Malignant melanoma
Justin M. Ko, MD, MBA, FAAD  Susan M. Swetter, MD
Susan M. Swetter, MD  Jonathan S. Zager, MD
Jonathan S. Zager, MD  Vernon K. Sondak, MD
Vernon K. Sondak, MD  Scott E. Woodman, MD, PhD
Scott E. Woodman, MD, PhD  Kim A. Margolin, MD
Kim A. Margolin, MD
Overview
Melanoma comprises a wide variety of malignant cell types arising from the skin, the mucous membranes, and the pigmented cells of the eye. While these tumors are all classified as melanoma and share a common molecular biology of pigmentation and biological resemblance to cells of neural crest origin, important distinctions in other molecular characteristics and patterns of exposure to ultraviolet light as a carcinogen determine their clinical natural history, including the response to therapeutic interventions. While the majority of melanomas are diagnosed at an early stage and curable with minimal surgery, melanoma has the potential for early and widespread dissemination via lymphatic and hematogenous routes. Surgery remains the mainstay of therapy for primary, regional, and many cases of single- or oligo-metastatic disease, but systemic therapies have dramatically improved the prognosis for metastatic melanoma, particularly immunotherapies that enhance existing cellular immunity. The rapid discovery of new molecular targets, immunotherapy combinations (including the use of radiotherapy), and understanding of the mechanisms of therapeutic resistance are likely to lead to even greater improvements in the prognosis for patients with melanoma in the near future.
Dermatologic principles in melanoma
Epidemiology and etiology
Melanoma incidence and mortality rates continue to rise worldwide, driven by increased ultraviolet (UV) light exposure (both natural and artificial sources) and differences in prognosis according to age and sex. In the United States, 76,380 new cases of invasive cutaneous melanoma will be diagnosed and approximately 10,130 deaths will occur in 2016.1 Incidence has been increasing steadily for the past 30 years, and since 2004, Caucasians have experienced an increase in incidence by 3% each year, faster than that of nearly all cancers.2 Western states with increased ultraviolet radiation (UVR) have a greater incidence.3 Incidence rates are rising more than twofold among young women (ages 15–39) and increasing even more sharply among middle-aged and older men.3, 4
The increase in melanoma incidence has been attributed to factors including increased intermittent UVR exposure in fair-skinned populations, higher rates of skin biopsies and screening resulting in the detection of thinner, more favorable lesions, and potential changes in the histologic interpretation of early evolving lesions from atypical melanocytic hyperplasia or severely dysplasia to melanoma in situ.5, 6 However, continued increases in the incidence rates of thicker tumors, including steep rises among individuals of lower socioeconomic status, points to a true increase in potentially fatal cases and lack of screening as a critical factor accounting for recent melanoma incidence trends.7
Risk factors
Environmental factors
The risk of developing melanoma is related to acute, intense, and intermittent exposure to UV light, but the relationship between UV exposure and melanomagenesis is not straightforward. Melanomas frequently occur in locations often covered by clothing, and indoor workers have been shown to have a higher incidence of melanoma on sun-exposed sites, observations that provide support for the notion that melanomas arise through different molecular pathways.8, 9 Melanoma incidence rates on sun-exposed and less exposed body areas also have different age peaks,10 around 55 years for melanomas on intermittently exposed sites such as the trunk and proximal extremities, which probably reflects a period of vulnerability to UV radiation early in life and a latency to full melanomagenesis followed by a decline in the incidence of melanomas attributable to those mechanisms.11 Conversely, melanomas on chronically exposed sites such as the face and distal extremities continue to rise with age. Signature DNA mutations induced by UV are found commonly in driver mutations identified in melanoma12 but not as often as in nonmelanoma skin cancers that are more directly related to chronic sun exposure, so alternate sources of mutagenesis are likely. Nevertheless, a randomized study in Australia showed that consistent daily application of both UVA- and UVB-filtering broad-spectrum sunscreen (compared with discretionary or nonuse) resulted in a decreased incidence of melanoma.13 Indoor tanning is now proven to be a major contributor to the increasing incidence of cutaneous melanoma among young women, with greater melanoma risk proportional to measures of tanning practices, including increasing years, hours, and sessions of indoor tanning.14, 15 Alarmingly, 76% of melanomas in fair-skinned participants were attributed to tanning bed use at young ages. A strong association of tanning bed use with recreational drugs,16 suggesting a common genetically mediated addiction, is consistent with animal studies showing sunlight-seeking behaviors mediated by opioid-related substances through endorphin receptors.17
Host factors
Although the process by which normal melanocytes transform into melanoma cells is not entirely understood, it is believed to involve progressive genetic mutations that alter cell proliferation, differentiation, and death and impact cellular susceptibility to the carcinogenic effects of UVR.
Melanoma is largely a disease of individuals of fair complexion, including those with red or blond hair and fair skin, who burn easily or have a history of severe sunburn, or who are unable to tan.18 Caucasians with an increased number of nevi or a tendency to freckle are also at increased risk for developing melanoma.19 The risk of cutaneous melanoma in the presence of increased numbers of common/typical nevi, large nevi, and/or clinically atypical nevi (CAN) on the body was confirmed in a pooled analysis of melanocytic nevus phenotype, even at different latitudes.20 Both prior personal history and family history are important risk factors for the development of melanoma.21 Solid-organ transplant populations are at far greater risk of squamous cell carcinomas and also develop more melanomas compared to the general population.22
Genetic predisposition and familial melanoma
The majority of melanomas appear to be sporadic, with only 5–10% of cases attributable to an identified familial predisposition.23 Familial melanoma is characterized by an increased risk of developing melanoma, a higher incidence of multiple primary melanomas, and typically an earlier age at onset.24 Specific genetic alterations have been implicated in the pathogenesis of familial melanoma. Mutations at the CDKN2A locus on chromosome 9p21, which codes for the tumor suppressor p16 and p14/ARF, account for about one-third of familial cases. Additional melanoma risk occurs in CDKN2A mutation carriers who express variants of the melanocortin receptor gene MCR1, which is associated with red hair, fair skin, and freckling. As mutations of either of these genes are present in only a subset of familial melanoma kindreds, other melanoma susceptibility genes likely exist. Formal recommendations for p16 mutation testing have been proposed in patients with a personal or family history of 3 or more invasive melanomas or cancer “events,” defined as 2 invasive melanomas and 1 pancreatic cancer in the patient or family members, or vice versa, which conveys a >20% risk of carriage.25 However, owing to the low frequency of mutations even among high-risk individuals and the lack of implications for dermatologic surveillance, genetic testing other than for research purposes is generally not recommended.
Atypical mole syndrome/phenotype
The presence of numerous CAN, also termed “dysplastic nevi,” is the most important clinical risk factor for melanoma. Compared with the general population, patients with CAN have a 2- to 15-fold elevated risk of developing melanoma, and risk increases with the number of CAN and/or personal or family history of melanoma.26 An atypical mole phenotype is characterized by numerous (>50–100) common nevi along with multiple (generally >5), large (>6–8 mm) nevi with color variegation, border irregularity, and asymmetric shape. Melanomas seldom arise in association with pre-existing atypical nevi, and over 70% of melanomas in patients with any type of melanocytic nevus (common, atypical, or congenital) are believed to develop de novo, although CAN with severe histologic dysplasia may more often be true melanoma precursors.27 Melanomas associated with atypical/dysplastic nevi are generally thin superficial spreading type, possibly due to increased skin surveillance in affected individuals.
Congenital melanocytic nevi (CMN)
Congenital melanocytic nevi (CMN) are evident in 1–6% of neonates and uncommonly transform into melanoma.28 Patients with “large or giant” congenital nevi (lesions >20 cm in diameter in an adult, >6 cm on the body of an infant, or >9 cm on the head of an infant) have a less than 5% lifetime risk of developing a melanoma29–32 with about half arising during the first few years of life.33 The risk of melanoma arising within small-sized (<1.5 cm) and medium-sized CMN is low and virtually nonexistent before puberty.34 Management of CMN hinges on many variables including ease of monitoring and potential psychosocial benefits and harms of surgical procedures.
Melanoma risk assessment
Several risk assessment tools have been used to target individuals at high risk for melanoma, in particular Caucasian men over 65 and individuals of lower socioeconomic status—two groups with the highest melanoma mortality.
Current risk assessment tools are derived from a large case–control study of 718 non-Hispanic white patients and 945 controls that involved inspection of the back for suspicious moles and asked two questions about complexion and history of sun exposure.35 Mild freckling and light complexion were demonstrated as risk factors for both men and women. In addition, >17 small moles and ≥2 large moles in men or ≥12 small moles on the backs of women were also significant risk factors. These data led to the Melanoma Risk Assessment Tool, which is available from the National Cancer Institute (http://www.cancer.gov/melanomarisktool/). The tool calculates absolute risk of melanoma over the next 5 years up to age 70.
Prognostic factors
A number of clinical factors affect patient prognosis including age, gender, and anatomic location of the primary tumor. In general, men, older age individuals, and those with melanoma on the head and neck tend to fare worse. A population-based study in France during 2004–2008 showed that male patients had thicker and more frequently ulcerated tumors. Older patients had thicker and more advanced melanomas, with more frequent head and neck location.36
While newer concepts in the taxonomy of melanoma suggest distinct molecular, genetic, anatomic, and UV-exposure-linked characteristics, growth kinetics of certain histological subtypes also appear to play a role in prognosis. The rapidly growing nodular subtype of melanoma tends to elude early detection based on clinical characteristics alone. Nodular melanoma (NM) comprises <15% of subtyped melanoma cases in the United States and Australia, but accounts for a disproportionate number of thicker tumors (>2 mm) and melanoma deaths compared with other histologic subtypes.
Newer molecular techniques such as gene expression profiling may soon assist in identifying thin melanomas with more aggressive behavior.37
Clinical presentation
Cutaneous melanoma can occur anywhere but is most common on the lower extremities and back in women and on the trunk in men. From a clinical standpoint, a new or changing “mole” or skin lesion is the most-common warning sign for melanoma. The so-called “ABCDEs” of early diagnosis pioneered by Rigel et al.38 are an easy mnemonic to improve recognition of the classic early signs of melanoma (Table 1). To simplify further, Weinstock39 succinctly focused the message by emphasizing that the most important warning sign is a new or changing skin lesion. The “ugly duckling” warning sign refers to a pigmented or clinically amelanotic lesion that looks different from the rest, which may be of value to identify melanomas that lack the classic ABCD criteria (e.g., nodular, amelanotic, or desmoplastic subtypes).40, 41
Table 1 ABCDEs: clinical features of melanoma
| A | Asymmetry—the two halves of the lesion do not match each other |
| B | Border irregularity—may appear ragged, notched or scalloped |
| C | Color variation—color is not uniform or lesion may be many colors displaying shades of tan, brown, or black. White, reddish, or blue-gray discoloration is of particular concern |
| D | Diameter—usually >6 mm (roughly the diameter of a pencil eraser) although melanomas may be smaller in size; any growth in a nevus warrants an evaluation |
| E | Evolving lesion—changes in size or color; critical for nodular or amelanotic melanoma, which may not exhibit the ABCD criteria above |
As histologic features of a primary melanoma are critical for melanoma staging and prognostication, proper initial biopsy of a suspicious lesion is paramount. An excisional biopsy with narrow clinical margins (1–3 mm) of normal-appearing skin around the pigmented lesion is preferred when possible to provide accurate diagnosis and histologic microstaging. An important exception to this rule is the lentigo maligna (LM) subtype of melanoma in situ, in which the risk of misdiagnosis is high if small or partial biopsy specimens are taken. The best diagnostic biopsy technique in this case is often a broad shave biopsy that extends into at least the papillary dermis, provides the opportunity to exclude microinvasive melanoma, and allows for optimal histopathologic interpretation of the tumor.
Pathologic features
With the exception of NM, the growth patterns of the other clinicopathologic subtypes are characterized by a preceding in situ (radial growth) phase that lacks the biologic potential to metastasize and may last from months to years before dermal invasion (vertical growth) occurs.
Dermal invasion confers metastatic potential, although the greatest risk occurs in the setting of a vertical growth (tumorigenic) phase.42 Immunohistochemical staining for lineage [S-100, human melanoma black 45 (HMB-45), melan-A/Mart-1] or proliferation markers (Ki67) may be helpful in some cases for histologic differentiation from melanoma simulators such as melanocytic nevi, Spitz nevi, cellular blue nevus, clear cell sarcoma, or malignant peripheral nerve sheath tumor.43 A study of melanoma biomarker expression in melanocytic tumor progression examined differential expression of melanoma biomarkers between nevi, primary melanoma, and metastases.44 Approaches combining Ki67/Anti-MART-1 (Melan-A) and HMB-45/MITF immunostains have also been shown to hold promise in the diagnosis of melanoma.45
A pathology report for melanoma should include cytomorphology and architecture, along with tumor thickness (Breslow depth), presence of ulceration, dermal mitotic rate (measured as number per mm2), and microsatellites and lymphovascular invasion, if present. Anatomic level of invasion (Clark’s level) has less prognostic significance and is now considered optional in pathology reporting.
