Fig. 8.1
Effects of hyperprolactinemia on the hypothalamic–pituitary–testicular axis
Another mechanism to explain the suppression of gonadal function in men with prolactinoma is a direct tumor mass effect on gonadotrophs. Large adenomas often cause deficiency of multiple anterior pituitary hormones, including the gonadotropins. However, a tumor mass effect appears to be less important than is hyperprolactinemia in the development of hypogonadism in most men with prolactinomas. In fact, there is no significant difference in the prevalence of testosterone deficiency between men with micro- or macroprolactinomas, either before or after medical treatment. Among patients with macroadenomas, however, testosterone levels are inversely correlated with tumor size, perhaps reflecting the increasing likelihood of panhypopituitarism as well as effects of high prolactin levels with larger tumors [17, 18].
Studies in male rats provide evidence that prolactin also affects the testes directly. Prolactin receptors are present in all stages of the cycle of the seminiferous epithelium, on spermatogonia and spermatocytes, and on rodent Sertoli and Leydig cells [19]. PRL modulates testosterone production by regulating the bioconversion of precursor steroids to testosterone under the influence of LH, and by upregulating LH receptor expression. However, there is no evidence so far for direct prolactin effects on the human testis.
The semen analysis in hyperprolactinemic men is characterized by fewer sperm implying spermiogenic arrest, and by reduced sperm motility and/or quality. In addition to the gonadotropin disturbance, hyperprolactinemia may produce structural changes in testicular cells expressing the androgen receptor [20]. On the other hand, male reproductive functions are normal in prolactin-KO and prolactin receptor-KO mice [21–23]. Thus, the functions of PRL in the testis remain uncertain.
Diagnosis
Young adult women with prolactinomas came to medical attention because of galactorrhea and irregular menses, but in men this tumor can be difficult to recognize. The symptoms and signs of hypogonadism (Table 8.1) may not become obvious until the tumor becomes large, or hyperprolactinemia and gonadotropin deficiency become severe. Therefore, the diagnosis of prolactinoma in men is often delayed until a visual field defect or other symptoms secondary to tumor pressure on surrounding structures occur (e.g., headaches, seizures, altered personality, cranial nerve palsies, symptoms related to the deficiencies of other pituitary hormones, hydrocephalus). With long-standing hypogonadism, the testes are usually soft but of normal size, and the semen analysis frequently reveals low semen volume, oligozoospermia or azoospermia [24]. Most men with hyperprolactinemia have low-normal serum levels of LH, testosterone and dihydrotestosterone, and low frequency and amplitude spontaneous LH secretory episodes [25] together with a normal increase in serum LH levels following GnRH administration [8]. The identification of a high prolactin level is the beginning of the diagnostic process; however, other causes of hyperprolactinemia should be excluded. Prolactin levels in patients with prolactin-secreting adenomas are usually higher than 100 ng/ml and tend to be proportional to the size of the tumor. Adenoma size is also correlated with hormonal dysfunction prevalence at presentation and following treatment [17].
Table 8.1
Clinical manifestations secondary to hypogonadism in men with prolactinoma
Infertility | Loss of pubic hair |
Decreased libido | Osteoporosis |
Impotence | Apathy |
Gynecomastia | Loss of muscle mass |
Galactorrhea | Decreased sense of well-being |
Lower PRL values are generally found with medication use, pituitary stalk compression, renal failure, cirrhosis, hypothyroidism and polycystic ovary disease [26]. Macroprolactin is a large molecular weight variant in which serum prolactin levels are increased because of binding to an IgG producing a large macromolecule with reduced hormone clearance; it is usually unassociated with clinical problems [27]. Computerized tomography (CT) and magnetic resonance (MR) allow for detailed noninvasive imaging of the pituitary gland. Both methods are effective in identifying large pituitary tumors; CT is better for defining bone erosions and calcified structures, but MR with gadolinium enhancement provides superior anatomical detail to detect prolactinomas and their relationship to neighboring structures. MR is more sensitive than CT in the identification of microadenomas [28].
