Fig. 6.1
Schematic presentation of sizes, locations of the carbohydrate side chains, and currently known mutations (red arrows) and functionally significant polymorphisms (green arrows) in the gonadotropin subunits, i.e., CGA, LHB, FSHB, and CGB. The numbers below the right ends of the bars indicate the numbers of amino acids in the mature subunit proteins. Symbols “Y” and “O” indicate the locations of N-linked and O-linked carbohydrate side chains, respectively. The amino acid numbering includes the signal peptides (24 amino acids for CGA, 20 for LHB, 18 for FSHB and 20 for CGB), but the bars only depict the mature peptides. For references, see Table 6.1 and the text
The CGA gene is located on chromosome 6q12.21, FSHB is found on chromosome 11p13, and the CGB/LHB gene cluster, consisting of one LHB and four CGB encoding genes and two apparent pseudogenes, is located on chromosome 19q13.32 [4, 5]. CGA consists of four exons, and LHB, CGB and FSHB are each composed of three exons. The crystal structures of deglycosylated hCG [6] and FSH [7] are similar and reveal that both subunits contain so-called “cystine knot structures” that are similar to some remotely related growth factor-type signaling molecules. Each subunit has an elongated shape, with two β-hairpin loops on one side of the central cystine knot and a long loop on the other side. The noncovalent interaction between the two subunits is stabilized by a segment of the β-subunit that extends like a “seatbelt” around the α-subunit and is “locked” by a disulfide bridge.
Gonadotropin Receptors
Like their ligands, the gonadotropin receptors are structurally related glycoproteins, belonging to class A of the large family of G protein-coupled receptors (GPCRs) (see Figs. 6.2 and 6.3) [8–10]. They each have a transmembrane domain that traverses the plasma membrane as seven α-helices connected by three extracellular and three intracellular loops. The glycoprotein hormone receptors form a subgroup within this gene family with a distinct long N-terminal extracellular domain, comprising approximately half the size of the receptor. The C-termini of the receptor proteins form short intracellular tails. Whereas the FSHR binds only FSH, LH, and hCG are structurally so similar that they bind to the same LHCGR. Both gonadotropin receptor genes are localized on chromosome 2p21 [8, 10], and they carry considerable structural similarity, especially in their transmembrane domains. The clearest difference is that the first 10 exons encode the extracellular domain of LHCGR, whereas this domain is encoded by 9 exons in the FSHR. The last long eleventh exon in LHCGR (tenth in FSHR) encodes the transmembrane and intracellular domains.
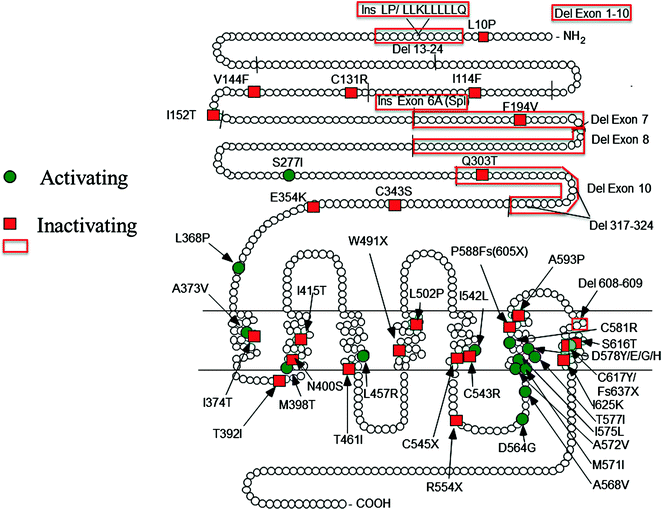
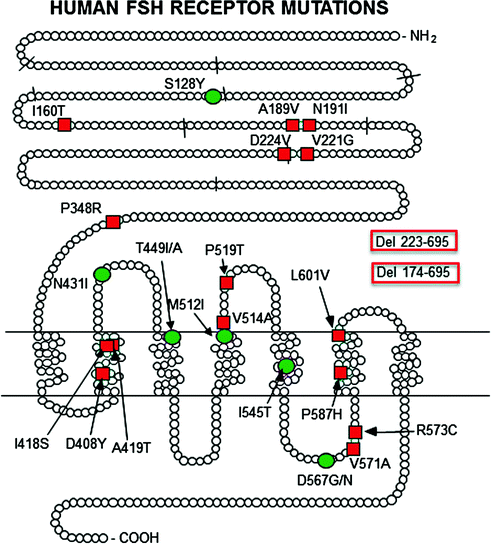
Fig. 6.2
Locations of the currently known mutations of LHCGR. Red squares and boxes indicate inactivating mutation and green circles activating mutations. The short lines across the peptide chain depict the exon boundaries. The original references are found in earlier reviews on the topic [9, 97]. The mutations detected thereafter are described in Refs. [99, 102, 147–149]. Fs frame shift, Ins insertion, Del deletion, Spl splice variants, X premature stop codon
Fig. 6.3
Locations of the currently known mutations of FSHR. Red squares and boxes indicate inactivating mutation and green circles activating mutations. The short lines across the peptide chain depict the exon boundaries. The original references are found in earlier reviews on the topic [9, 117]. The mutations detected thereafter are described in Refs. [125, 130–157]. Del deletion