Atypical melanocytic lesions in children and adolescents in particular may be difficult to distinguish from true melanoma, including atypical Spitz tumors and Spitz or melanocytic tumors of uncertain malignant potential (STUMPs or MelTUMPs, respectively). As such, molecular techniques such as comparative genomic hybridization (GGH) and fluorescence in situ hybridization (FISH) have been utilized to assist in the determination of malignant versus benign behavior.46–49 Newer gene expression profiling techniques may be of further value of molecular testing for challenging atypical melanocytic neoplasms and to aid in prognosis and risk stratification,50–53 and some of these findings may yield prognostic data sufficiently robust to support their inclusion in future melanoma staging systems.
Clinicopathologic subtypes
The four major classical “histogenetic” subtypes of primary cutaneous melanoma are based on histopathologic findings, anatomic site, and degree of sun damage. These include superficial spreading melanoma, NM, lentigo maligna melanoma (LMM), and acral lentiginous melanoma. In addition, there are other rarer variants (<5% of melanomas), which include (1) desmoplastic/neurotropic melanoma, (2) mucosal (lentiginous) melanoma,53 (3) blue nevus-like melanoma, (4) melanoma arising in a giant/large congenital nevus, and (5) melanoma of soft parts (clear cell sarcoma).
Superficial spreading melanoma
Superficial spreading melanoma accounts for nearly 70% of cutaneous melanoma, commonly displays the ABCDE signs, and is the most-common subtype in individuals aged 30–50 years, as well as those with atypical/dysplastic nevi. It is most common on the trunk in men and women and on the legs in women.
Nodular melanoma
NM is the next most-common melanoma subtype, and occurs in 15–30% of patients, most commonly on the legs and trunk in men and women. It typically presents as a dark brown-to-black papule or dome-shaped nodule with rapid growth over weeks to month, which may ulcerate and bleed with minor trauma. This subtype is responsible for most thick melanomas at diagnosis.54, 55
Lentigo maligna melanoma
LMM incidence is rising in the United States.56 It is typically located on chronically sun-damaged skin (head, neck, and arms) of fair-skinned older individuals (average age 65 years), slowly growing over years to decades. The in situ precursor lesion termed LM is typically a longstanding large >1–3 cm flat (macular) lesion, demonstrating pigmentation ranging from dark brown to black, although white or hypopigmented areas are common within LM. Dermal invasion denoting progression to LMM is characterized by the development of raised brown-black nodules within the in situ lesion.
Acral melanoma
Acral melanoma is the least-common subtype of melanoma in white persons (2–8% of melanoma cases), although the most-common subtype of melanoma in darker-complexioned individuals (i.e., African American, Asian, and Hispanic persons), representing 29–72% of melanoma cases in these populations. Because of delays in diagnosis, it may be associated with a worse prognosis.57, 58 Acral lentiginous melanoma occurs on the palms, soles, or beneath the nail plate (subungual melanoma), which may manifest as diffuse nail discoloration or a longitudinal pigmented band (melanonychia striata) within the finger or toenail. Pigment spread to the proximal or lateral nail folds is termed the Hutchinson sign, which is a hallmark for subungual melanoma.
Less-common subtypes
Desmoplastic melanoma is a less common but important melanoma subtype, given its predilection for older age individuals, clinical features similar to nonmelanoma (keratinocytic) skin cancer, and potential indication for adjuvant radiation therapy following wide excision (depending on tumor thickness, perineural invasion, and margin status). Amelanotic melanoma (<5% of melanomas) can occur with any subtype and often mimics basal cell or squamous cell carcinoma, dermatofibroma, or a ruptured hair follicle. It occurs most commonly in the setting of the nodular or desmoplastic subtype or melanoma metastasis to the skin, presumably because of the inability of these poorly differentiated cancer cells to synthesize melanin pigment.
Genetics and molecular pathology
Significant complexity exists in the clinical and histopathological presentation, cells of origin (epithelium associated versus nonepithelium associated), causative relationship to UV radiation, age of onset, somatic mutations, and germline genetic predisposition. Melanoma is not a homogeneous disease but instead is composed of biologically distinct subtypes, the phenotype of which is driven by underlying genetic alterations.59
One growth factor pathway that has garnered considerable attention as related to melanoma is the RAS–RAF–MAPK–ERK (mitogen-associated protein kinase, MAPK) signaling cascade. A unique mutation at position 1799 of the gene for the serine–threonine kinase BRAF in the MAPK pathway—occurring in about half of cutaneous melanomas—results in a substitution of glutamine (about 75–80% of cases) or lysine for valine (most of the remaining cases) at position 600 which confers constitutive activity, resulting in hyperproliferation and resistance to apoptosis among cells driven by this oncogenic mutation.60 The biology of melanoma in cells dependent on a BRAF mutation is also impacted by coexisting mutations or other alterations of gene expression in linked pathways, particularly the PTEN/AKT/PI3K/mTOR pathway that is critical in the control of metabolic sensors and the cancer/microenvironment nutrient balance. Activating mutations of NRAS, as well as less-common oncogenes such as c-kit (covered in greater detail below), result in downstream activation of both of these pathways.
Most BRAF-mutated melanomas arise in intermittently sun-exposed skin, while melanomas from chronically sun-exposed areas have a lower incidence of BRAF mutations and occasionally carry a mutation or amplification of c-KIT, a receptor tyrosine kinase that61, 62 may be altered in about 15–20% of acral and mucosal melanomas.61–63 Activation of c-kit results in the stimulation of both the MAPK and PI3K–AKT pathways, producing both proliferative and survival advantages.64
The observation that BRAF mutations noted in benign melanocytic nevi65 typically arising by early adolescence occurred with the same frequency as in non-CSD melanoma led Bastian and colleagues to propose that patients with acquired nevi and non-CSD melanomas may have a particular susceptibility for developing BRAF-mutated melanocytic neoplasms at relatively low doses of UV radiation. This concept was supported in subsequent studies that demonstrated a germline variation in the melanocortin receptor 1 (MC1R) to significantly contribute to this susceptibility.66, 67 More specifically, variants of MC1R were shown to strongly increase the risk for non-CSD melanomas with BRAF mutation.
None of the oncogenes or tumor suppressor genes identified in melanoma are thought to be solely responsible for melanoma pathogenesis, and some appear to be mutually exclusive due to overlapping downstream functions. For instance, NRAS, as mentioned above, activates Raf kinases in response to growth factor receptor activation and harbors activating mutations in 15–20% of melanomas, but almost never occurs with a BRAF mutation.67 The loss of the p16 tumor suppressor is a relatively frequent event in melanoma, and there is significant overlap with BRAF mutation.68 PTEN mutations and deletion have been described in a minority of melanomas and appear to coincide with BRAF mutation (Figure 1).69
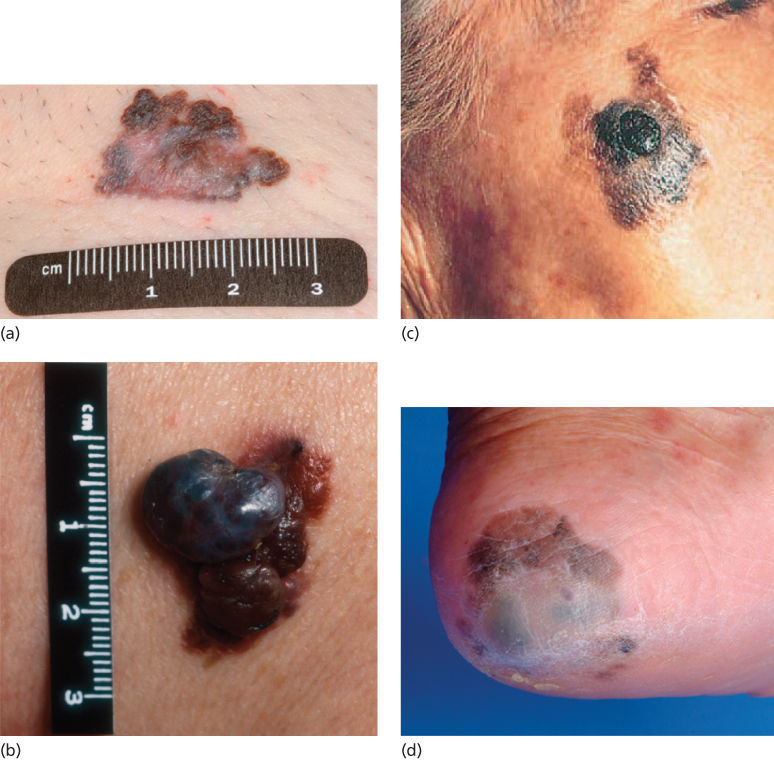
Figure 1 (a) Superficial spreading melanoma. Note the irregular borders, a variegation in color, and size >6 mm. (b) Nodular melanoma. (c) LMM with a nodular area of accelerated growth. (d) Acral lentiginous melanoma of heel.
Source: Courtesy of Jeffrey E. Gershenwald, MD.
Surgical management of melanoma
Management of primary cutaneous melanoma
Surgery remains the mainstay of treatment for primary cutaneous melanoma, and the specific recommendations have not changed significantly over the last decade. Primary melanoma is treated by wide excision with a defined margin of adjacent normal-appearing skin. The margin of resection is dependent on the Breslow depth of the tumor and the site of the primary. Margin widths are measured from the biopsy scar or residual pigment at the time of surgery; it is not expected to equate to a histopathologic measurement on the resection specimen. A histologically negative margin, however, is always the goal in excising the primary tumor. Current recommendations for width of the excision in invasive melanoma (Table 2) are supported by randomized trials, summarized in Table 3.71–74 For melanoma in situ, the recommended margin is 0.5–1 cm.75 For invasive melanomas ≤1 mm in depth, 1 cm margins are recommended and are associated with low rates of local recurrence. For melanomas between 1 and 2 mm, 1–2 cm margins are recommended, taking into consideration cosmetic or functional outcome. For melanomas >2 mm, 2 cm margins are recommended, whenever feasible.75 A meta-analysis found that margins >2 cm are unnecessary even for thick primaries, and margins should not be <1 cm for any invasive melanoma.76
Table 2 Recommended margins of wide excision for cutaneous melanoma based on primary tumor thickness and location
| Breslow thickness | Primary site | Recommended excision margin (cm) |
| Melanoma in situ | Anywhere on the skin | 0.5–1 |
| 0.01–1.00 mm | Anywhere on the skin | 1 |
| 1.01–2.00 mm | Head/neck, distal extremitya | 1 |
| Trunk or proximal extremityb | 2 | |
| >2.00 mm | Head/neck, distal extremitya | 1 |
| Trunk or proximal extremity | 2 |
a Subungual primary tumors may require distal digital amputation.
b If a skin graft would be required to reconstruct the excision defect, it is acceptable to take a 1 cm excision margin.
Note: Local anatomic constraints and specific patient factors may justify minor deviations from the standard margin recommendations.
