM/ATR and Chk1/Chk2 signaling cascade that blocks entry into mitosis and extends G2. Eventually, the cells switch to a state resembling G1 in which the DNA replication inhibitor geminin, which prevents re-replication in G2, is degraded and Cdt1, which is required for origin licensing, is re-expressed. Thus, in the face of persistent telomere or other DNA damage, cells skip mitosis entirely and “endocycle” between G1 and S phase, producing tetraploid cells. Cell fusion is also an additional mechanism by which tetraploidy is generated.
It has been hypothesized that the presence of an additional complement of normal chromosomes may enhance fitness by buffering against the effects of deleterious mutations. In addition, tetraploidy itself may enhance genomic instability by the presence of extra centrosomes. As previously described, work has shown that extra centrosomes promote CIN and chromosome mis-segregation through a transient multipolar spindle intermediate.161,162 Further evidence for this tetraploid intermediate model has also come from the study of progressive dysplasia in Barrett’s esophagus, which reveals that early loss of p53 is correlated with the development of tetraploidy and subsequent aneuploidy.182 Moreover, experimental inhibition of cytokinesis in p53-null cells results in the generation of whole-chromosome aneuploidy, chromosome rearrangements, and rapid tumor formation in a mouse breast cancer model.179 Finally, recent studies suggest that genetic inhibition of cytokinesis or viral induction of cell fusion can promote tumorigenesis.
Finally, aneuploidy associated with APC loss has been shown to result from a combination of defects in mitosis and apoptosis that results in an early stage of tetraploidy and polyploidy.183,184 Given that APC loss occurs early in colorectal cancer development and is associated with the majority of the 85% of sporadic cancers with CIN, it is possible that genomic instability in this setting is related to the formation of a tetraploid intermediates.86 However, this remains an open issue because little genomic instability has been detected at early stages in APC-deficient mouse models.185 Finally, the mechanism by which MAD2 overexpression generates aneuploidy appears to involve mitotic slippage, resulting in the formation of tetraploid cells.173 Because RB pathway inactivation is fundamental to tumorigenesis, it is possible that associated up-regulation of MAD2 promotes aneuploidy more generally in cancer cells via the production of unstable tetraploid cells.
What is the Mechanism of Tumorigenesis?
There are multiple theories of how whole-chromosome aneuploidy could promote tumorigenesis. Extra copies of chromosomes could provide an advantage under certain selective pressures by increasing the expression of a single gene, or a combination of multiple genes on the aneuploid chromosome. This type of mechanism has been described for budding yeast to adapt to defects in cytokinesis, as well as the acquisition of drug resistance by Candida albicans.186,187 An alternative explanation for the tumor-promoting activity of aneuploidy is that the extra chromosomes buffer cancer cells against the effects of deleterious mutations in essential and haploinsufficient genes.
Another theory proposes that the driving force of tumorigenesis is the inherent instability of aneuploid karyotypes.169 According to this theory, a chromosome mis-segregation event, which is initiated by either a carcinogen or spontaneously, generates additional karyotypic evolution by destabilizing the proteins that segregate, synthesize, and repair chromosomes. Sporadically, such evolutions generate new cancer-causing karyotypes, which are stabilized by selection for their oncogenic function. Thus, in this model, CIN is generated in an autocatalytic fashion. A variation on this theme suggests that aneuploidy can cause protein imbalances that generate additional, non–whole-chromosome genomic instability, such as the acquisition of transforming mutations. In support of this model, structural chromosome abnormalities, such as nonreciprocal translocations, dicentric chromosomes, and double-minute chromosomes, are often noted alongside numerical chromosome abnormalities.179 It is unclear, though, whether these structural rearrangements are directly linked to the mis-segregation events.
Aneuploidy may also contribute to tumorigenesis through loss of heterozygosity. The possible roles for LOH in tumorigenesis include the duplication of oncogenes, loss of tumor suppressors, or acquisition of improper epigenetic patterns.77 As discussed earlier, it has been demonstrated in mice that Bub1 insufficiency can drive tumor formation through tumor suppressor gene LOH.78
PERSPECTIVES AND IMPLICATIONS FOR CANCER THERAPEUTICS
Oncogenesis represents an evolutionary process by which cells acquire successive genetic alterations that facilitate growth, survival, and ultimately properties that allow for dissemination to distant sites. Genomic instability can facilitate tumor development by accelerating the accumulation of such growth-promoting mutations, but this potentially comes with the cost of acquiring deleterious mutations that can impair fitness. Thus, the outcome of genomic instability can be cancer, but it can also be tissue degeneration, cell death, and aging. As detailed in this chapter, a wide variety of mechanisms can result in the generation of genomic instability. At one end of the spectrum, mutations in proteins such as the RecQ helicases, which play critical roles in normal genome maintenance, result in predominantly degenerative disease, manifesting premature aging phenotypes in addition to cancer development. On the other hand, mutations in mismatch repair genes, such as MLH1, MSH2, and BRCA, which have more limited and overlapping roles in the DNA damage response, result in normal development but a tissue-specific predisposition to cancer. Thus, destabilization of the genome can vary in the degree to which cancer promotion or tissue degeneration is favored. A major challenge for the field now is to elucidate the specific mechanisms and genetic interactions that tip the balance in one direction or the other.