Gonadotropins bind to the extracellular domain of their cognate receptors. This domain contains several leucine-rich repeats that are found in various proteins responsible for protein–protein interactions. The domain is glycosylated in an N-linked fashion, but the role of these structures in receptor function is not clear. The extracellular and transmembrane domains are connected by a short hinge region that may influence ligand specificity, as shown with a human mutation of LHCGR lacking this region [11] and subsequent in vitro mutagenesis studies [12]. The transmembrane domain, with its seven membrane-spanning α-helices and intracellular and extracellular loops, resembles other GPCRs and is crucial for signal transduction. In particular, the third intracellular loop, the sixth transmembrane domain, and the cytoplasmic tail are closely involved in G protein coupling and signal transduction. The crystal structure of the gonadotropin receptors (FSHR, and structurally related TSHR) has been partially unraveled [13–15].
The gonadotropin receptors are primarily coupled to a G protein that activates adenylyl cyclase and elevates intracellular cyclic adenosine monophosphate (cAMP) levels [8, 10]. These receptors also activate other signaling pathways, however, including phosphatidylinositide turnover, intracellular Ca2+, mitogen-activated protein kinases, and activation of the Gi protein [8, 10]. The alternate signaling pathways are often activated at higher hormone concentrations and receptor densities in target cells, and their functions may be limited to special circumstances in which gonadotropin levels are elevated, e.g., ovulation, pregnancy, or constitutively activating receptor mutations (see sections on activating mutations). A schematic presentation of the signaling cascades mediating gonadotropin action is shown in Fig. 6.4.
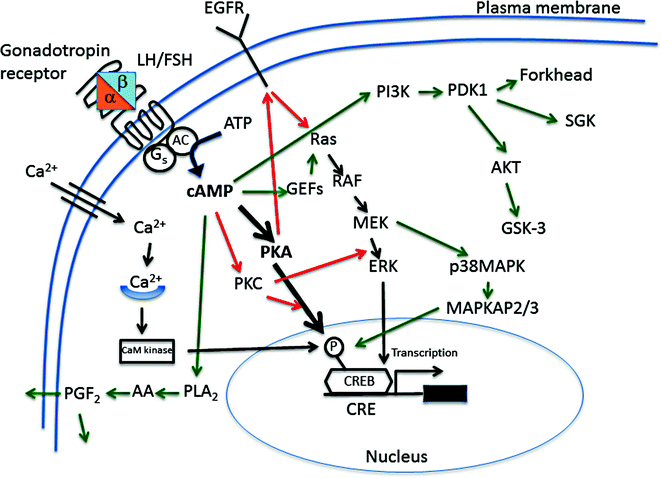
Fig. 6.4
Main signaling cascades employed in LH/hCG and FSH action. Black arrows depict pathways common for both hormones, red arrows those specific for LH/hCG and green arrows those specific for FSH. AA arachidonic acid, AC adenylyl cyclase, ATP adenosine trisphosphate, CaM calmodulin, cAMP cyclic adenosine-3’:5’-monophosphate, CRE cAMP response element, CREB cAMP response element binding protein, EGFR epidermal growth factor receptor, ERK extracellular signal-regulated kinase, Forkhead transcription factor, GEF guanine nucleotide exchange factor; G s stimulatory guanine nucleotide binding protein; GSK-3 glycogen synthase kinase-3, MEK mitogen/extracellular signal-regulated kinase, PDK1 phosphoinositol-dependent protein kinase, MAPK mitogen-activated protein kinase, MAPKAP, MAPK-activated protein kinase; PGF 2 prostaglandin F2, PI3K phosphoinositol 3 kinase, PKA protein kinase A, PKC protein kinase C, PLA 2 phospholipase A2, RAF rapidly accelerated fibrosarcoma–ser/thr specific protein kinase, Ras small GTPase family, SGK serum glucocorticoid-regulated kinase
Gonadotropin Secretion and Actions Throughout the Male Life Span
Physiological studies have clarified in great detail the role of gonadotropins in the development and mature functions of male reproduction. In the fetal period, placental hCG, a superagonist of pituitary LH, is responsible for the stimulation of testosterone production by fetal Leydig cells, which is essential for masculinization of the male reproductive organs [16]. This occurs in collaboration with the Sertoli cell product, anti-Müllerian hormone (or Müllerian-inhibiting substance), and another Leydig cell product, insulin-like factor-3 (INSL3) that regulate, in the male fetus, the involution of the Müllerian ducts and the transabdominal phase of testicular descent, respectively [17, 18]. Although FSH secretion begins in the fetus, and fetal testes express FSHR [19], FSH’s role in the fetal period is uncertain. Data from animal models indicate that its first role is the stimulation of Sertoli cell proliferation in the prepubertal testis [20]. Fetal pituitary gonadotropin secretion begins at the end of the first trimester of pregnancy, with high levels in mid-pregnancy, followed by a decline toward the end of gestation, apparently resulting from development of feedback regulation [21]. A secondary increase in gonadotropin levels (often termed “minipuberty”, see Chap. 4) occurs during the first 4–6 mo of postnatal life, which is accompanied by a peak in testosterone production [22] and by Sertoli cell proliferation [23]. Spermatogenesis, however, remains quiescent, apparently because of lack of androgen receptors in Sertoli cells of the postnatal testis [24]. No functional role has been demonstrated for this period of the hypothalamic–pituitary–gonadal activation, and it may represent an adaptation to extrauterine life and the hormonal milieu following elimination of placental hormones.