Adapted from Sondak and Gibney 2014.70
Table 3 Summary of ASCO-SSO recommendations for sentinel lymph node biopsy in melanoma
|
SLNB, sentinel lymph node biopsy.Adapted from Wong et al, 2012.85
Management of the regional lymph nodes
The role of sentinel lymph node biopsy
The presence of occult tumor deposits within clinically negative lymph nodes (defined by the AJCC staging system as “microscopic” disease) is a key predictor of outcome in clinical stage I and II melanoma,77 and evidence suggests even tiny nodal micrometastases have clinical relevance.78, 79 Melanoma micrometastases in the regional lymph nodes cannot reliably be detected by any imaging modality including PET–CT80, 81 or ultrasonography.82, 83 Sentinel lymph node biopsy (SLNB) can identify micrometastases with low morbidity.84 In 2012, an evidence-based assessment of the indications for SLNB in melanoma was issued jointly by ASCO and the Society of Surgical Oncology (SSO) (Table 4).85
Table 4 Summary of selected final results of the Multicenter Selective Lymphadenectomy Trial 1 (MSLT-1)
| Result | Comment | |
| Feasibility | At least one sentinel node was identified in 99.5% of patients undergoing SLNB | SLNB highly feasible in a worldwide experience |
| Yield and false-negative rate | The sentinel node was positive in 19% of patients with melanomas ≥1.2 mm: 16% for melanomas 1.2–3.5 mm and 33% for melanomas >3.5 mm Nodal recurrence occurred in 5.9% of patients with a negative sentinel node: 4.8% for melanomas 1.2–3.5 mm and 10.3% for melanomas >3.5 mm | SLNB is an effective staging procedure with an acceptable false-negative rate |
| Prognostic significance | A positive sentinel node was associated with ∼2.5-fold increases in disease recurrence and death from melanoma for melanomas 1.2–3.5 mm; 10-year melanoma-specific survival was 85% for patients with a negative sentinel node versus 62% for patients with a positive sentinel node | Sentinel node status is the strongest known prognostic indicator in clinically node-negative intermediate-thickness melanoma |
| Survival impact of SLNB | There was a nonsignificant 3% increase in 10-year melanoma-specific survival for intermediate-thickness melanoma patients randomized to SLNB versus observation | SLNB does not significantly affect survival for all patients subjected to the procedure |
| Relapse-free survival impact | Patients randomized to SLNB had a statistically significant improvement in relapse-free survival compared to observation | SLNB significantly reduces melanoma recurrence for intermediate and thick melanomas, mostly by decreasing subsequent nodal relapse |
| Impact on node-positive patients | Patients with intermediate-thickness melanoma and positive nodes who were randomized to SLNB (with completion lymphadenectomy) had statistically significantly improved distant metastasis-free survival and melanoma-specific survival compared to observation arm patients who failed clinically in the regional nodal basin; there were no significant differences for patients with thick melanomas and positive nodes between the SLNB and observation arms | Early treatment of intermediate-thickness node-positive melanoma by radical lymphadenectomy improves outcomes significantly; patients with thick node-positive melanomas may be at such high risk of distant disease that timing of lymphadenectomy loses importance |
SLNB, sentinel lymph node biopsy.
Adapted from Sondak and Gibney 2014.70 Original data from Morton.
The panel found that the strongest evidence supported SLNB for patients with intermediate-thickness melanomas, based on interim results from the prospective randomized Multicenter Selective Lymphadenectomy Trial I (MSLT-1).77 Key ASCO–SSO SLNB recommendations are summarized in Table 3. The mature results of MSLT-1 (Table 5)86 and retrospective institutional series89–92 also support SLNB for patients with clinically node-negative thick melanomas (>4 mm). Controversy remains regarding the use of sentinel node biopsy in patients with thin melanomas (<1 mm), which were not adequately assessed in the MSLT-1 study nor included in the prospective but nonrandomized Sunbelt Melanoma Trial.93–95
Table 5 High-risk features for selecting T1 melanomas for sentinel lymph node biopsy
| High-risk criterion | Impact on likelihood of finding a positive node | Comment |
| Thickness 0.76–0.99 mm | In a registry series of 1250 patients with melanomas ≤1 mm selected by a wide variety of criteria to undergo SLNB, metastases were detected in 6.3% of 891 melanomas ≥0.76 mm but in only 2.5% of 359 melanomas ≤0.75 mm. No metastases were detected in sentinel nodes from patients with melanomas <0.5 mm.87 In a large contemporary single-institution experience from a center where SLNB was routinely offered to patients with melanoma ≥0.76 mm without requiring any other high-risk feature to be present, 8.4% of patients had a positive sentinel node86 | Most patients with thin melanomas and a positive sentinel node are found in this upper end of the T1 thickness spectrum; very few unselected patients with melanomas <0.76 mm are at sufficient risk of nodal metastasis to justify SLNB |
| Ulceration | 18.3% of patients in a registry series87 and 23.5% of patients in a single-institution series88 who had ulcerated T1 melanomas had a positive sentinel node | Relatively rare finding in T1 melanomas (present in <10% of cases) but perhaps the highest risk factor for sentinel node positivity in thin melanoma |
| Mitotic count | Mitotic count ≥1/mm2 was not predictive of nodal metastases in a registry series87 but was predictive in a single-institution series88 | Presence of even one dermal mitosis in a T1 melanoma upstages the tumor from T1a to T1b in the current AJCC staging system. It remains unclear whether the presence of dermal mitoses is sufficient to justify SLNB, especially in melanomas <0.76 mm |
| Patient age | — | Younger patients have a higher risk of positive sentinel nodes across all tumor thickness categories as well as more years at risk for nodal recurrence |
| Clark level | Clark level was a significant predictor of sentinel node status in a registry series87 but not in a single-institution series88 | Clark level IV melanomas are more likely to be at the thicker end (≥0.76–1.00 mm) of T1 where most nodal metastases are encountered. The value of SLNB for Clark IV melanomas <0.76 mm has not been demonstrated |
SLNB, sentinel lymph node biopsy.
Data from Han 201287 and Han 2013.88
Today, the majority of newly diagnosed cutaneous melanomas are T1 lesions (≤1 mm in thickness), with a low overall risk of nodal metastasis or death from melanoma.96 Recommending sentinel node biopsy for all patients with T1 melanomas is not cost-effective.95 ASCO–SSO recommends that SLNB for thin melanoma be considered in selected cases with “high-risk” features (Table 6).85
Table 6 Summary of follow-up guidelines for patients with completely resected melanoma from various national and international organizations. These guidelines have not been prospectively validated and hence should only be considered as recommendations
| Stage | Physical examination | Imaging |
| Stage I | Every 3–12 months × 5 years with annual follow-up beyond 5 years as clinically indicated | No imaging needed unless clinically indicated |
| Stage II | Every 3–6 months × 2 years, then every 3–12 months for an additional 3 years with annual follow-up beyond 5 years as clinically indicateda | Cross-sectional body imaging considered for higher risk patients: CT and/or PET/CT, to screen for recurrent disease. Consider brain MRI annuallyb |
| Stage III–IV | Every 3–6 months × 2 years, then every 3–12 months for an additional 3 years with annual follow-up beyond 5 years as clinically indicatedc | Cross-sectional body imaging should be considered for higher risk patients: CT and/or PET/CT, to screen for recurrent disease. Consider brain MRI annuallyb |
a Consider more frequent and longer follow-up in high-risk stage II patients with thick and/or ulcerated primary tumors.
b Ultrasonography can be considered to evaluate regional nodal basins.
c Consider more frequent and longer follow-up in high-risk stage IIIb/c patients.
Abbreviations: CT, computed tomography; CXR, chest X-ray; PET/CT; positron emission tomography/computed tomography.
Adapted from Fields and Coit 2011.145
On the basis of the large retrospectively collected registry data, SLNB seems well justified for many patients with melanomas 0.76–1.00 mm, but not for the majority of patients with melanomas <0.76 mm in thickness.96 In addition, patient age, preference, and comorbidities need to be considered in the decision regarding SNLB.
A few other clinical situations pertaining to the use of SLNB deserve mention. Desmoplastic melanomas in general appear to have a lower risk of nodal metastases,97, 98 and some authors advocate abandoning SLNB in this histologic type.99 In recent series, the risk of nodal metastases has been shown to be high enough to justify routine consideration of SLNB in all patients with desmoplastic melanomas ≥1 mm in thickness.99 Pediatric melanoma patients have a higher incidence of nodal metastases, yet an apparent better overall prognosis compared to adults, and the role of SLNB in these patients remains controversial, especially for the so-called atypical melanocytic proliferations of childhood.100–103
Completion lymphadenectomy for sentinel node-positive disease
NCCN guidelines,75 strongly supported by decades of clinical experience, call for routine performance of a therapeutic lymphadenectomy in all melanoma patients with clinically positive nodes and no radiographic evidence of distant metastases. The routine use of completion lymphadenectomy after a positive SLNB is more controversial,104 although it is recommended by the ASCO/SSO guidelines as standard of care in the absence of clinical trial participation.85 The MSLT-1 trial showed that outcomes for patients with intermediate-thickness melanoma undergoing completion lymphadenectomy were superior to those for patients who recurred in the nodal basin,86 but by the nature of the trial, the contribution of the completion lymphadenectomy over and above removal of the sentinel node could not be assessed. Nonsentinel nodes are only found to have tumor involvement in a minority of cases,107 and at least some sentinel node-positive patients do well for an extended period of time even without undergoing completion lymphadenectomy.86 The morbidity of lymphadenectomy, especially severe lymphedema, has been shown to be less for completion lymphadenectomy after a positive SLNB than for therapeutic lymphadenectomy after nodal recurrence.109 A prospective randomized trial comparing ultrasound surveillance of the lymph node basin to immediate completion lymphadenectomy for sentinel node-positive patients, MSLT-2, recently completed accrual with results pending (clinicaltrials.gov NCT00297895).
Radical lymphadenectomy for macroscopic nodal disease
In contrast to the situation for SLNB identified microscopic disease, a therapeutic radical lymph node dissection is virtually always indicated for clinically evident, resectable nodal metastasis; systemic therapy and/or radiation is not an adequate substitute for surgical treatment of the clinically positive nodal basin.75
Management of in-transit and locoregional recurrent melanoma
Wide excision is indicated for biopsy-proven local, satellite, or in-transit recurrence in the absence of distant disease. Full radiographic staging (with PET/CT or CT and brain MRI or CT) should be performed before curative-intent excision of the recurrent tumor.
Intra-arterial regional perfusion therapies
Hyperthermic isolated limb perfusion (HILP) and isolated limb infusion (ILI) are methods by which an extremity with unresectable locally recurrent or in-transit metastatic melanoma is isolated from systemic circulation and high-dose chemotherapy administered intra-arterially with limited systemic exposure. HILP is performed by directly dissecting and cannulating the major vessels of the extremity and circulating chemotherapy via a cardiopulmonary bypass machine, which allows increased temperature and an oxygenated perfusate. HILP can achieve concentrations within the limb 15–25 times higher than would be tolerated systemically, with limb temperatures reaching 39–41°C. By virtue of the increased concentration and the potential synergistic effects of hyperthermia, drugs such as melphalan, with little or no activity if administered systemically, become highly effective regional agents.110, 111 Response rates in the range of 80–90% with complete response (CR) rates as high as 60–70% have been reported.111–114 Melphalan is the most-common agent used in the United States, while melphalan plus tumor necrosis factor alpha (TNF-α) is often used in Europe.115 The value of TNF-α has not been clearly demonstrated, as a large multicenter randomized trial found no statistically significant differences in either overall or CR rates or survival and the addition of TNF-α was associated with significantly higher regional toxicity.114
Isolation of the limb during HILP substantially minimizes the risk of systemic toxicity from the high-dose chemotherapy; however, significant morbidity may still occur. The most-common morbidity is lymphedema and has been reported to occur in 12–36% of patients. Severe regional toxicities including compartment syndrome in up to 5% of cases as well as limb loss in up to 3.3% of cases have been described.115, 116
ILI is the less-invasive counterpart to HILP and is a low-flow infusion conducted in a mildly hyperthermic, acidotic, and hypoxic environment. Catheters are percutaneously placed into the artery and vein of the uninvolved limb and advanced into the vessels of the involved limb proximal to the extent of disease, avoiding the need for open surgical cannulation with its attendant morbidity. The chemotherapy is manually circulated for 30 min.117 Regional CR rates after ILI are in the range of 23–44% with partial response (PR) rates in the range of 27–56%.117, 118 Although these results are lower than reported after HILP, the two techniques have not been prospectively compared in a head-to-head manner in a clinical trial setting. In a recent retrospective comparison of HILP and ILI (ILI = 94, ILP = 109), the overall response rate was 53% for ILI versus 80% for ILP (p > 0.001). Median overall survival (OS) was 46 months for ILI versus 40 months for ILP (p = 0.31). There were no differences in age, sex, or N stage between groups; however, BOD was higher for the ILI group (high BOD 58 vs. 44%, p = 0.04).119 Importantly, ILI can be readily repeated if necessary and appears to have less regional toxicities than HILP (erythema and edema of the skin are among the most-common side effects with tissue loss seen <1% of the time) and virtually no systemic toxicities.116–119
There is some debate regarding whether ILI or HILP should be employed first in the treatment of in-transit melanoma, and this decision is based in part on the presence of nodes requiring node dissection or a high burden of disease in the limb, both of which might be better addressed with HILP, although the data are lacking to support this theory. In view of the relative morbidities and effectiveness, HILP is usually reserved as a salvage procedure for patients who progress rapidly after ILI without any evidence of distant metastasis. Repeat ILI can be attempted in patients who had a good initial response to ILI but eventually progressed.
Intralesional and topical therapies
Intralesional therapy in melanoma has several advantages over regional or systemic therapy. Local drug administration allows for delivery of an increased concentration of the agent and reduced regional and systemic exposure, potentially increasing efficacy and lowering toxicity. Moreover, alterations in the tumor microenvironment can be immunogenic and induce local immune responses that result in the “bystander effect,” where uninjected distant lesions exhibit a response, as has been reported with multiple different types of intralesional therapies.