Another important consideration is whether conditions leading to genomic instability are present in cancers at diagnosis or are transient, hit-and-run events. For example, inherited cancer syndrome mutations such as mismatch repair gene defects in HNPCC can speed up the acquisition of critical growth-promoting mutations, but, once transformed, the fitness of the tumor cells may be compromised by the ongoing mutator phenotype. Likewise, in sporadic tumors, loss of repair proteins such as ERCC1 in NSCLC may initially promote tumorigenesis, but the presence of these defects in mature tumors may provide a point of attack for certain chemotherapeutic agents. For instance, recent studies suggest the ERCC1-deficient lung tumors are more sensitive to the cytotoxic agent cisplatin.92 However, some genome-destabilizing events are transient. For instance, some FANC genes are methylated and silenced early in cancer progression, leading to genomic instability.188 However, later in tumorigenesis, these genes may be reactivated, resulting in a tumor with a stable genome. Also, although short telomeres can produce a crisis accompanied by gross chromosomal rearrangements, rampant aneuploidy is suppressed by telomerase re-expression. Similarly, cytokinesis failure and tetraploidy may be transient early events. If the major genome destabilization is transient, cancer cells may be aneuploid, but stably aneuploid. Indeed, it is fairly common for every cell in an aneuploid tumor to have the same abnormal karyotype; metastases and recurrences can have the same abnormal karyotype as the primary tumor. Ill-defined adaptations may enable many tumors to tolerate their altered genomes.
Understanding the mechanisms of genome destabilization that are operative in specific tumors will likely have important consequences for cancer therapeutics. Traditional cytotoxic chemotherapy combinations have largely been derived empirically. Many cytotoxic agents, such as platinum chemotherapies, induce cancer cell killing through DNA damage, with a therapeutic window that is relatively narrow. Tumor cell killing is at least in part correlated with p53 expression and the ability of cancer cells to undergo apoptosis in response to damaging agents. Indeed, the recent observation that restoration of wild type p53 function in mouse models of oncogenesis induces spontaneous tumor regression highlights the fact that some tumors become “addicted” to p53 loss.18,189 However, the heterogeneity of response within tumor types also suggests that genome-destabilizing mutations present in the cancer genome may sensitize certain subtypes to specific cytotoxic agents. The apparent sensitivity of ERCC1-deficient NSCLC to platinum-based chemotherapy highlights this point.92 Moreover, topoisomerase I inhibitors such as the camptothecins have been shown to have enhanced sensitivity in the setting of defects in the multiple protein components that respond to DSBs.190 Similarly, spindle checkpoint defects, if they become fixed in cancer cell populations, may modulate the response to microtubule-based agents such as taxanes and vinca alkaloids.191,192
This idea, in which defects in one pathway facilitate sensitivity to DNA-damaging agents, or alternatively, predispose to cell death in response to targeted inhibition of another pathway, relates to the concept of synthetic lethality.193,194 Originally defined in yeast, extension of this concept to targeted cancer therapy may ultimately result in improved selective cancer cell killing with a wider therapeutic window. An elegant example of this approach has been demonstrated in vitro for BRCA1- and BRCA2-deficient cells.93,94 Deficiency or inhibition of PARP1 in normal cells results in impairment of the BER response, causing lesions that would normally be repaired by BER to be channeled into the HR pathway. Exposure of cells lacking BRCA1 and BRCA2 to PARP inhibition results in the lethal accumulation of DNA damage. Thus, PARP inhibition appears to be selectively toxic for BRCA-deficient cancer cells, with potential efficacy in other contexts in which HR or even other types of DNA damage responses are impaired.
The profound sensitivity of BRCA mutant cells to PARP inhibition has led to the development of a number of clinical trials to test the efficiency of this approach.195 A recent phase 1 study reported that the orally active PARP inhibitor olaparib (AZD2281) is well tolerated and has few of the adverse effects associated with conventional chemotherapy.196 Furthermore, objective antitumor activity was reported in patients with BRCA1 or BRCA2 mutations, all of whom had ovarian, breast, or prostate cancer. PARP inhibitors are also being used in combination regimens, as inhibition of PARP can potentiate the effects of numerous DNA-damaging agents, such as temozolomide and irinotecan.