The prepubertal phase of male development is typified by low circulating gonadotropin levels, but they are not absent, and ultrasensitive assays can detect pulsatile LH secretion especially at night [25]. Prepubertal testes also express both gonadotropin receptors, as shown indirectly by their functional responses to stimulation by hCG or FSH [26].
Puberty is the period of reawakening of gonadotropin secretion (see Chap. 4). At this time, both hormones reach adult levels and the pulsatile secretion pattern. LH activates Leydig cell growth, proliferation, and steroid production. FSH stimulates Sertoli cell proliferation and the metabolic functions (in concert with testosterone) that are needed for the initiation and maintenance of spermatogenesis. Whether FSH is mandatory for the pubertal initiation and adult maintenance of human spermatogenesis is controversial, but it is clear that qualitatively and quantitatively normal spermatogenesis is possible only in the presence of all endocrine and paracrine factors, including FSH, that are functioning in the pituitary–gonadal axis [27].
Human testicular function is maintained through old age. However, mean testosterone levels and spermatogenesis decline with aging [28–30]. This phenomenon of “andropause” or “late-onset hypogonadism” (LOH) and the controversies of its testosterone replacement therapy has recently received a great deal of attention (see Chap. 17).
When considering the influence of gonadotropin or gonadotropin receptor mutations on male reproductive functions, specific phenotypic effects can be identified during all stages of the male life span.
Gonadotropin Subunit Mutations
Common α-Subunit (CGA) and hCGβ-Subunit (CGB)
Mutations in the gonadotropin subunit genes are extremely rare, most likely because of the deleterious effects they would have on reproduction (see Table 6.1). Probably for the same reason, not a single patient with a germ line mutation in the CGA has been detected. Such mutations would lead to defective hCG, LH, FSH, and TSH function, a condition that would probably be lethal, particularly because of lack of hCG. Nevertheless, CGA knockout mice are viable, though hypogonadal and hypothyroid [31], but because this species does not produce chorionic gonadotropin, the gestation regulation may be under different hormonal control. Only one somatic CGA point mutation (E56A) in the ectopically produced hCG of a carcinoma has been described in the literature (see Fig. 6.1) [32]. Rare non-synonymous substitutions have been detected in CGB5 and CGB8 at higher frequency in women with recurrent miscarriages [5]. Despite the phenotypic association, only mild functional alterations were detected in the mutant hormones in vitro [33]. One relatively common (4.5% allelic frequency) amino acid-altering point mutation, V79 M, has been detected in heterozygous form in apparently asymptomatic subjects (see Fig. 6.1) [34]. This mutation hampers α/β dimer formation in vitro and, if present in homozygous form, could block sufficient bioactive hCG synthesis and be lethal. Curiously, additional studies have been unable to find this polymorphism in 5 European and Mexican populations [35, 36]. Milder forms of CGB mutations could, if they exist, cause intrauterine undermasculinization of male fetuses because of hCG’s crucial role in the stimulation of fetal testicular testosterone synthesis. However, because CGB is expressed by 4 duplicate genes, mutation in a single CGB gene may not be functionally significant.