Bacille Calmette-Guèrin (BCG) was one of the first intralesional therapies used for in-transit metastases. In that initial landmark series, 90% of injected cutaneous lesions regressed and 17% of patients also had regression of uninjected lesions. Some patients remained disease free for years after completion of the BCG injections.120 Interleukin-2 (IL-2) has also been used for intralesional injection in melanoma; the toxicity profile of IL-2 appears to be better than that of BCG. There is a risk of local reactions, and flu-like symptoms following injections are very common (85%).121 Intralesional treatment with IL-2 is relatively expensive. PV-10 is a 10% solution of rose bengal, a water-soluble xanthine dye used for decades as an intravenous diagnostic agent for assessing hepatic function and topically by ophthalmologists, making it a potentially low-cost treatment that has shown efficacy when “repurposed” for intralesional administration.122 Responses have been reported in patients refractory to previous systemic ipilimumab, anti-PD1 and vemurafenib, with evidence of bystander effects in uninjected lesions.123
Talimogene laherparepvec (TVEC, previously known as OncoVEX), is a genetically modified oncolytic herpes virus incorporating the coding sequence for human granulocyte-macrophage colony-stimulating factor (GM-CSF). Oncolytic viruses are designed to selectively replicate in tumors, thereby infecting and destroying cancer cells and inducing immune responses that target the cancer cell. Expression of the GM-CSF gene (GM-CSF) in tumor cells is expected to recruit and activate antigen-presenting cells and immune effectors that mediate potent antitumor T cell cytotoxicity. Tumor destruction in this fashion may also induce T cells capable of circulating and exerting antitumor effects in nearby and distant uninjected metastases. The phase III OPTiM study included 436 patients randomized in a 2 : 1 manner to intralesional TVEC or subcutaneous GM-CSF alone, respectively. There was a 26.4% objective response rate for TVEC compared to a 5.7% for GM-CSF (p < 0.001). There was a statistically significant increase in the primary endpoint of durable response rate (DRR), defined as a partial or CR lasting for 6 months or more. DRR was 16.3% for TVEC and 2.1% for GM-CSF (p < 0.001). Overall survival results also favored TVEC, although without statistical significance.126 This promising form of viral-mediated immune gene therapy is now being tested in combination with immune checkpoint blockers and other immunomodulatory interventions, and other oncolytic viruses with or without transgenes are also under investigation for melanoma and other malignancies.124
Imiquimod and diphencyprone (DPCP) are topically applied, rather than injected intralesionally. This approach is particularly appealing for patients with multiple small cutaneous nodules. Topical application of imiquimod, with or without additional agents such as topical 5-fluorouracil, resulted in regression of up to 90% of treated superficial lesions.125, 126 DPCP is a contact sensitizer that induces contact hypersensitivity; in a cohort of 50 patients treated with DPCP, 23 patients (46%) achieved a CR and an additional 19 patients (38%) showed a PR. The side effect profile of DPCP is tolerable, with skin reactions such as blistering and irritation being the most-common side effects.127
Radiation therapy
In patients with in-transit or regional recurrence, radiation may offer a benefit. Treatment protocols are not well defined but the potential for symptom control makes radiation an option for selected patients with unresectable locoregional melanoma128 and is now under active investigation as an immunomodulator, detailed later in this chapter.
Surgical metastasectomy for stage IV melanoma
Patients with isolated, resectable distant metastatic melanoma are candidates for surgical management aimed at removing all known sites of disease. Identifying which patients will benefit from metastasectomy requires good clinical judgment and a thorough preoperative staging that includes PET–CT of the body and MRI of the brain to rule out occult sites of metastasis.129
There has been only one prospective (albeit nonrandomized) trial evaluating surgery for patients with stage IV melanoma, the S9430 trial conducted by the Southwest Oncology Group.130 Patients with stage IV melanoma were enrolled as soon as the determination of potential resectability was made, before actually undergoing surgery. This allowed the investigators to estimate resectability rate and define relapse-free and overall survival after complete resection. Among the 77 study patients, 3 patients had no evidence of melanoma in the resected specimen (the suspected metastatic deposit was either a second primary malignancy or a benign finding) and 2 patients had only stage III disease. An additional 8 patients were not able to have all disease resected. Therefore, 64 patients (88.9%) were in fact resected to a disease-free state. After a median follow-up of 5 years, all but 6 (9.4%) had recurred, with a median relapse-free survival (RFS) of approximately 5 months. Median overall survival was 21 months with an estimated 12-month survival of 75% and survival at 4 years was 31%.130 Although small, this prospectively derived and with otherwise unselected patients, the resectability and survival rates reported in S9430 are the most representative data available for comparing surgical and nonsurgical approaches to stage IV melanoma. It is possible that using the more sensitive current imaging methods to find patients with resectable single or oligometastases would lead to more favorable outcomes if the study were done now. However, new systemic therapy approaches to metastatic melanoma are becoming so effective that a reconsideration of the role of surgery in front-line management may be appropriate, and of great importance is the investigation of the potential for neo-adjuvant therapies from among the molecularly targeted agents and the new immunotherapies, particularly PD-1 blocking agents that work relatively quickly and may render a formerly unresectable lesion more amenable to surgery.
The role of adjuvant therapy for stage II and III disease
Immunotherapy
Immunotherapy has been the adjuvant treatment approach evaluated most extensively in melanoma, and the most extensively evaluated adjuvant immunotherapy has been interferon-α (IFN-α), in a variety of doses, schedules, and formulations. High-dose IFN-α is a 1-year adjuvant therapy regimen involving two components: a 1-month “induction” phase administering 20 million units of IFN-α2b/m2 of body surface area intravenously 5 days per week for 4 weeks, followed by an 11-month “maintenance” phase administering 10 million units/m2 subcutaneously three times a week. Three randomized trials have demonstrated that high-dose IFN-α is associated with improved RFS, and two trials showed improved OS as well.131, 132 In the United States, high-dose IFN-α is approved for use in the adjuvant therapy of thick, node-negative and node-positive melanoma. To date, no studies of high-dose IFN-α have defined subsets of patients that clearly respond better or worse to treatment. The relative importance of the induction and maintenance phases has not been adequately defined, but it does appear that the 4-week induction phase alone is inadequate.133
Adjuvant IFN-α regimens involving lower doses of IFN-α (3–10 million units administered three times a week not adjusted for body surface area and without the initial intravenous component) have also been tested. Low-dose IFN-α is approved for adjuvant use in some European countries and has been advocated for use in intermediate-thickness, node-negative melanoma as well.134 In the absence of direct comparisons between high- and low-dose IFN-α regimens, meta-analyses have been performed to assess the overall and comparative efficacy of adjuvant IFN-α therapy. Meta-analysis results clearly support that IFN-α therapy delays recurrence, with an overall RFS improvement of 17% (hazard ratio for recurrence 0.83) in the latest analysis, but do not identify clear differences between high- and low-dose regimens in efficacy. Meta-analysis results also show a statistically significant improvement in OS for adjuvant IFN-α, with a 9% (hazard ratio for survival 0.91) improvement seen.135
Pegylated IFN-α was approved in the United States as an alternative adjuvant treatment for stage III melanoma in 2011, based on the results of a single randomized trial.136 This trial demonstrated a statistically significant improvement in RFS, in keeping with trials of standard IFN-α, but did not show an OS benefit. An interesting observation was made, however, that the subset of patients with sentinel node-positive melanoma from an ulcerated primary showed a dramatic survival benefit (41% improvement) when treated with pegylated IFN-α. Although some other IFN-α trials have seen a similar benefit in this subset,137, 138 this is best considered an unproven but provocative finding pending definitive replication. Pegylated IFN-α has not been directly compared to high-dose IFN-α, but appears to have fewer serious side effects with pharmacologic properties that might make it a more favorable agent for the maintenance phase. Unlike high-dose IFN-α, pegylated IFN-α is intended to be given subcutaneously for 5 years (3 mcg/kg/week), and the 1-month intravenously administered induction phase is replaced with a 2-month long subcutaneously administered induction phase at a higher dose (6 mcg/kg/week).
Other adjuvant systemic therapies under investigation
The availability of new agents with documented survival benefits in unresectable metastatic melanoma has spurred trials of these agents in the adjuvant setting. It remains to be seen if these agents will be equally or more effective than available IFN-α regimens, and how the toxicities of these new drugs will be tolerated by patients who are disease free and possibly cured at the time of treatment. Nonetheless, it is likely that the advances in understanding melanoma biology and the regulators of the antitumor immune response that have profoundly changed treatment for stage IV melanoma will eventually translate into new, more effective and hopefully less toxic approaches to the prevention of recurrence by adjuvant therapy.
A multiagent biochemotherapy regimen consisting of IFN-α, IL-2, dacarbazine, vinblastine, and cisplatin was compared to high-dose IFN-α in a randomized trial showing a statistically significant RFS advantage but no difference in OS.139 Although this is the first randomized trial to ever show a statistically significant advantage over high-dose IFN-α, the toxicity of this regimen limits its use, and the lack of an OS advantage argues against this becoming a new standard for the adjuvant treatment of melanoma.
Ipilimumab has been shown to improve survival in stage IV melanoma (detailed later in this chapter), with less toxicity than is associated with biochemotherapy, making it a logical candidate to evaluate in the adjuvant setting. Preliminary data from a randomized trial comparing ipilimumab at 10 mg/kg (a dose higher than that approved for use in the treatment of unresectable metastatic melanoma) to placebo showed statistically significantly improved RFS relapse for the ipilimumab arm.140 Unfortunately, the toxicity of the regimen was unexpectedly severe. Data regarding the impact of this adjuvant ipilimumab regimen on OS are pending. Importantly, the relative impact of adjuvant ipilimumab over that provided by IFN-α is currently unknown. Given the toxicity encountered with adjuvant ipilimumab at 10 mg/kg, there is also a need to assess the risk–benefit ratio relative to the standard 3 mg/kg ipilimumab dose. A randomized trial has been conducted to evaluate ipilimumab at 10 and 3 mg/kg compared to high-dose IFN-α (E1609, NCT01274338). This trial has completed accrual and results are eagerly awaited. The safety and tolerability of anti-PD1 antibodies appear to be superior to both IFN-α and ipilimumab, making these antibodies ideal candidates for evaluation as adjuvant therapy. The results of completed but not yet mature randomized trials of ipilimumab versus nivolumab (Checkmate 238, NCT 02833906) and of pembrolizumab versus placebo (EORTC Keynote 054. NCT 02362594) will likely define future adjuvant systemic therapy recommendations.
Additional trials investigating the activity of adjuvant BRAF inhibitors alone or in combination with MEK inhibitors have completed accrual and await outcome data. Legitimate concerns have been raised about the tolerability of these agents in the adjuvant setting, particularly considering the potential for rapid development of resistance and for secondary malignancies to arise in patients taking these agents, and their routine use is not recommended in the adjuvant setting.141
Adjuvant radiation to the resected nodal basin
Criteria for identifying patients at high risk of regional recurrence in the resected nodal basin after radical lymphadenectomy include multiple (≥4) or matted tumor-involved lymph nodes, the presence of extranodal extension of tumor in at least one node, and large size (≥3 cm) of any one lymph node. In a prospective trial, 217 eligible patients possessing at least one of these criteria were randomized to observation or postoperative nodal basin irradiation after lymphadenectomy.108 After a median follow-up of 40 months, nodal basin relapse occurred in 34 of 108 patients (31.5%) in the observation arm, confirming the high risk of regional recurrence in these patients. Nodal basin relapse was significantly reduced in the adjuvant radiotherapy group compared with the observation group, but no differences were noted for RFS when all sites of relapse were included or for OS.108 This confirmed the ability of postoperative radiation to significantly decrease recurrence within the nodal basin in patients at high risk of such recurrence, and radiation should be considered for selected high-risk cases. It will also be important to study the interaction of adjuvant locoregional radiotherapy with immune checkpoint blockade in the adjuvant therapy of high-risk melanoma.
Surveillance for high-risk melanoma patients
A variety of algorithms have been proposed regarding the frequency and nature of the follow-up evaluation of melanoma patients after surgical treatment.142–144 To date, no studies have documented that surveillance of patients after surgical treatment of melanoma with any imaging or laboratory studies (including chest X-rays, PET/CT, or MRI scans) are cost-effective. One study evaluating the results of follow-up after surgery in stage melanoma III patients found that half of all recurrences were detected by patients, and that “neither more intense nor more frequent follow-up is associated with discovery of resectable first relapses.”142 While most proposed follow-up algorithms involve more frequent follow-up in the initial years after surgery, the conditional probability of recurrence of stage I and II melanoma is actually fairly constant over the first decade after surgery.144 Education is key and should be focused on both the patient and family as well as the primary care physicians, dermatologists, and surgeons alike, and melanoma centers may want to train and even “certify” providers in their catchment area. Patients should be willing to return to the melanoma center for evaluation of suspected recurrence, because properly diagnosing recurrence (e.g., documenting nodal recurrence by needle aspiration cytology instead of open biopsy) is important to successful treatment. Importantly, we need to individualize the follow-up, identifying which patients are best suited to have the bulk of their surveillance outside the melanoma center and which ones should return more frequently.