Therapy
Current therapeutic options for patients with prolactinomas include observation, medical therapy with dopamine agonists (e.g., bromocriptine, cabergoline), transsphenoidal or transcranial surgery, and conventional or stereotactic radiotherapy.
The first-line treatment of prolactinoma is generally pharmacotherapy with a dopamine agonist [29–32]. These drugs were developed based on the finding that prolactin synthesis and secretion are regulated by the tonic inhibitory effects of hypothalamic dopamine via dopamine D2 receptors on lactotrophs. Bromocriptine mesylate is a semisynthetic ergot alkaloid and long-acting dopamine receptor agonist. It normalizes prolactin levels in 80–90% and 60–75% of patients with micro- and macroprolactinomas, respectively [31]. Generally, the therapeutic doses are in the range of 2.5–15 mg/day, and most patients are successfully treated with 7.5 mg or less. However, doses as high as 20–30 mg/day may be necessary for patients who demonstrate resistance. Headache, nasal congestion, nausea, vomiting and dizziness are common side effects. Quinagolide is a more selective D2 agonist with only mild antagonistic effects at α1-adrenoceptors. It is better tolerated, and 100 times more potent than bromocriptine and readily crosses the blood–brain barrier. This drug induces an excellent response both in normalizing prolactin levels and in restoring gonadal function, with normalization of libido and improvement in the ejaculation and in nocturnal penile tumescence and rigidity parameters. Usual doses are 0.15–0.6 mg/day. However, at least 6–12 months of treatment is needed to obtain a significant improvement in seminal fluid parameters and circulating testosterone levels. Quinagolide is not available in the USA.
The most commonly used dopamine agonist is cabergoline because of its prolonged duration of action and lower incidence of side effects. Tumor shrinkage and/or disappearance were observed in 60–88% of cases [33]. Cabergoline appears to be more efficacious than bromocriptine in normalizing prolactin levels and in restoring gonadal function. Normalization of serum prolactin levels was followed by restoration of gonadal function in all patients with microprolactinomas, and in most patients with macroadenomas [34]. Moreover, the beneficial effects occurred more rapidly than with bromocriptine or quinagolide [35]. In cabergoline-treated patients, the percentage of immature germ cells was lowered and sperm viability, swollen tails and penetration into bovine cervical mucus were increased significantly during the first 3 months of treatment [36]. Generally, the median starting dose of cabergoline was 1 mg/week in patients with macroprolactinomas and 0.5 mg/week in those with idiopathic hyperprolactinemia or microprolactinomas. The dosage may be increased up to 1–3 mg twice weekly with a goal of PRL <50 ng/ml. Dosage increases should occur every 4 weeks. In addition, cabergoline may be effective in patients resistant to bromocriptine or quinagolide [31]. Furthermore, treatment with cabergoline and androgen replacement therapy may improve the metabolic profile and reduce the prevalence of metabolic syndrome in male hyperprolactinemic patients with concomitant testosterone deficiency [37].
Approximately 24 and 11% of patients demonstrate resistance to bromocriptine and cabergoline, respectively [31]. So far, there is no evidence that somatostatin receptor agonists are effective in patients with prolactinomas resistant to dopamine agonists.
Surgery may be necessary in men who have not tolerated or have not responded to medical therapy, with invasive macroadenomas and visual field impairment with no immediate response to dopamine agonists, or in patients with complications, such as intra-tumoral hemorrhage or cerebrospinal fluid fistula [38, 39]. A transsphenoidal or endoscopic transnasal approach is the preferred technique for microadenomas and most macroadenomas, while craniotomy is rarely necessary. Surgical success rates are highly dependent on the size of the tumor and the experience and skill of the surgeon. Pituitary tumors are almost always benign adenomas, and the preservation or restoration of anterior pituitary function, including gonadal function, is an important goal of surgery.
Because of the excellent therapeutic responses to medical therapy and surgery, radiotherapy is generally not considered a primary treatment for prolactinomas, but is reserved for patients who are refractory to medical and surgical therapy. Hypopituitarism, including hypogonadism, is a frequent side effect following radiotherapy.
In the uncommon case of hyperprolactinemia and hypogonadism induced by prolactinoma that is resistant or intolerant to medical therapy, and with contraindications to surgery, gonadotropin therapy can be used to induce virilization and advance spermatogenesis sufficiently to achieve fertility [40, 41].