Table 6.1
List of currently known mutations and polymorphisms in human LHB and FSHB genes
Gene | Location | Type | Amino acid change | Symptoms in patient(s)a | Functional effect | Reference |
|---|---|---|---|---|---|---|
LHB | Exon 3 | Inactivating missense | Q74R | Infertility, delayed puberty | No binding to receptor | [37] |
LHB | Exon 2 | Inactivating missense | G56D | Infertility, delayed puberty | Disrupted dimerization and secretion | [38] |
LHB | Exon2/Intron 2 boundary | Inactivating splice site mutation | Inhibition of intron 2 splicing | Sexual infantilism Secondary amenorrhea, infertility | Disrupted dimerization and secretion | [39] |
LHB | Exon 2 | Inactivating deletion | Deletion of amino acids 30–32 (HPI) | Testosterone–induced puberty, normal spermatogenesis off–treatment | Reduced stability and secretion, low bioactivity | [40] |
LHB | Exon 2 (signal peptide)/Intron 2 | Inactivating compound heterozygous splice site mutation/deletion | Inhibition of intron 2 splicing/Deletion of amino acids 7-10 (LLLL) | Hypogonadism, infertility Secondary amenorrhea, infertility | Disrupted secretion | [41] |
LHB | Exon 2 | Polymorphism with 2 missense mutations | W28A/I35T | Delayed tempo of puberty Slightly suppressed fertility | Increased bioactivity in vitro Short half-life in vivo | |
Association with PCOS? | ||||||
LHB | Exon 3 | Polymorphism, missense | G122S | Endometriosis | Not detected | [68] |
LHB | Exon 3 | Polymorphism, missense | A17T | Not studied | Slight decrease in signaling activity | [69] |
LHB | Exon 2 | Polymorphism, missense | M14I | Not studied | Unknown | [70] |
LHB | Exon 2 | Polymorphism, missense | H10R | Not studied | Unknown | [70] |
FSHB | Exon 3 | Inactivating, 2-bp deletion → frame shift | V79fs105X | Prim. amenorrhea, infertility Azoospermia | Truncated protein, disrupted secretion | |
FSHB | Exon 3 | Inactivating, missense | C69G | Delayed puberty, hypogonadism | Faulty tertiary structure and disrupted dimerization | [72] |
FSHB | Exon 3 | Inactivating, missense | C100R | Azoospermia | Faulty tertiary structure and disrupted dimerization | [78] |
FSHB | Exon 3 | Inactivating, nonsense | Y94X | Prim. amenorrhea, infertility Azoospermia | Disrupted secretion and loss of bioactivity | |
FSHB | Exon 3 | Inactivating, nonsense | Y94X | Prim, amenorrhea, infertility | Disrupted secretion and loss of bioactivity | [75] |
FSHB | Exon 3 | Inactivating, 1-bp deletion → frame shift | A97fs126X | Prim, amenorrhea, infertility | Disrupted stability and secretion | [76] |
FSHB | Exon 3 | Polymorphisms, silent | No change (Y94) | High FSH, association with PCOS+obesity | Studied | [158] |
LHβ Subunit(LHB) Mutations
Five reports describing inactivating mutations of the LHB gene have so far been published, comprising a total of 6 affected men and 2 women [37–41] (Table 6.1 and Fig. 6.1). The male subjects have similar phenotypes with delayed puberty, sexual infantilism, and infertility. The only exception is the male described by Achard et al. [40], who appeared to have normal spermatogenesis (see below). The men have undetectable LH, elevated FSH and anti-Müllerian hormone, low testosterone, and normal or low-normal inhibin B and estradiol levels. There is an elevated FSH but no LH response to GnRH stimulation. Testicular size is reduced (5–12 mL) and histological examination revealed [38, 41] tubular hypoplasia and greatly reduced spermatogenesis. Normal pubertal maturation can be induced in these men by testosterone treatment. hCG treatment induces a normal serum testosterone response, and long-term hCG treatment results in testicular enlargement and normal virilization, but spermatogenesis appears to recover only partially, and the men may remain infertile.