Table 6 summarizes the follow-up guideline for patients with completely resected melanoma from various national and international organizations. These guidelines have not been prospectively validated and hence should only be considered as recommendations. (Adapted from Fields and Coit.142)
Uveal melanoma and rare melanomas of the eye
Uveal melanoma (UM) is the most-common primary intraocular cancer of the eye in adults and represents 5% of all melanomas. The term “UM” is used for melanomas that arise within the uveal tract (i.e., iris, ciliary body, or choroid), while the broader term “ocular” melanoma includes such sites as conjunctiva and eyelid, which behave like cutaneous melanoma. UMs are further designated as anterior (iris) or posterior (ciliary body and/or choroid) chamber tumors. Anterior chamber UM is rarer (<10% of UMs) and tends not to metastasize, whereas approximately half of patients diagnosed with posterior chamber UM will develop metastases.
Local therapy for UM employs radioactive plaque, proton therapy, or enucleation of the primary UM tumor. In a randomized study comparing 128I plaque brachytherapy with enucleation, 85% of patients receiving radiation retained their eye, and 37% had visual acuity over 20/200 in the irradiated eye 5 years after therapy. No survival difference was seen between the radioactive plaque and enucleation groups.145 Thus, a radiotherapeutic approach has become the strategy of choice for primary UM treatment, with enucleation reserved for the remaining <10% of cases in which radiotherapy is not possible (e.g., bulky disease, technically difficult tumor location, patient preference). Because most patients do not undergo enucleation, fine-needle aspiration techniques are now often used to obtain the pathologic and molecular diagnostic information that is critical to prognosis.
UM metastases are hematogenous, because the uveal tract does not appear to contain clear lymphatic channels with draining lymph nodes. The liver is ultimately involved in 95% of metastatic UM cases and is the sole site of metastatic UM in approximately 50% of patients. The most common other sites of metastatic UM are lung (24%), bone (16%), and skin (11%). Unlike cutaneous melanoma, UM infrequently metastasizes to lymph nodes or brain. The clinical course of patients with UM is highly dependent on disease progression in the liver. The median survival after diagnosis of patients with liver metastases is approximately 4–6 months with a 1-year survival of approximately 10–15%. Patients with metastases limited to extrahepatic sites have a median survival of approximately 19–28 months with a 1-year survival of approximately 76%. Thus, hepatic- versus extrahepatic-only disease may represent distinct biological entities.146
Approximately 90% of UM harbor activating, mutually exclusive, recurrent mutations in the g-protein alpha q (GNAQ) or 11 (GNA11) subunit genes. Nearly all GNAQ/11 gene mutations localize to a hotspot in exon 5 (Q209), although a small number localize to exon 4 (R183). GNAQ/11 gene mutations are early tumorigenic events that are insufficient to initiate metastatic disease, but some of the downstream pathways in UM resulting from additional molecular aberrations, such as protein kinase C and others, may be targetable with investigational agents in development.147 Additional recurrent missense mutations in SF3B1 (altering the R625 amino acid position in most cases) or EIF1AX (exons 1 or 2) genes occur in equal proportion in approximately 40% of UM tumors and are essentially mutually exclusive with each other. Truncating and nontruncating mutations are observed in the nuclear ubiquitin carboxy-terminal hydrolase BAP1 gene located on chromosome 3p21.1. Genetic aberrations in BAP1 tend to co-occur with monosomy 3, resulting in the loss of heterozygosity of BAP1. Both BAP1 gene mutations and monosomy 3 tend not to co-occur with SF3B1 or EIF1AX gene mutations, and the former are associated with a high risk of developing metastatic UM.148 Consistent with BAP1 mutations leading to loss of tumor suppression, there have been multiple reports of germline BAP1 mutations within families that result in a high incidence of UM.149 Another recurrent chromosomal aberration observed in UM is 8q copy number gain (often with 8p loss), which tends to accompany monosomy 3 and is associated with a shorter time to relapse. Conversely, 6p copy number gain (often with 6q loss) is usually present in tumors that lack monosomy 3 or 8q copy number gain and is associated with a low risk of metastasis. Less-frequent chromosomal aberrations, such as 1p and/or 16q loss, have also been described. With the exception of BAP1, the identification of specific genes that correlate functionally with these chromosomal aberrations is less clear.150
Multiple anatomic, histologic, and molecular features within primary tumors are associated with poor prognosis: (1) location in ciliary body (poorer) > choroid ≫ iris (rarely metastasize); (2) greater tumor size; (3) extrascleral invasion; (4) epithelioid > spindle cell histology; (5) higher mitotic rate; (6) presence of monosomy 3 ± chr 8q gain; and (7) class 2 ≫ class 1b ≫ class 1a gene expression profile (a 15-gene expression profile that accurately predicts outcome and does not require chromosomal analysis). However, the strongest risk factors for metastasis are copy number profile (e.g., presence of monosomy 3/chr 8q gain) and/or a class 1b/2 gene expression profile.151, 152
Metastatic UM has proven essentially recalcitrant to traditional chemo- and immunotherapeutic approaches. Given the current inability to therapeutically target the aforementioned molecular aberrations characteristic of UM, there has been a major focus on targeting the “effector” molecules associated with these genetic alterations. The most successful systemic approach to date has been to target the MEK–MAPK pathway that is clearly activated by GNAQ/11 mutations. A phase II randomized clinical trial showed that treatment of metastatic UM patients with single-agent selumetinib (a small molecule MEK inhibitor) resulted in tumor regression in 50% and RECIST responses in 15% of cases. A significant difference in median progression-free survival following treatment with selumetinib (15.9 weeks) compared to dacarbazine (7 weeks) was shown, although without an improvement in overall survival.153 Targetable effectors of mutant BAP1/monosomy 3 are still under investigation. Retrospective analyses of metastatic UM patients from multiple centers treated with ipilimumab suggest that long-term tumor response was observed in a subset (∼5%), as well as prolonged stable disease (SD) in a slightly larger subset of advanced disease patients.154 More recently, therapy of metastatic uveal melanoma with antibodies blocking PD-1 or PD-L1 was reported from a multi-institution retrospective series and also showed very low activity,155 further evidence that the successful therapy of this melanoma variant requires strategies that focus on its unique molecular biology and targets in its immune tumor microenvironment.
For patients with liver-only or -dominant metastatic UM, multiple therapies specifically directed at the liver have been explored: isolated or percutaneous hepatic perfusion of chemotherapy, transarterial chemoembolization (TACE), radioembolization or cryoembolization, or selective internal radiation therapy (SIRT). Liver-directed therapy studies tend to have low patient numbers, but when taken in aggregate, suggest relatively high tumor response, prolonged time-to-progression, and/or survival in the population of patients upon whom they have been employed. Nine studies (totaling 209 patients) using TACE (mostly cisplatin-based) reveal a 2% CR, 24% PR, and 33% SD rate when taken in aggregate. Eight pooled studies (totaling 277 patients) show hepatic intra-arterial chemotherapy (mostly employing either a fotemustine or melphalan-based regimen) to have a 9% CR, 22% PR, and 29% SD rate to this approach. Finally, four studies (totaling 59 patients) treated with hepatic arterial perfusion reveal an 8% CR, 44% PR, and 10% SD rate (reviewed in Ref. 156).
There may be a role for surgical resection of metastatic UM if there is a relatively stable and safely resectable solitary lesion(s). In a study in which 61 patients had surgical resection in addition to chemotherapy, a 22-month median overall survival was observed among patients who could undergo resection with curative intent compared to 10 months among those who could not. Variations in patient referral and selection criteria are important factors in all of these outcomes, which must be validated by performing well-controlled prospective randomized trials.
With one of the most predictive sets of molecular markers of metastatic risk, the relatively recent identification of clear genetic drivers within primary UM tumors, and a wide spectrum of clinically relevant approaches (molecular inhibitors, immunotherapies, radiation and surgery) now available, there is tremendous excitement that truly effective therapies for UM in both the adjuvant and advanced settings are within practical reach.
Melanoma of the conjunctival surface of the eye is rare and may complicate primary acquired melanosis. Recent reports demonstrating the presence of typical activating BRAF mutations in 29% and NRAS mutations in 18% of a cohort of 78 conjunctival melanomas provide further evidence of the close relationship of melanomas in this site to cutaneous melanomas, with case reports of clinical responses to vemurafenib that corroborate the importance of BRAF as an oncogenic driver in conjunctival melanoma.157, 158
Biology and therapy of advanced melanoma
The diagnosis of metastatic melanoma may be triggered by new symptoms or signs in a patient with a history of melanoma (about half of the presentations) or a new but asymptomatic finding on radiographic surveillance scans, based on guidelines and practice that take into account the initial stage, time-dependent risk of relapse, and likelihood that intervention will change the natural history of the disease. Skin and soft tissue are more common sites of initial metastatic disease than visceral sites, and a minority of patients have widespread metastatic disease secondary to hematogenous spread, such as is common with mucosal melanomas.
Uncommonly, advanced melanoma may present as a solitary or multiple metastases—even in the central nervous system—without a known history or evidence of a concurrent new primary melanoma. It has been postulated that immune-mediated control or regression of a missed primary controlled by a local immune response explains this phenomenon, which also has a slightly more favorable prognosis than matched cases with a known primary.159 Melanoma spreads widely and spares few organs or sites, with a particular affinity for the brain, where it is often the immediate or major contributing cause of death due to bleeding and edema. Other sites of melanoma metastasis that are rare for other cancers include the small and large intestine and even the heart. Characteristics of primary melanoma that pose increased propensity to specific sites of metastasis are under investigation. For example, the CCL25 and CCR9 chemokine: chemokine receptor interaction has been reported in intestinal metastases160 and increased activity of the phosphatase and tensin homolog (PTEN)-related pathway is associated with a higher rate of eventual metastasis to the brain (reviewed in Ref. 161). Many other molecular alterations, most recently the expression of a variant CD44v6 molecule, a receptor for hyaluronic acid that is highly expressed by brain,162 have been reported to impact the risk and/or the biological behavior of established brain metastases (reviewed in Ref. 163).
Molecularly targeted therapy for melanoma
Metastatic melanoma is divided into three substages based on modest differences in the survival curves for M1a, limited to skin and soft tissue; M1b, involving lung with/without skin/soft tissue; and M1c, metastatic to any visceral organ and/or with elevation of the serum lactate dehydrogenase (LDH) above the institutional upper limit of normal.94 The differences among these subsets are being eclipsed by a rapid growth in understanding of the molecular basis of different melanomas such as detailed by Bastian’s group.61 However, except for notable examples such as the melanomas that carry actionable oncogenic driver mutations (BRAF, c-kit, NRAS) and UM, which is routinely excluded from most clinical trials of immunotherapy, clinical trials for advanced melanoma have included unselected patients, with prestratification in some instances for known prognostic variables such as the LDH and the performance status.
Melanoma, together with lung cancer, has the highest frequency of mutations per cell among all human malignancies, which is largely attributable to frequent C→T transitions resulting from solar UV exposure.12 A few mutations result in “driver” oncogenes (such as activating BRAF v600E or v600K mutations or NRAS mutations at G12, G13 or Q61) and/or important “passenger” alterations that collaborate to varying degrees in oncogenesis (such as PTEN loss of function, which occurs in at least 20–30% of melanomas, often in association with BRAF mutations,164 and many others such as TP53 and p16INK4a and p14ARF12). Simultaneous mutation of BRAF and NRAS is rarely found in primary melanomas untreated with BRAF inhibitors. The importance of the PTEN pathway and its downstream mediators of a wide variety of proliferative, metabolic and antiapoptotic functions is illustrated by a large number of mechanisms for pathway activation, including hypermethylation of the PTEN promoter and rare mutations of AKT or PI3kinase isoforms (reviewed in Ref. 165).