It is noteworthy that, in patients treated with dopamine agonists for hyperprolactinemia, especially those with large tumors and panhypopituitarism, hypogonadism can persist despite normalization of prolactin levels, so that testosterone replacement therapy becomes necessary in addition to dopamine agonist therapy [15].
Acromegaly
Physiopathology of Hypogonadism
Acromegaly is characterized by the excessive production of growth hormone (GH). More than 99% of cases of acromegaly result from a tumor of the pituitary gland secreting either GH alone, or both GH and prolactin. Gangliocytomas of the hypothalamus or pituitary, and ectopic neuroendocrine tumors secreting GH-releasing hormone (GH-RH), account for less than 1% of cases. Patients with acromegaly have a considerable burden of complications and coexisting morbidities. In particular, acromegaly is associated with cardiovascular and metabolic diseases such as hypertension, glucose intolerance or diabetes, cardiomyopathy and sleep apnea which all increase the risk of morbidities and mortality [42].
Hypogonadism has been reported in 49–70% of acromegalic patients [43]. Secretion of prolactin by the tumor together with GH, stalk disruption by suprasellar growth, and destruction of gonadotrophs by tumor have been implicated in the development of hypogonadism. However, Katznelson et al. [44] described low testosterone levels in 39% of men with somatotroph microadenomas, most of whom had normal prolactin levels. Therefore, other factors may contribute to the pathogenesis of hypogonadism in acromegaly. GH excess is often associated with a low level of sex hormone-binding globulin (SHBG), so that free testosterone levels should be measured to accurately diagnose testosterone deficiency in men with acromegaly. Hypogonadism occurs more often in patients with macroadenomas than in those with microadenomas because of the tumor mass effect on surrounding normal tissue.
GH appears to regulate testicular function by stimulating the local production of IGF-1 which modulates steroidogenesis and spermatogenesis. GH receptors are located in both the male and female gonads and genitalia [45]. Furthermore, GH is important in the development of the male and female reproductive systems, as demonstrated by the expression of GH receptors not only on the testis and ovary but also on Wolffian and Müllerian duct-derived structures [46]. GH administration to rats increases Leydig and Sertoli cell responses to FSH and LH [47]. However, high levels of GH and IGF-1 induce severe testicular alterations and disrupt the hypothalamic–pituitary–testicular axis. Repeated administration of very high doses of GH in dogs reduced LH and testosterone levels as well as testicular weight with germ cell degeneration and epithelial atrophy [48]. In a human study, administration of GH (0.01–0.03 mg/day/kg) for 3 weeks to eight healthy male volunteers increased estradiol levels and decreased anti-Mullerian hormone, inhibin B and LH levels, whereas the GH antagonist pegvisomant reduced only estradiol [49]. The extent of testicular damage appears to be related to the severity of acromegaly. In fact, serum GH and IGF-1 levels correlated negatively with serum dihydrotestosterone levels (Fig. 8.2). Several mechanisms have been proposed to explain these findings, including a change in androgen metabolism [50], FSH and LH suppression by increased somatostatin tone, a decrease in the serum concentration of SHBG, and lactogenic effects of GH separate from those of prolactin [51]. In addition, prostatic dysfunction, leading to alterations of the seminal fluid, may occur in men with acromegaly. In fact, GH/IGF-1 excess causes prostate enlargement with a high prevalence of prostatic abnormalities [52].
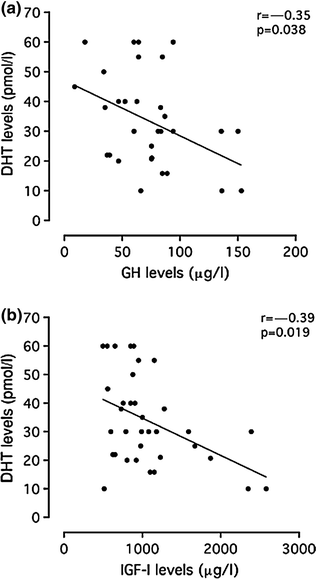
Fig. 8.2
Relationship between serum GH and IGF-1 levels to serum dihydrotestosterone (DHT). Reprinted from Colao et al. [57]
Diagnosis
The symptoms of hypogonadism in men with acromegaly are similar to those of men with prolactinoma and include oligozoospermia and reduced sperm motility. These manifestations, like acromegalic-specific clinical features (Table 8.2), progress slowly. Therefore, the diagnosis of acromegaly is often delayed for as many as 5–15 years.