The man described by Achard et al. [40] represents a remarkable exception. Despite profound inactivation of LH through a 3-amino acid deletion (Table 6.1), the man had normal spermatogenesis. His other features, such as absent virilization, undetectable serum LH, and low serum testosterone levels, were as in the other men with LH inactivation. It was concluded that in this man, the residual LH activity was able to stimulate intratesticular testosterone levels sufficiently to maintain quantitatively normal spermatogenesis. Upon testicular biopsy, intratesticular testosterone was 20-fold reduced, suggesting that a high intratesticular testosterone concentration may not be necessary for spermatogenesis. The low residual testosterone, in conjunction with elevated FSH, may be sufficient to maintain spermatogenesis. Similar observations have been made on mice with disrupted LHCGR function [42, 43]. Perhaps the only reason why the intratesticular testosterone concentration is so high may be because this organ is the site of testosterone synthesis.
Different types of LHB mutations have been detected in affected individuals, including point mutations, deletions in the signal peptide and mature protein encoding regions, and splice site mutations. The severity of the phenotype is in good agreement with the severity of the inactivation of LH function when studied in vitro by expression of the mutated gene product (Table 6.1). Heterozygous family members, both men and women, seem to be asymptomatic, indicating that one functional LHB gene can encode sufficient LH synthesis.
These rare cases clarify some points about pituitary LH’s developmental role. Because the affected men were apparently normally masculinized at birth and had descended testes, pituitary LH is not needed for the stimulation of testicular testosterone or INSL-3 production in utero. Indeed, fetal testicular testosterone production is initiated seemingly autonomously but becomes subsequently dependent on placental hCG [16, 44], and fetal pituitary LH plays a minimal role in utero. In contrast, the endocrine function of the postnatal testes is critically dependent on pituitary LH, as demonstrated by the total absence of mature Leydig cells in testicular biopsy samples and of spontaneous puberty in its absence.
The two women with homozygous LHB inactivation [39, 41] have normal puberty, followed by secondary amenorrhea and infertility. Like men, their LH is undetectable, but FSH is normal to high, estradiol and progesterone are at the follicular level, and testosterone, androstenedione, dehydroepiandrosterone sulfate, and prolactin levels are normal. Apparently theca cell androgen production without LH stimulation is sufficient to provide substrate for FSH-stimulated granulosa cell estrogen production, which explains the normal puberty of the affected females. However, LH stimulation is crucial for ovulation, which explains the infertility of the affected women. In fact, the phenotype of the female LHB inactivation is identical with inactivating LHCGR mutation (see below).
LHB Polymorphisms
Several polymorphisms have been detected in the LHB gene (see Table 6.1 and Fig. 6.1), and their occurrence has been studied in relation to various phenotypic signs of male and female gonadal dysfunction, including hypogonadism and infertility [9, 45]. We concentrate below on findings that concern the effects on male reproductive function.
Of particular interest is the W8R/I15 N polymorphism resulting from two point mutations that are in linkage disequilibrium [46–48]. This variant (V) LH is common worldwide, with considerable ethnic variability in carrier frequency, from 0% in western India to 53.5% in aboriginal Australians [49, 50]. It was originally detected as an immunologically anomalous form of LH in a woman with normal reproductive function but undetectable LH using a specific combination of monoclonal antibodies to detect immunoreactive LH [51]. It has been shown to display increased bioactivity in vitro but to have a shortened circulating half-life [52–54]. However, there is also a report indicating a longer half-life of the variant in the circulation [55]. The discrepancy is likely due to differing experimental conditions used in the studies.
The polymorphism affects variably circulating LH concentrations. No difference was found in LH levels in 73–94-year-old Dutch men who were WT or V–LHB carriers [56], but significantly lower serum LH concentrations were detected in V–LHB carriers in a large cohort of 40–79-year-old men from 8 European countries [57]. In another study on young Estonian men (mean age 20.1 yrs), V–LHB carries were reported to have higher LH levels than WT controls [58]. Likewise, in women, both higher and lower concentrations of serum LH have been reported in V–LHB carriers [48, 59–61]. The best explanation for the discrepant findings is provided by the fact that V–LHB has eight additional point mutations in its promoter region [62]. They increase the transcriptional activity of the variant promoter by approximately 50% in vitro. However, the promoter could respond in vivo differently to various physiological and pathophysiological conditions, resulting in differences in the relative amounts of the two LH forms [63].