The molecular drivers currently targetable by therapeutic agents approved for melanoma are limited to BRAF-activating mutations, which are tested by either pcr-based or gene sequencing methods166 for the mutations encoding BRAF v600E or one of the less-common activating mutations at residue 600 of the BRAF serine–threonine kinase. BRAF v600E has a change from valine to glutamic acid (V→E) and accounts for approximately 75% of cases, while the next most common is valine to lysine (V→K) in about 17%. Other amino acid substitutions account for the small number of remaining cases.167 Tumors carrying V600K are associated with more advanced age, chronic sun damage, and a somewhat less favorable outcome even with BRAF inhibition.168 There are many assays available for next generation genomic sequencing that detect additional alterations with therapeutic implications, for example, activating c-kit mutations in about 20% of mucosal and 15% of acral melanomas with occasional responsiveness to imatinib170–172 and related tyrosine kinase inhibitors, and NRAS mutations, which have shown responsiveness to MEK and cyclin-dependent kinase (CDK) 4/6 inhibitors inhibitors although not as sensitive as BRAF-mutated melanoma172 These assays are also valuable for discovery of new mechanisms of resistance, especially when performed sequentially on biopsies before and during therapy. Also of interest is the recent observation of BRAF gene fusions with several other molecular species in Spitzoid nevi that provide unique targets for several agents already in testing.173
Disease regression occurs in the majority of patients treated with single-agent BRAF or MEK inhibitors (Table 7) and can be improved with the combination of inhibitors, which have shown survival benefit over single-agent BRAF inhibition. As some of the toxicities of BRAF inhibitors occur via paradoxically-enhanced signaling from upstream pathways that also activate MEK,174 these effects are less prominent during combined therapy; furthermore, the secondary low-grade cutaneous proliferations (keratoacanthoma and squamous cancers) attributed to mutant HRAS activation upstream of MEK175 are also reduced during combination therapy. Such combinations also yield higher response rates and additional disease regression, as shown on the “waterfall” plots of maximum regression in individual subjects. Longer progression-free and overall survival has also been reported with these combinations over BRAF inhibition alone.176–179 Toxicities of single-agent therapy with either BRAF or MEK inhibitors are generally tolerable but may require dose reduction, brief drug holiday, or a switch in agent(s). They are briefly summarized in Table 7, but further management guidelines are available from recent experience181, 182 and will undoubtedly emerge with more experience, particularly since the regulatory approval of these agents has led to their more widespread use.
Table 7 Antitumor activity and toxicities of molecularly targeted agentsa
| DRUG (References) | Vemurafenib178 | Vemurafenib177 | Dabrafenib176 | Trametinib179 | Dab + Tram176 | Dab + Tram177 | Vemu + Cobimetinibb,178 |
| Patient number | 239 | 352 | 212 | 214 | 211 | 352 | 254 |
| Objective response (%) CR (%) | 45 4 | 51 8 | 51 9 | 22 2 | 67 10 | 64 13 | 68 10 |
| Progression-free survival (median, mo) | 6.2 | 7.3 | 8.8 | 4.8 | 9.3 | 11.4 | 9.9 |
| TOXICITIESc | |||||||
| All grade 3 (%) | 49 | 57 | 34 | Grade 3 or 4 8 | 32 | 48 | 49 |
| All grade 4 (%) | 9 | 1.4 | 3 | 3 | 1 | 13 | |
| Fever | 22 | 21 | 28 | — | 51 | 53 | 26 |
| Fatigue | 31 | — | 35 | 26 | 35 | — | 32 |
| Headache | — | — | 29 | — | 30 | — | — |
| Nausea or emesis | 24 | 15 | 26 | 18 | 30 | 29 | 10 |
| Chills | — | 8 | 16 | — | 30 | 31 | — |
| Arthralgia | 40 | 51 | 27 | — | 24 | 24 | 32 |
| Diarrhea | 28 | 38 | 14 | 43 | 24 | 32 | 17 |
| Rash | 35 | 43 | 22 | 57 (acneiform 19) | 23 | 22 | 17 |
| Hypertension | — | — | 14 | 15 | 22 | — | — |
| Peripheral edema | — | — | 5 | 26 | 14 | — | — |
| Increased transaminase | 18 | — | 5 | — | 11 | — | 18 |
| Photosensitivity | 15 | 22 | — | — | — | 4 | 28 |
| Hyperkeratosis | 29 | 25 | 32 | — | 9 | 4 | 10 |
| Keratoacanthoma | 20 | KA or SCC 18 | 21 | — | — | KA or SCC 1 | 1 |
| Squamous cancer | 11 | 9 | — | — | 3 | ||
| Alopecia | — | 26 | 26 | 17 | — | 2 | |
| Hand-foot syndrome | — | 25 | 27 | — | 5 | 4 | — |
| Decreased LVEF | — | — | 2 | — | 4 | 8 | — |
a From randomized trials.
b Thirty percent of patients had asymptomatic grade 1–2 creatinine phosphokinase elevation.
c Reversible transient drug-induced retinopathy, rarely reported.
Acquired resistance to single MAPK inhibitors occurs after a median of 6 months and somewhat later (median 8–11 months) with combined MAPK inhibitors (Table 7), although a fraction of patients may enjoy prolonged control.183 The mechanisms of resistance as well as protection against resistance are becoming evident from the many clinical trials that provide tissue for analysis before therapy and during therapy or at the time of progression. Reactivation of MEK signaling occurs in most cases of acquired resistance to MAPK inhibition, via alterations in BRAF (gene amplification or truncation mutations); secondary mutations in NRAS with the downstream consequence of increased CRAF heterodimerization with blocked mutant BRAF to reactivate downstream signaling; rare MEK mutations or related (COT1) activation; and several receptor kinase alterations (Figure 2) that also activate the PI3kinase/AKT pathway.182, 183 An earlier-onset form of therapy resistance, termed “adaptive” by Kugel and Aplin, involves shifts in metabolic or receptor tyrosine kinase signaling that confer growth advantage or resistance to apoptosis and may also be targetable with small molecule inhibitors or antibodies but have been less extensively studied than acquired mechanisms.184 Therapeutic trials to delay or prevent the emergence of resistance will be critical, as established resistance is difficult to overcome with any form of targeted therapy. For example, the activity of combination MEK plus BRAF inhibitor after BRAF inhibition fails is modest (only 15% response, PFS <4 months185), and current efforts are focused on preventing or delaying resistance by improving front-line therapy and molecular typing. The use of intermittent dosing, based on strong preclinical data188 and clinical anecdotes, is also under investigation (clinicaltrials.gov NCT02196181).
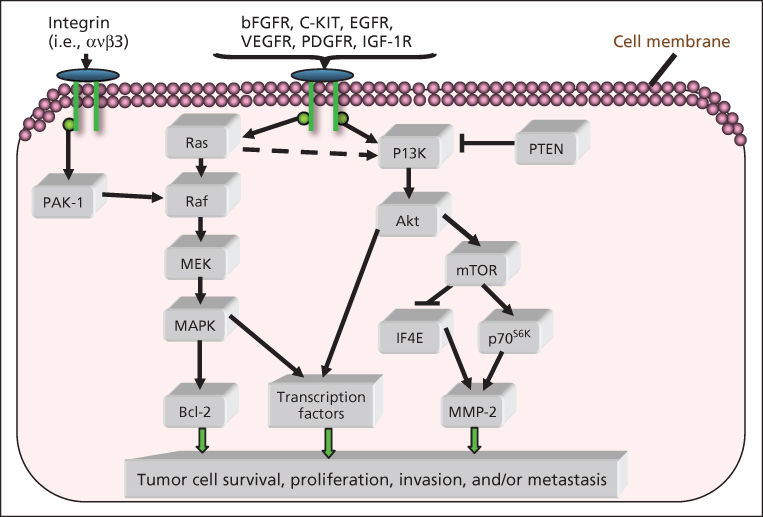
Figure 2 Multiple molecular pathways, such as the RAF/MEK/MAPK and PI3K/AKT pathways, have been found to support melanoma proliferation and survival. Understanding these pathways will enable the development of targeted therapies for melanoma.
The choice between immunotherapy and targeted agents for the first-line therapy of patients whose melanoma carries a BRAF activating mutation remains unclear, as the kinetics of benefit are so different. MAPK inhibitors work quickly and relieve symptoms within days to weeks, while immunotherapy responses may require weeks to months, particularly ipilimumab, which has shown several patterns of response, including both delayed responses after objective progression as well as the appearance of new lesions before an overall objective response.189 Until the completion of planned drug-sequencing trials (see below) to answer this question, it is generally recommended that patients with a BRAF-mutated melanoma and rapidly growing, symptomatic or very high tumor burden be treated first with combination MAPK inhibitors. This critical question is being tested in a large U.S. cooperative group trial (NCT02224781). Patients with advanced melanoma carrying an activating BRAF mutation are randomized to initial therapy with dabrafenib plus trametinib versus ipilimumab plus nivolumab. Upon progression, patients are crossed over to the opposite treatment. The trial results will define the optimal initial therapy and sequence among these patients, as well as important predictors (which may range from simple clinical tests such as serum LDH to complex immunologic parameters in blood or tumor) of benefit from each regimen. Additional unresolved questions include whether and how to continue targeted agents in patients requiring palliative radiotherapy (which can interact to enhance skin and liver toxicities) or surgery for single sites of symptomatic progression during disease control in other sites.
Immunotherapy for advanced melanoma
Interleukin-2 and other cytokines
IL-2 is an immunomodulatory cytokine with pleiotropic effects on immune effectors that has activity against melanoma when given at high pharmacologic doses. Most recent data show a 15–25% rate of objective response and an RFS plateau of 20%.190 To date, clinical criteria for safety and the availability of an expert team to administer high-dose IL-2 and the associated supportive care are used to select patients for this form of treatment; although many dose and schedule alterations and the addition of a variety of toxicity modulators have been studied, no modification of the original regimen has improved its unfavorable therapeutic index. The toxicities of high-dose IL-2 result from a generalized capillary leak syndrome mediated by small molecule vasoactive agents such as nitric oxide and a reversible systemic inflammatory response syndrome with multiorgan dysfunction that may also be attributed to the effects of IL-2-induced inflammatory mediators such as tumor necrosis factor and interferon-γ. Studies are now underway to identify immunologic or genetic predictors of benefit (clinicaltrials.gov NCT01288963) that will potentially complement those under investigation for selection of other immunotherapies (detailed below) as single agents or, more likely, in combination or carefully sequenced strategies. Other trials test the activity of regimens that combine or sequence IL-2 with immune checkpoint blockers as well as the potential role for newer cytokines, particularly those that stimulate cytotoxic T cells but, unlike IL-2, do not also stimulate regulatory T cells or activation-induced cell death. The importance of collecting blood and tumor tissue before and, whenever possible, during therapy for the elucidation of biomarkers of response and resistance (and possibly of toxicity) has emerged as a critical element likely to lead to better selection criteria for first and subsequent therapies.
Immune checkpoint inhibitors in advanced melanoma
Ipilimumab is a fully human antibody to CTLA4, a molecule that provides an “immune checkpoint” on activated effector T cells by engaging with the ligand B7.1 on tumors or on dendritic cells and removing it from the activating receptor CD28. In the presence of anti-CTLA4, activation is restored, and a pre-existing but ineffective immune response against tumors is enhanced. CTLA4 blockade also promotes homing of effector T cells to tumor and elimination of Treg cells.190, 191 When used at the approved dose of 3 mg/kg × 4 doses at 3-week intervals, ipilimumab provides objective response in 10–15% of patients, with less than partial regressions or disease stabilization in another 10–15%.192 The survival appears to plateau after 2.5 years at about 20%, so these patients may be cured by a durable and effective immune response (reviewed in Ref. 193). Likely as a result of additional mechanisms of action, the patterns of response to CTLA4 blockade may be atypical and delayed, making it more difficult to know when to switch a patient to second-line therapy upon the appearance of progression. However, with the advent of new checkpoint blocking agents particularly antibodies that block the PD-L1/PD-1 interaction (see below and Table 8, Refs ), practical recommendations for the use of single-agent ipilimumab are evolving rapidly. For melanoma and some other malignancies, the role of combined or optimally-sequenced CTLA4 and PD-1 or PD-L1 blockade are now the most critical clinical questions. Remaining questions include the survival benefit and safety/tolerability use of adjuvant ipilimumab (covered earlier in this chapter) and the therapeutic index of higher doses and more prolonged dosing schedules of CTLA4 blockade as well as novel combinations with other immunomodulatory agents (see Figure 3). For example, the addition of GM-CSF (daily for 2 weeks of every 3) to a 10 mg/kg dose of ipilimumab, given every 3 weeks × 4 and then every 12 weeks, was recently shown to both enhance survival and reduce ipilimumab’s toxicities,192 an intriguing result that warrants further study for both advanced disease and in the adjuvant setting. Although the BRAF mutation has been associated with a higher likelihood of relapse and slightly more aggressive metastatic disease,199 it does not appear to impact the activity of ipilimumab in melanoma.200
Table 8 Activity and toxicities of selected CTLA4 and PD-1 blocking antibodies from Phase III trials for advanced melanoma
| Drug (references) | Nivolumab194 | Pembrolizumab195,a | Ipilimumab195 | Ipilimumab196 | Nivolumab + | Nivolumab196 | Pembrolizumab197,b | |||||||
| Ipilimumab196 | ||||||||||||||
| Patient number | 206 | 277 | 256 | 315 | 314 | 316 | 178 | |||||||
| ORR (%) | 40 | 33 | 12 | 19 | 58 | 44 | 38 | |||||||
| Overall survival | 1-yr | 79% | 1-yr | 74% | 1-yr | 58% | NR | NR | NR | NR | ||||
| Toxicity (%)c, grades | All | 3–4 | All | 3–4 | All | 3–4 | All | 3–4 | All | 3–4 | All | 3–4 | All | 3–4 |
| Fatigue | 20 | 0 | 21 | 0 | 15 | 1 | 28 | 1 | 35 | 4 | 34 | 1 | 21 | 1 |
| Colitis/diarrhea | 16 | 1 | 17 | 2.5 | 23 | 3 | 33 | 6 | 44 | 9 | 19 | 2 | 8 | 0 |
| Dermatitis/pruritus/rash | 17 | 0.5 | 15 | 0 | 25 | 1 | 35 | 2 | 40 | 5 | 26 | 0.6 | 21 | 0 |
| Hypophysitis/other endocrinopathyc | NR | NR | 0.4 | 0.4 | 1 | 2 | 4 | 0 | 15 | 0.3 | 9 | 0 | 5 | 0 |
| Hepatitis | NR | NR | 1 | 1 | 1 | 0.4 | 4 | 0.6 | 18 | 8 | 4 | 1 | NR | NR |
| Pneumonitis | NR | NR | 0.4 | 0 | 0 | 0 | NR | NR | NR | NR | NR | NR | NR | NR |
| Nephropathy | NR | NR | 0 | 0 | 0.4 | 0.4 | NR | NR | NR | NR | NR | NR | NR | NR |
| Fever | NR | NR | NR | NR | NR | NR | 7 | 0.3 | 18 | 0.6 | 6 | 0 | NR | NR |
| Arthralgia | 6 | 0 | 9 | 6 | 5 | 0.8 | 6 | 1 | 10 | 0.3 | 8 | 0 | 7 | 1 |
| Nausea | 16 | 0 | 10 | 0 | 9 | 0.4 | 16 | 0.6 | 26 | 2 | 13 | 0.6 | 4 | 0 |
a Pembrolizumab 10mg/kg every 3 weeks.