Table 8.2
Specific clinical features of acromegaly
Recent acral growth | Osteoarthrosis |
Prognathism and malocclusion | Kyphosis |
Frontal bossing | Carpal tunnel syndrome |
Widened spaces between the teeth | Proximal muscle weakness |
Increased breadth of the nose | Fatigue |
Generalized visceromegaly | Deep and resonant voice |
Cardiomyopathy | Hyperhidrosis |
Hypertension | Psychological disorders |
The specific clinical manifestations of acromegaly are caused by excessive GH/IGF-1 secretion. In addition, pituitary tumor mass effects (headaches, hypopituitarism and visual disturbances) are common, because of the high prevalence of macroadenomas (about 75% of somatotroph adenomas).
It is now known that an elevated random serum GH level is not absolutely necessary for the diagnosis of acromegaly. By contrast, serum IGF-1 levels are invariably high in acromegaly. If the IGF-1 level is increased, and a random GH level is normal, an oral glucose tolerance test (75 g) is used to establish the diagnosis. A normal response is a decrease in the serum GH level to <1 μg/l, but acromegalic patients do not suppress normally, and 20% of patients have a paradoxical GH rise [53].
After the clinical and endocrine diagnosis of acromegaly is made, radiological evaluation of the pituitary gland should be performed, preferably by MR with gadolinium enhancement to localize and delineate the tumor from the surrounding tissues. GH-secreting tumors tend to be larger than prolactinomas at the time of the radiographic evaluation, perhaps due to a later clinical presentation.
Therapy
The treatment goals for men with hypogonadism secondary to acromegaly are the normalization of serum GH and IGF-1, and prolactin levels when hyperprolactinemia is present, and the preservation and restoration of gonadotropin secretion and testicular function.
Trans-sphenoidal adenomectomy is the preferred treatment for GH-secreting tumors, inducing an endocrine remission in 80% of patients with microadenomas, and 50% of those with macroadenomas in experienced centers [54]. It is likely that many centers do not achieve these success rates, however, which is a matter of concern. Surgery relieves the compression of adjacent structures, including the pituitary stalk and surrounding pituitary cells. If gonadotropin secretion is normal prior to surgery, gonadal function can be restored if GH secretion normalizes. Tominaga et al. [55] reported serum testosterone concentrations of less than 300 ng/dl (10.5 nmol/L) in 14/20 men (70%) with active acromegaly. Postoperatively, the serum GH level normalized in 14 patients, while the testosterone concentration returned to normal in 11 patients (79%), restoring gonadal function. This increase in testosterone levels may result from an increase of SHBG production [56], or an increase in LH secretion [57].
The three classes of medical therapy for acromegaly are based on receptor targets of the GH/IGF-1 axis: pituitary somatostatin receptor subtypes, pituitary dopamine D2 receptors and peripheral GH receptors [58].
Somatostatin analogues normalize IGF-1 levels in 60–70% of patients and achieve tumor shrinkage in about 40% of patients. Long-acting analogues of somatostatin are available and improve compliance. Octreotide-LAR consists of octreotide incorporated into microspheres of a biodegradable polymer. It is injected intramuscularly at a dose of 10, 20 or 30 mg (every 4 weeks). Slow-release lanreotide is a similar preparation and is administered intramuscularly at a dose of 30 mg (every 1–2 weeks) or 60 mg (every 4 weeks). Lanreotide Autogel achieves the same effect as slow-release lanreotide, but is longer acting. It is administered by deep subcutaneous injection at a dose of 120 mg (every 6–8 weeks). Pasireotide is being evaluated in patients with acromegaly. Based on differences in binding affinity and functional activity, pasireotide may have a stronger inhibitory effect on GH secretion with tumors that express somatostatin (sst) receptors other than sst2 [59]. Important side effects of somatostatin analogues are gallstone formation, which is generally silent, abdominal cramps, and decreased glucose tolerance, especially with pasireotide [60–66]. Suppression of GH and IGF-1 levels by somatostatin analogue therapy for 6 months was associated with a significant increase in LH and total testosterone and dihydrotestosterone levels; in most hypogonadal men, there was also improvement in sperm number and motility [61].