The significance of V-LH as a contributing factor to the variability of reproductive functions remains elusive. Most of the existing studies associate the LH variant with various mild reproductive disorders in men and women [reviewed in 9, 45]. In women, it has mild, although statistically significant, associations with fertility problems and ovarian steroidogenesis, including certain types of polycystic ovary syndrome, pregnancy complications, premature ovarian failure, and breast cancer occurrence. Many of these findings have been made in Japan, which, along with its lower frequency in Asian populations [49, 50] may indicate ethnic differences in its phenotypic expression.
In the male, several findings associate V-LH to reproductive function. When pubertal progression was compared in WT boys and heterozygotes for V–LHB, there was no difference in the age of onset of puberty, which is mainly determined by the reawakening of hypothalamic gonadotropin-releasing hormone (GnRH) secretion. Instead, the tempo of pubertal progression was significantly slower in boys who were heterozygous for the V–LHB allele [64]. When its occurrence was studied in cryptorchidism, the allele frequency in cryptorchid and healthy boys was similar, but when the frequency in cryptorchid boys was correlated with gestation duration, it increased significantly with increasing gestation duration, from 6% in boys born before week 30–45% in boys born beyond week 42 of pregnancy [65]. The final phase of testicular descent, from the inguinal region to the scrotum, depends on testicular testosterone production in late gestation. The tropic stimulus for this activity is provided by the combined action of hCG and pituitary LH, and when the duration of pregnancy is prolonged, the role of pituitary LH becomes more important. If this hormone is less active, as V-LH is, then testosterone production may be compromised, predisposing the post-term boys carrying the V–LHB allele to cryptorchidism. There is also a report describing an association of V-LH with familial and sporadic prostate cancer [66], but no association of V-LH has been found with male infertility [67]. Finally, V-LH is more prevalent in older men who are obese and have low testosterone levels [56].
These findings, together with those in women, indicate that V-LH represents a genetic variant of the hormone with borderline significant effects on a variety of reproductive functions in men and women. Most importantly, the clinician must be aware of its occurrence, because some immunoassay kits for LH are unable to detect V-LH, which may explain unexpectedly low LH levels.
FSHβ Subunit (FSHB) Mutations
Six women [71–76] and three men [73, 74, 77, 78] with a total of five different inactivating mutations of the FSHB gene have so far been described in the literature (Fig. 6.1 and Table 6.1). All women had similar phenotypes, with slightly variable severity, including sexual infantilism, lack of follicular development, and infertility, which are readily explained by the lack of FSH action on granulosa cell estrogen production and progression of follicular maturation beyond the early stages.
Of the reports on the 3 men with inactivating FSHB mutations, the one from Sweden described a 32-year-old man of Serbian origin who presented with azoospermia, normal puberty and male physical characteristics, and a selective FSH absence [78, 79]. His serum testosterone level was low–normal (11 nmol/L), and LH was slightly elevated (12 IU/L). These findings reflect the small size of his testes (3 mL/6 mL) and the missing FSH effect on their growth at puberty. Genetic analysis demonstrated a homozygous T to C mutation, predicting a C100R substitution in the FSHB protein. It was postulated that the elimination of a cysteine would impair formation of the proper intramolecular disulfide bond of FSHB. This would result in abnormal tertiary structure during FSHB synthesis, with extensive intracellular degradation, inability to dimerize with the CGA, defective glycosylation, and, finally, inability to form biologically active hormone.
The second male, reported from Israel [77], was an 18-year-old with slight delay of puberty, small testes, azoospermia, and a plasma FSH concentration below 0.5 IU/L. Total testosterone was low (4.5 nmol/L), and LH was increased (24.5 IU/L), indicating a defect in testosterone biosynthesis. DNA sequencing revealed the same homozygous 2-bp deletion in codon 79 that was found previously in female patients [71–73]. The mutation gave rise to a completely altered amino acid sequence between codons 79 and 105 of the FSHB chain, which was followed by a premature stop codon, and lack of translation of amino acids 106–129. Consequently, the translated FSHB protein was truncated and unable to associate with common α-subunit to form bioactive or immunoreactive α/β dimers.