b Pembrolizumab 2mg/kg every 2 weeks.
c NR = not reported
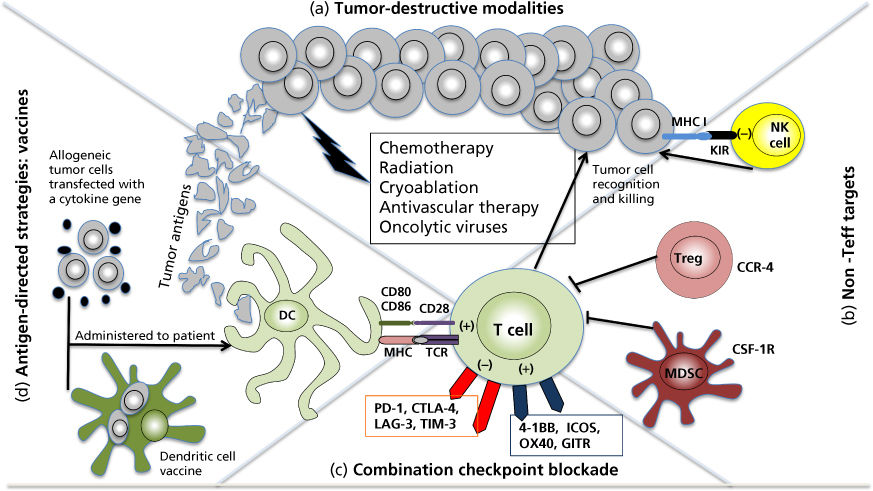
Figure 3 Combination strategies with cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) blockade—(a) conventional and novel therapies may effi- ciently destroy tumor and liberate tumor-associated antigens (TAAs), thereby enhancing antigen presentation and tumor-specific adaptive immunity; (b) novel-targeted agents may inhibit the suppressive effects of regulatory T cells (Treg) and myeloid-derived suppressor cells (MDSCs) or enhance innate immu- nity via natural killer (NK) cell-killer immunoglobulin-like receptor (KIR) activity; (c) immune checkpoints and costimulatory receptors can be targeted in combination with CTLA-4 to enhance T-cell function; (d) adaptive immunity to known TAAs can be enhanced via vaccine strategies employing peptides, whole proteins, whole cells, DNA, or virus-based vectors. CSF-1R, colony-stimulating factor 1 receptor; DC, dendritic cell; GITR, glucocorticoid-induced tumor necrosis factor receptor (TNF) receptor-related protein; ICOS, inducible T-cell costimulator; LAG-3, lymphocyte activation gene 3; MHC, major his- tocompatibility complex; PD-1, programmed cell death 1; TCR, T-cell receptor; Teff, effector T cell; TIM-3, T-cell immunoglobulin and mucin-containing domain 3.
Source: Funt et al. 2014.198 Reproduced with permissions of UBM Medica.
Hypofractionated or other schemes of radiation may also overcome resistance to ipilimumab and other immunomodulatory therapies via multiple mechanisms that include enhanced antigen and MHC expression, reduction of suppressive cells and molecules, and induction of inflammatory substances that promote dendritic cell function, which in turn enhances tumor antigen-specific responses, and many clinical trials have been initiated to study these effects in melanoma and other tumors.201–203 Important interactions among these modalities are shown in Figure 3.
Ipilimumab’s toxicities are immune-based and include pruritus, rash, fatigue, and diarrhea (about 1/3 to half of patients), while colitis requiring intervention is less common (7–10%) but can cause bleeding and even bowel perforation (<1%). Hypophysitis may result in adrenal, thyroid, and reproductive hormone insufficiency requiring replacement hormone therapy and sometimes a brief course of therapeutic glucocorticosteroid for local inflammation, especially if vision is compromised by compression of the optic chiasm by the inflamed pituitary. Immune-related hepatitis and rare cases of other organ involvement such as neuropathy have been reported. The first line of therapy for most immune-related adverse events is generally a brief course of therapeutic glucocorticoids followed, if necessary, by anti-tumor necrosis factor antibody (for rare cases of steroid-resistant hepatitis, mycophenolate mofetil is preferred) (reviewed in Ref. 204). While an association between autoimmune reactions and tumor regression has been reported for CTLA4 antibody therapy,205 and the generation during therapy of a higher circulating lymphocyte count has also correlated with therapeutic benefit, these associations are imperfect and do not provide insight into pretreatment biomarkers that can be used to foresee the likelihood or either benefit or toxicity. However, a recent report details the presence in pretreatment melanoma biopsies of common exomic mutation sequences creating immunogenic MHC class I-binding tumor neoepitopes from the tumors of patients who benefited from CTLA4 blockade.206 This report, along with others that demonstrate benefit associated with antigen-specific immune responses to melanoma in patients benefiting from ipilimumab,207 lends support to the concept that immune checkpoint inhibition exploits existing antitumor immune responses, which in many cases are now believed to be tumor-specific mutations, presumably derived from the damage induced by carcinogens of known importance in tumorigenesis.208 Thus, combinations with other immunomodulators that provide costimulation, reduce negative signaling in the tumor microenvironment, or enhance the functions of antigen-presenting cells are under investigation. Insights from further study of pre- and posttreatment tumor and circulating cells and their gene expression and patterns of activation may also lead to better selection criteria for initial therapy in individual patients.
PD-1 (for programmed death), has two important ligands, PD-L1 (expressed on a wide variety of cells and variably inducible in tumors, generally by cytokines produced by infiltrating lymphocytes) and PD-L2 (predominantly expressed on hematopoietic cells). Ligand engagement of PD-1 triggers negative signaling in effector T cells, rendering them unresponsive to further antigen stimulation and eventualy apoptotic, a cascade that, in the case of tumor expressing PD-L1, may be one mechanism of tumor resistance to immune control. This may be of particular importance in “adaptive resistance,” a term used to describe the induction of PD-L1 on tumor cells by γ-interferon produced by infiltrating CD8 lymphocytes, which may set the stage for effective immunotherapy by PD-1 or PD-L1 blocking antibodies.209–211 Such antibodies, fully human or extensively humanized, have recently been studied in patients with advanced melanoma and several other malignancies, and the results of these studies, which showed objective responses in about 25–30% of patients previously treated with ipilimumab and, if BRAF mutant, with a BRAF inhibitor, led to the approvals of pembrolizumab and nivolumab in late 2014 for advanced pretreated melanoma. However, the randomized trial that demonstrated a dramatic progression-free and overall survival benefit of nivolumab over dacarbazine (hazard ratio for death 0.42, 1-year survival 73% vs 42%) in the first randomized study for patients with untreated BRAF wild-type melanoma194 (Figure 4), quickly led to the routine use of these antibodies in the first-line setting, even for patients with BRAF mutant melanoma.
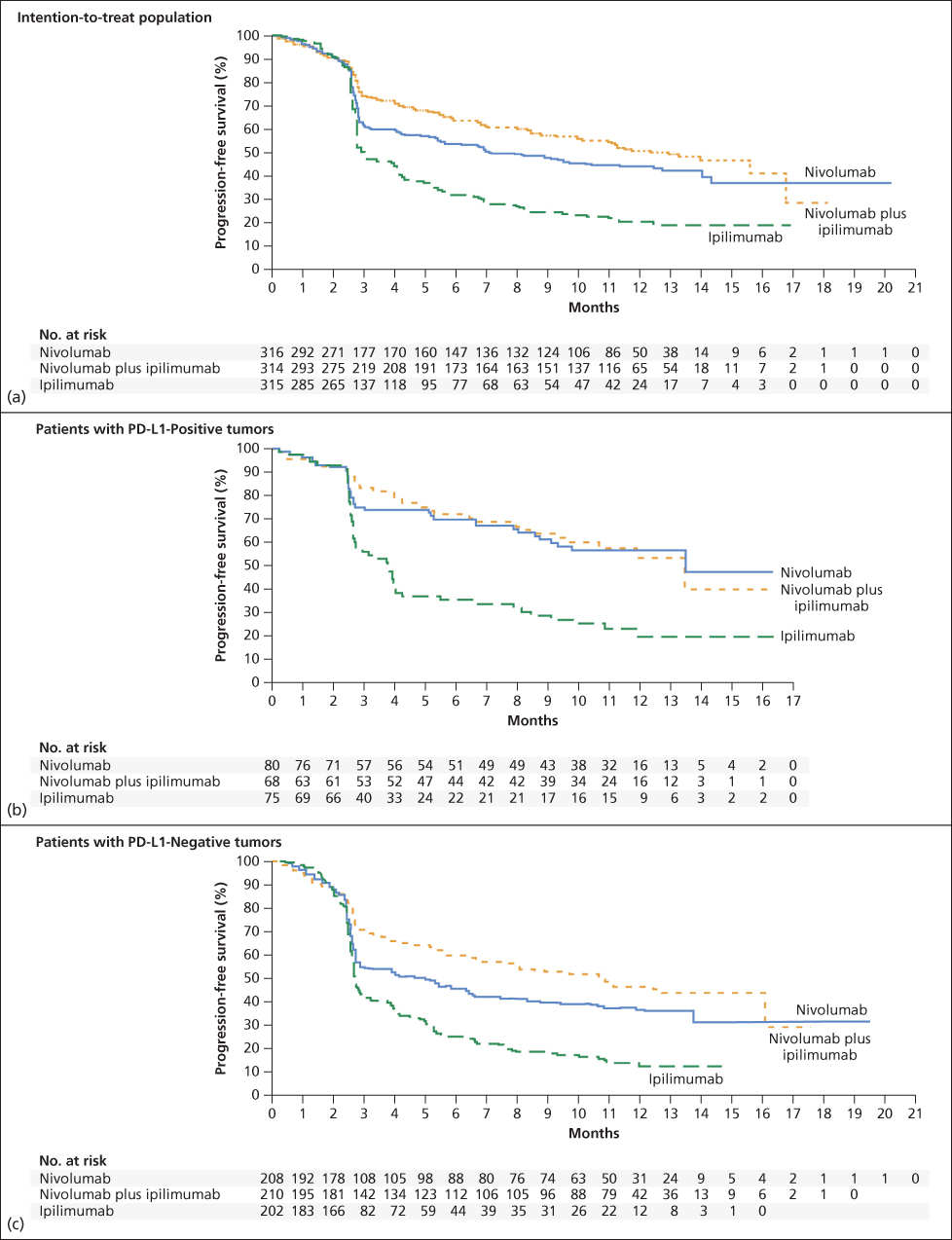
Figure 4 PFS, nivolumab plus ipilimumab versus either single agent. (a) Kaplan–Meier curves for progression-free survival in the intention-to-treat population. The median progression-free survival was 6.9 months (95% CI, 4.3 to 9.5) in the nivolumab group, 11.5 months (95% CI, 8.9 to 16.7) in the nivolumab-plus-ipilimumab group, and 2.9 months (95% CI, 2.8 to 3.4) in the ipilimumab group. Significantly longer PFS was observed for nivolumab plus ipilimumab than for ipilimumab (hazard ratio for death or disease progression, 0.42; 99.5% CI, 0.31 to 0.57; P < 0.001) and for nivolumab versus ipilimumab (hazard ratio, 0.57; 99.5% CI, 0.43 to 0.76; P < 0.001). (b) PFS for patients with PD-L1-positive tumors, and (c) PFS for patients with PD-L1-negtive tumors. Source Larkin et al 2015.196 Reproduced with permission of the New England Journal of Medicine.