The concept of synthetic lethality has also been applied to tumors harboring mutations in PTEN, MSH2, VHL, and RAS.197–201 The work with RAS, for example, suggests that targeting the NF-kB signaling pathway might be one strategy to treat K-RAS mutant tumors.200 Furthermore, this concept might be further generalizable. For example, certain genes in yeast are required for the survival of polyploid cells, with deletion of these genes resulting in so-called ploidy-specific lethality.202 Identification of similar targets in human cancer cells may facilitate the design of targeted agents that selectively impair the growth of tumors with increased numbers of chromosomes or centrosomes.203
Finally, the advent of genomic technologies and large-scale characterization of cancer genomes will allow for a more refined view of carcinogenesis and enhanced subclassification of tumors. Knowledge of genome-destabilizing pathways that promote oncogenesis but impair fitness in specific tumors may eventually allow better tailoring of therapies in individual patients.
Selected References
The full list of references for this chapter appears in the online version.
1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100:57.
4. Weinberg R. The Biology of Cancer. New York: Garland Science; 2006.
10. Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature 2004;432:316–323.
16. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009;9:153–166.
23. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009;461:1071–1078.
32. Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 2010;10:51–57.
36. Bartkova J, Rezaei N, Liontos M, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006;444: 633–637.
38. Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature 2001;410:1111–1116.
46. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature 2009;458:719–724.
49. Greenman C, Stephens P, Smith R, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007; 446:153–158.
50. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008;455: 1061–1068.
53. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009;462: 739–744.
55. Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010;17:225–234.
56. Ley TJ, Mardis ER, Ding L, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008;456:66–72.
59. Pleasance ED, Cheetham RK, Stephens PJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010;463:191–196.
60. Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009;10:57–63.
63. Maher CA, Kumar-Sinha C, Cao X, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature 2009; 458:97–101.
68. Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med 2009;60:167–179.
69. Lin C, Yang L, Tanasa B, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009;139:1069–1083.
72. Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010;463:899–905.
75. Tuna M, Knuutila S, Mills GB. Uniparental disomy in cancer. Trends Mol Med 2009;15:120–128.
78. Baker DJ, Jin F, Jeganathan KB, van Deursen JM. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell 2009;16:475–486.
80. McKenna ES, Roberts CW. Epigenetics and cancer without genomic instability. Cell Cycle 2009;8:23–26.
82. Sharma SV, Lee DY, Li B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010;141:69–80.
89. Cleaver JE, Lam ET, Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet 2009;10:756–768.
92. Olaussen KA, Dunant A, Fouret P, et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med 2006;355:983–991.
94. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434:913–917.
100. Moldovan GL, D’Andrea AD. How the Fanconi anemia pathway guards the genome. Annu Rev Genet 2009;43: 223–249.
105. Knipscheer P, Raschle M, Smogorzewska A, et al. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 2009;326:1698–1701.
110. Chu WK, Hickson ID. RecQ helicases: multifunctional genome caretakers. Nat Rev Cancer 2009;9:644–654.
121. Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 2003;4:712–720.
127. Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis 2010;31:9–18.
128. Artandi SE, Chang S, Lee SL, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000;406:641–645.
130. Maser RS, Choudhury B, Campbell PJ, et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature 2007;447:966–971.
139. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature 1997;386:623–627.
150. Schvartzman JM, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer 2010;10:102.
151. Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008;14:111–122.
157. Liu D, Vader G, Vromans MJ, et al. Sensing chromosome bi-orientation by spatial separation of aurora B kinase from kinetochore substrates. Science 2009;323:1350–1353.
161. Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009;460:278–282.
162. Silkworth WT, Nardi IK, Scholl LM, Cimini D. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One 2009;4:e6564.
165. Cimini D, Howell B, Maddox P, et al. Merotelic kinetochore orientation is a major mechanism of aneuploidy in mitotic mammalian tissue cells. J Cell Biol 2001;153: 517–527.
175. Weaver BA, Silk AD, Montagna C, et al. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 2007;11:25–36.
177. Williams BR, Prabhu VR, Hunter KE, et al. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 2008;322:703–709.
179. Fujiwara T, Bandi M, Nitta M, et al. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005;437:1043–1047.
189. Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009; 136:823–837.
193. Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 2005;5: 689–698.
195. Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol 2008;26:3785–3790.
200. Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009;462:108–112.
201. Scholl C, Frohling S, Dunn IF, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell 2009;137:821–834.
202. Storchova Z, Breneman A, Cande J, et al. Genome-wide genetic analysis of polyploidy in yeast. Nature 2006;443: 541–547.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