The third man with inactivating FSHB mutation, described from Brazil [73, 74], had had normal puberty but presented with infertility resulting from azoospermia. His serum testosterone at the age of 30 yr was normal (26 nmol/L), FSH was low, and LH was elevated. His testes were small (12 mL), and testicular biopsy revealed Leydig cell hyperplasia and sparse, small seminiferous tubules with germinal cell aplasia, peritubular fibrosis, and few Sertoli cells. The C to A mutation in codon 94 of his FSHB gene brought about a premature stop codon (TAA). Because the phenotype of the female patient reported with this mutation was less severe than those of women with the other FSHB mutations [72–74], it was hypothesized that the missing amino acids 94–129 of FSHB may not bring about complete inactivation of FSH action.
Conspicuously, all 3 men with the FSHB mutation were azoospermic. As will be elaborated in the section on FSHR mutations, this is not the case with the five men reported to carry homozygous inactivating FSHR mutations [80]. They each displayed variable suppression of spermatogenesis but were not azoospermic. Neither are the knockout mice for FSHB [81] or FSHR [82, 83] azoospermic, and, moreover, the phenotypes of women with inactivating FSHB and FSHR mutation were practically identical [71–76, 84]. The more severe phenotype of the ligand mutation in men is curious also for the reason that animal data show the opposite [85], most likely because of the slight constitutive activity of FSHR in the absence of ligand. Why then are the three men with FSHB mutation azoospermic? This will be discussed in more detail in connection with FSHR mutations.
FSHB Polymorphisms
One functionally significant single nucleotide polymorphism (SNP) of FSHB-211G/T [86] has been recently identified and appears to affect both male and female reproductive function. In a study from Estonia, The TT phenotype carrier men had highly significantly lower serum FSH concentrations compared to GG men [87], and in infertile men, FSH was even lower [88]. A similar attenuating effect of the T allele was observed with the high FSH levels in Klinefelter syndrome patients [89]. The apparent mechanism for the reduced FSHB expression is attenuated transcriptional activity through an impaired LHX3 binding site [90]. The association of the T allele with lower FSH levels has been confirmed in other populations [91–93] and is associated with lower serum inhibin B and testosterone levels, smaller testes, and a lower sperm concentration. This polymorphism has been suggested as a pharmacogenomics tool to predict the response of men with idiopathic oligozoospermia to FSH therapy [94]. Indeed, significant improvement in sperm parameters following FSH treatment has been demonstrated in the minor allele carriers [91]. In women, curiously, both increased and decreased levels of FSH have been associated with the T allele [95, 96]. The combination of the -211G/T polymorphism with a common FSHR polymorphism in improving the pharmacogenetic prediction will be discussed later.
Gonadotropin Receptor Mutations
Gonadotropin receptor mutations are also quite rare. There are both activating and inactivating mutations in these genes, with widely varying phenotypic effects. Several mechanisms can underlie receptor inactivation: (1) decreased synthesis of receptor protein; (2) aberrant intracellular processing; (3) reduced or absent ligand binding activity; (4) impaired or absent signal transduction; (5) inability to anchor to the plasma membrane; (6) inability to dimerize, if needed, for signal transduction; (7) dominant negative effect, i.e., formation of non-functional dimers, or (8) increased degradation. Unlike the activating mutations, the types of inactivating mutations vary from point nonsense and missense mutations to various size insertions and deletions. In this case, only homozygotes or compound heterozygotes for two different inactivating mutations produce a clear phenotype because the normal function of a single receptor allele supplies a sufficient number of functional receptors to sustain normal gonadotropin action.
Activating receptor mutations can be classified into four categories: (1) receptor activation in the absence of ligand hormone; (2) increased sensitivity of the receptor to its normal ligand; (3) relaxed specificity of the receptor to ligands; and (4) acquired novel functions of the mutated receptor. All activating gonadotropin receptor mutations detected have been missense point mutations that are almost invariably localized to the transmembrane region or its vicinity (see Figs. 6.2 and 6.3). These mutations produce a structural alteration, activating constitutively the signal transduction machinery of the transmembrane region. Most clearly, receptor activation without ligand explains the altered function of these mutations. Abnormal phenotypes occur in heterozygous individuals because activation of the translation product of a single allele is sufficient to initiate the altered functional response.