Dopamine agonists are less effective than are somatostatin analogues in normalizing IGF-1 levels (10–30% of patients) and reducing the tumor mass (<20% of cases). However, dopamine agonists are effective in those GH-secreting tumors that also secrete prolactin, or immunostain positively for prolactin. In addition, combined treatment with somatostatin analogues and dopamine agonists induces a greater suppression of IGF-1 levels compared with drug alone and improves testicular function [62].
Pegvisomant is the only GH receptor antagonist available for clinical use [63]. It has been shown in clinical trials to normalize IGF-1 levels in up to 97% of patients and can improve comorbidities such as insulin resistance. Furthermore, because the efficacy of pegvisomant is not dependent on tumor somatostatin receptor expression, pegvisomant effectively inhibits IGF-1 secretion in patients who are non-responders or partial responders to somatostatin analogues. GH secretion is not inhibited during pegvisomant therapy, and pegvisomant does not treat the tumor, necessitating regular monitoring for pituitary tumor growth.
Radiation therapy should be considered for patients with contraindications to pituitary surgery, or those whose operation and/or medical treatment has failed. Radiotherapy induces a slow response and may damage normal pituitary tissues, leading to hypopituitarism, and worsening hypogonadism in 50% of patients 10 years after irradiation. Stereotactic radiation surgery (SRS), either in single or hypofractionated protocols, has undergone rapid technical improvement. With this approach, the temporal lobe, the optical system and the normal pituitary are exposed to less radiation. SRS may be associated with better biochemical remission, and estimates of hypopituitarism range from 6 to 30% [64].
Cushing’s Disease
Physiopathology of Hypogonadism
Cushing’s disease is characterized by chronic glucocorticoid excess secondary to hypersecretion of ACTH and other proopiomelanocortin peptides. It is commonly caused by a pituitary corticotroph adenoma.
Gonadal and sexual dysfunctions are common in men with Cushing’s syndrome [65–69]. High levels of glucocorticoids decrease serum testosterone levels through several mechanisms (Fig. 8.3). Excessive production of glucocorticoids may produce gonadotropin deficiency by acting at the pituitary and hypothalamic levels. Two regions of the mouse GnRH promoter (distal and proximal negative glucocorticoid response elements) regulate transcriptional repression by glucocorticoids. Glucocorticoid receptors appear to induce glucocorticoid repression of GnRH gene transcription by virtue of their association within a multiprotein complex at the negative glucocorticoid response element [70]. In addition, elevated levels of glucocorticoids directly suppress testicular function, inducing apoptosis in Leydig cells [71], inhibiting LH receptor signal transduction, decreasing the oxidative activity of 11beta-hydroxysteroid dehydrogenase and impairing Leydig cell steroidogenesis [72].
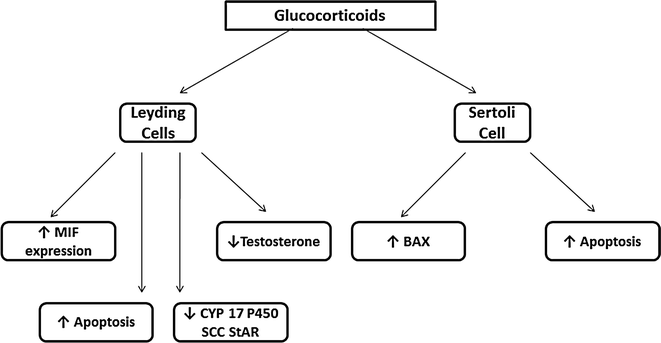
Fig. 8.3
Mechanisms involved in the induction of hypogonadism in men with Cushing’s disease
Diagnosis
Since the description by Dr. Harvey Cushing at the beginning of the twentieth century, it is well known that hypogonadism is common in active Cushing’s disease [73] (Table 8.3).