The toxicity data for PD-1 blocking antibodies have demonstrated the expected superior therapeutic ratio over single-agent ipilimumab, which has lower benefit and higher immune-related side effects (data summarized in Table 8). Most toxicities affect the same tissues targeted by ipilimumab’s immune-related adverse effects but appear to be less frequent and severe; unique toxicities such as pneumonitis, nephritis, and neuritis have been reported in occasional patients on PD-1 axis blockade, and rare case reports describe other unusual events likely to represent loss of immune tolerance resulting from this form of checkpoint blockade. However, also shown in Table 8, while the combination of CTLA4 and PD-1 blockade has shown higher response rates and progression-free survival over single-agent ipilimumab (further detailed below), the toxicities of the combination are much more frequent, of higher grade, and include events not reported previously for either agent alone. The advantage of combined blockade over PD-1 blockade alone is also under intense investigation, as the retrospective subset analyses of this ground-breaking study suggested that for high PD-L1-expressing tumors, the benefits of single agent nivolumab are comparable to those of combined therapy, while for tumors without PD-L1 expression, the optimal therapy may require both agents. It has been postulated that blocking PD-L1 rather than PD-1 would leave intact the immune tolerance dependent on PD-L2:PD-1 interactions. Two PD-L1 antibodies currently under investigation in melanoma and other malignancies (Atezolizumab and Durvalumab) have demonstrated preliminary activity and may be combinable with other agents of interest in melanoma, including bevacizumab (antivascular endothelial factor, which has immunopotentiating properties), MAPK inhibitors, and other immunomodulatory agents that costimulate cytotoxic cells or inhibit local suppressive cells or molecules in the tumor microenvironment.
Combination therapy with concurrent ipilimumab and nivolumab showed particularly dramatic antitumor effects confirmed in a phase III study comparing the combination with each of the single agents in untreated advanced melanoma.197 Several recent reports have described potential biomarkers of benefit from PD-1/PD-L1 antibodies in both melanoma and non-small-cell lung cancer, including the expression of PD-L1 on tumor and/or stromal cells, the presence of a brisk CD8 lymphocyte infiltrate, and oligoclonality of the T-cell receptors among the tumor-infiltrating CD8 cells in melanoma,215, 216 and the explosion of clinical settings in which these agents are now undergoing evaluation is certain to reveal additional insight into predictive factors and mechanisms of activity and failure. Other checkpoint blocking antibodies and related immunomodulatory agents also hold promise in melanoma, including agonistic antibodies that costimulate T cells through ligands associated with activation (agonistic anti-CD137, anti-OX40, anti-CD40, anti-CD27) and dendritic cells (Toll receptor ligands and tumor vaccines antigens bound to dendritic cell-targeting antibodies). Other strategies include the inhibition of other immunosuppressive signals in the tumor microenvironment, such as indoleamine dioxygenase or signaling receptors on suppressive tumor-associated macrophages.
Sequential or simultaneous administration of molecularly targeted and immunotherapeutic agents
The data from retrospective series suggested that patients with BRAF-mutated melanoma who started with targeted therapy and then received ipilimumab at the time of progression had more unfavorable outcomes than patients who started with immunotherapy and crossed over to targeted therapy at the time of treatment failure.217, 218 While this outcome may have resulted from a true treatment-sequence effect, it may also reflect the assignment of patients with more aggressive disease to targeted therapy first, and patients with less aggressive tumor to immunotherapy first. An ongoing randomized clinical trial to test this question of optimal sequence, comparing the two combination regimens with the highest reported activity in melanoma (detailed below) as first-line therapy with a crossover to the alternative therapy at progression, will answer this critical question of the optimal first-line therapy and sequence of therapies for these patients, who comprise approximately half of those with advanced melanoma arising in common cutaneous sites (clinicaltrials.gov NCT 02224781). Concurrent therapy with MAPK inhibitors while initial combination therapy with vemurafenib and ipilimumab proved excessively toxic,219 many other combinations of molecularly targeted therapy with immunotherapy are under investigation, based in part on promising data supporting the importance of BRAF mutation in resistance to immunotherapy and its inhibition in promoting immune response (reviewed in Ref. 220).
Cytotoxic agents and combinations with immunotherapy for advanced melanoma
Until the advent of high-dose IL-2, therapy for metastatic melanoma was of very limited benefit, with low rates of objective response to single-agent cytotoxic therapies and limited response durations for both IL-2 and cytotoxic agents or combinations. The standard and only FDA-approved drug was dacarbazine, with response rates in the 10–12% range, and an oral equivalent of this drug, temozolomide, has often been used with similar outcomes but better tolerance.221 Although expectations were high for temozolomide as treatment for patients with brain metastases due to its central nervous system penetration and its widespread use in primary brain tumors, the low activity of dacarbazine and temozolomide in melanoma tempered enthusiasm, and it is now used predominantly for patients who have failed or cannot tolerate the other agents detailed above. Alternative multiagent regimens have been more toxic and without proven benefit despite reports of higher response rates, particularly to “biochemotherapy” combinations with at least 3 cytotoxic agents and 2 immunotherapies, usually IL-2 and IFN-α.222 Nanoparticle-albumin-bound paclitaxel or unmodified paclitaxel as a single agent or in combination with carboplatin are cytotoxic regimens with modest activity against melanoma but have not demonstrated survival benefit in prospective trials.223
Melanoma metastatic to the brain
Melanoma has the highest propensity of any adult solid tumor to spread hematogenously to the brain, and due to its vascular nature and its high growth rate in many cases, it poses serious threats to the survival and well-being of patients. Surgical resection may be required for diagnosis and is often necessary for immediate relief of the complications of edema, bleeding, and rapidly progressive neurologic deficits. The impact of whole-brain radiation given in standard or alternative dose and fractionation schedules has been minimal and cannot be distinguished from the benefits of simply treating patients with glucocorticosteroids.224 Stereotactic radiotherapies (SRS) have the most favorable outcomes despite the lack of randomized, controlled comparisons,225 with the maximum size and number of lesions amenable to treatment varying by center and type of modality (gamma-knife or cyberknife). Whole-brain radiotherapy is of limited palliative benefit and may be offered to patients with numerous lesions or failure of prior therapy, including systemic agents, which are increasingly showing activity against melanoma brain metastases (molecularly targeted agents226 and ipilimumab227). Ongoing investigations test double MAPK inhibition for melanoma metastatic to the brain, including preoperative therapy of resectable disease to allow post-therapy analysis of responding and nonresponding metastases (clinicaltrials.gov NCT01978236). Despite the lack of direct penetrance of the intact BBB by these therapies,228 their level of activity against melanoma brain metastases is in the range reported outside of the central nervous system, probably due to several factors, including the loss of BBB integrity resulting from tumor growth and inflammation as well as, in the case of immunotherapies, peripherally activated lymphocytes that gain access and migrate to the melanoma metastasis in the CNS.229, 230 Further understanding of the process of brain metastasis for the prevention and selection of optimal therapies will depend on factors that include the molecular characteristics of the primary site as well as changes occurring as melanoma cells experience other metastatic niches before the brain. Investigation of earlier-stage disease has provided limited and heterogeneous insights, but recent reports suggest the importance of the PTEN/AKT/PI3 kinase pathway either intrinsically or via signals from nearby astrocytes.163 It will be important to study the effects of other checkpoint inhibitors such as PD-1 and PD-L1 antibodies in patients with melanoma metastatic to the brain, and such studies are ongoing (clinicaltrials.gov NCT02085070 and NCT02320058).
Tumor-infiltrating lymphocyte (TIL) therapy in melanoma
The ability to isolate, expand, and infuse large numbers of autologous tumor-derived cytotoxic T lymphocytes, usually derived from dissected fragments of excised tumor and expansion of predominantly CD8 lymphocytes in IL-2-containing medium into patients with advanced melanoma, has been under investigation for over 20 years and appears to be improving. Using regimens that include lymphodepleting high-dose chemotherapy and, in some cases, total body irradiation to provide homeostatic cytokines and high-dose IL-2 to stimulate in vivo expansion and survival of antitumor effector cells, response rates in the range of 50% with durable complete remissions in 20% of patients have been reported among patients experiencing adequate TIL (tumor-infiltrating lymphocyte) growth and able to complete the entire course of therapy.231 Further manipulations have included pretreatment with BRAF inhibitors, “young” TIL cells, and immune checkpoint blockers, and it is likely that the need for HDIL-2 in these regimens will be replaced by more tolerable factors to expand and maintain the infused cytotoxic cell product. Recent observations regarding the immunobiology of TIL cells in melanoma are consistent with the findings detailed earlier for effective melanoma immunotherapy with PD-1 checkpoint blockade, including the presence of multiple immune checkpoints (PD-1, LAG3, TIM3) and costimulatory molecules (4-1BB, ICOS) on CD8 effector T cells and oligoclonality of T-cell receptor beta gene sequences on CD8 cells with antigen specificity for tumor-specific mutations.232
The recent advent of multiple new targeted agents and immune checkpoint inhibitors will provide ample opportunities to study the role of TIL cell therapies both for patients who fail to achieve durable benefit from the former agents and as part of multicomponent strategies for optimal therapy of advanced melanoma. While advanced melanoma will continue to be a lethal disease for a fraction of patients, the rapid emergence of highly active molecularly targeted therapies and immunotherapies with mechanisms of action and resistance that are more successfully probed and manipulated promise to counter the threat of this constellation of diseases and to continue to lead the way in new therapeutic discoveries.
Summary
Melanoma is rising in incidence and mortality in a pattern reflecting exposure to ultraviolet light (both natural and artificial) and other carcinogens that have not been fully elucidated. Familial risk factors include rare mutations of cyclin kinase genes and more common polymorphisms of genes such as the melanocortin receptor that control pigmentation of the skin and hair. Other risk factors include large numbers of moles or atypical nevi. Among the clinicopathologic subtypes, nodular melanoma has the most unfavorable prognosis owing to more rapid growth, the absence of an initial noninvasive radial growth phase, and a propensity to be nonpigmented, resulting in thicker tumors at diagnosis. Surgical excision guidelines include the need for adequate margins around the primary site to reduce the probability of local relapse; sentinel node biopsies guided by lymphoscintigraphic- and lymphatic-tracking dyes at the time of wide local excision; and completion lymph node dissection for patients with one or more involved sentinel nodes. Imaging guidelines are often tailored to the calculated risk of relapse using the 2009 AJCC staging system based on Breslow thickness, ulceration, mitotic rate, and number and size of nodal metastases. Metastasis occurs via predominantly hematogenous spread for uveal and mucosal melanoma and some cutaneous melanomas, while lymphatic spread is also common with cutaneous melanomas. Surgical management of metastatic melanoma depends on the number and location of metastases but can be associated with prolonged relapse-free survival. Metastatic melanoma is divided by prognosis into skin/soft tissue only, lung, and visceral metastasis and/or elevation of serum lactate dehydrogenase. Metastasis to the brain is more common in melanoma than any other malignancy and carries the most unfavorable prognosis, although improvements have resulted from stereotactic radiosurgery, and new molecularly targeted therapies as well as immunotherapies have shown activity against brain metastases. Melanoma is an immunogenic tumor that has been successfully treated with high-dose interleukin-2, ex vivo expanded tumor-infiltrating lymphocytes, and antibodies that block the CTLA4 and PD-1/PD-L1 immune checkpoints. Combinations with radiation therapy have also shown promise through multiple modulations of suppressive factors in the tumor immune microenvironment. Molecularly targeted agents, particularly those that inhibit sequential steps in the mitogen-activated protein kinase pathway in melanomas carrying an oncogenic activating mutation of BRAF, have also improved survival, and new agents to prevent or overcome resistance as well as to target other molecular drivers are under investigation. The challenge of brain metastases, likely originating from cells with molecular features predisposing them to traffic to and thrive in the brain, is still an important cause of melanoma death despite some responsiveness to targeted and immunotherapies and remains under intense investigation.
Related posts:




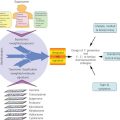

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