LHCGR Mutations
Inactivating Mutations
About 30 different LHCGR mutations have been reported (Fig. 6.2). In the human male, LHCGR function is essential at all ages for testicular androgen production. Therefore, these mutations influence male sexual differentiation, development, and mature functions from the fetal period through adulthood. LHCGR mutations thus form one etiological subclass of 46, XY disorders of sexual development (DSD). The receptor inactivation varies in completeness, and the phenotype of affected males depends on its extent [9, 10, 97]. In the mildest forms, the phenotype is hypospadias and/or micropenis (Leydig cell hypoplasia, LCH Type 2), and in the completely inactivating form, the male phenotype is complete male to female sex reversal (LCH Type 1) [9, 97]. The latter group presents with near-total to total lack of male type sexual differentiation in utero and during postnatal development. The phenotype resembles the complete form of androgen insensitivity syndrome but notably differs by the lack of pubertal breast development, which, in androgen insensitivity, is explained by the conversion of testosterone to estradiol. On histological examination, the testes of the individuals with complete LHCGR inactivation are totally devoid of mature Leydig cells, which explain the lack of testosterone production and absent masculinization. However, it is apparent that even in complete LHCGR inactivation, some testicular steroidogenesis is possible, because epididymides and vasa deferentia are present [9]. The explanation for these structures may be low, but functionally sufficient, constitutive steroidogenesis in precursor fetal Leydig cells. It could induce in paracrine fashion, partial masculization of the Wolffian ductal structures. This has also been shown with LHCGR knockout mice in which low basal androgen synthesis in their testes can stimulate spermatogenesis, a response which can be eliminated by antiandrogen treatment [42].
In women, LHCGR inactivation causes a milder phenotype with normal intrauterine female sex differentiation and a seemingly normal FSH-dependent component of ovarian function [9]. Affected women present with primary amenorrhea and normal primary and secondary sex characteristics, increased gonadotropin levels, and a lack of ovarian response to hCG treatment. Histological study of ovarian tissue revealed all stages of follicular development, with the exception of preovulatory follicles and corpora lutea [98]. These observations emphasize the importance of LH for ovulation and formation of corpora lutea. LH may also be needed for the maturation of antral to preovulatory follicles. A very interesting recent study reported on successful pregnancy of a woman with inactivating LHGCR mutation following ovum donation [99], indicating that the extragonadal LHCGR expression, demonstrated in a variety of male and female organs, is redundant for fertility.
Activating Mutations
The first gonadotropin receptor mutations detected were those activating constitutively the LHCGR [100, 101], perhaps because of their striking phenotype. The syndrome caused by this mutation, i.e., familial male-limited precocious puberty (FMPP), is a gonadotropin-independent form of precocious puberty, also called “testotoxicosis”. Boys with FMPP begin to exhibit signs and symptoms of puberty between 0.5 and 3 yr of age. If they remain untreated, puberty progresses rapidly, resulting in premature epiphyseal closure, compromised adult height and associated behavioral problems of premature puberty. For the treatment, various antihormonal therapies have been used (medroxyprogesterone, ketoconazole, cyproterone acetate, anastrozol, bicalutamide) [102] to reduce virilization and advancement of bone age. Men with this syndrome appear to be fertile.
FMPP has been known for a long time and was originally assumed to result from a circulating nongonadotropic factor that induces premature LH-independent activation of Leydig cell function. However, the subsequent detection of point mutations in the LHCGR gene, which, in transfection studies, caused constitutive activation of the LH signaling pathway in the absence of ligand, provided a logical molecular pathogenesis for the syndrome. Today, more than 10 activating mutations of the LHCGR gene are known (see Fig. 6.2), and they all (except for one) are localized in or near the transmembrane region of the LHCGR, which is pivotal for LH signal transduction. The mutations change the conformation of the transmembrane region of the receptor such that it assumes, at least partially, an activated conformation in the absence of ligand. The consequence is premature activation of Leydig cell testosterone production before the pubertal onset of LH secretion. In cell transfections, the mutated receptor protein usually displays an approx. tenfold increase in basal cAMP production, whereas maximal stimulation is often suppressed. The latter alteration has no physiological importance, because at the circulating levels of LH, receptor occupancy remains relatively low. Whether other signaling pathways that are initiated by LHCGR activation (see Fig. 6.4) are also activated has not been systematically studied.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