Table 8.3
Clinical features of Cushing’s syndrome
Centripetal obesity | Proximal myopathy |
Buffalo hump | Abdominal pain |
Moon face | Psychiatric disturbances |
Facial plethora | Hypertension |
Purple striae | Backache, vertebral collapse, fracture |
Skin thinning | Polydipsia, polyuria |
Easy bruising | Renal calculi |
Acne, oily skin | Hyperpigmentation |
Skin infections | Male hypogonadism |
Lethargy | Hirsutism, oligomenorrhea |
Males have loss of libido, impotence, oligozoospermia and histological damage to the testes. Basal LH and FSH levels are commonly decreased, the response to GnRH is impaired, and testosterone levels are low. The hypogonadotropic hypogonadism in male patients is reversible and improves after hypercortisolism is suppressed or blocked [65, 68].
When Cushing’s syndrome is suspected, initial laboratory testing to detect hypercortisolism includes measurement of 24-hour urinary free cortisol excretion, and/or the 1 mg overnight dexamethasone suppression test [74]. The sensitivity and specificity of the first test in detecting cortisol excess are 95 and 98%, respectively. Because of the difficulty in obtaining 24-hour urine collections in many outpatients, some physicians use the 1 mg overnight dexamethasone suppression test with a diagnostic cutoff of 1.8 μg/dL in the morning cortisol level (sensitivity 98% and specificity 80%). Another diagnostic approach is the late-night (11 PM) salivary cortisol determination [75]. Cortisol is highly stable in saliva, and the level is positively correlated with free serum or plasma cortisol levels and is independent of the rate of salivary flow.
Once the diagnosis of Cushing’s syndrome is established, the source of the excess cortisol production needs to be determined. The plasma level of ACTH in the late afternoon is useful in identifying the ACTH-dependent pathologic state. At this time of day, plasma ACTH levels exceed 10 ng/L in Cushing’s disease, while plasma ACTH levels are generally suppressed in Cushing’s syndrome secondary to adrenal disease. The high-dose dexamethasone test (8 mg) and the CRH test can be performed to obtain a clearer differential diagnosis. In Cushing’s disease, serum cortisol levels and 24-hour urinary free cortisol excretion are generally suppressed after the 8-mg dexamethasone test, and ACTH levels rise following CRH stimulation.
Pituitary ACTH-secreting adenomas are most often microadenomas with a diameter <5 mm in 45% of cases, and only 10–20% of patients with Cushing’s disease have a macroadenoma [76]. While MR imaging is required in all patients with ACTH-dependent Cushing’s syndrome, pituitary MR imaging is often normal in patients with corticotropinomas. MR should be performed with thin overlapping sections and high field strength (1.5 T) magnets. In the absence of a detectable pituitary tumor, bilateral inferior petrosal venous sinus and peripheral vein catheterization with simultaneous collection of samples for measurement of ACTH should be performed to localize ACTH production to the pituitary and exclude the ectopic production of ACTH [77]. Stimulation with CRH, together with bilateral and simultaneous sampling, increases the sensitivity of this procedure.
Therapy
The hypogonadotropic hypogonadism characteristic of male Cushing’s disease is a reversible phenomenon [65, 68]. In fact, cure of hypercortisolism restores the decreased plasma testosterone, FSH and LH levels to normal and improves sexual and gonadal dysfunctions.
Trans-sphenoidal adenomectomy, after accurate preoperative localization of the corticotroph adenoma, represents the treatment of choice for Cushing’s disease patients, but can be technically challenging [78]. The remission rate for microadenomas has ranged from 48.7 to 100%, whereas for those with macroadenomas, it has ranged from 30.8 to 100%. A trans-sphenoidal approach was also successfully used in ACTH-producing adenomas located in the pituitary stalk. A recent meta-analysis reported that 11.5–47.4% of patient will experience recurrence of their disease as long as 10 years after surgery, so that follow-up must be life-long [79].
Transcranial surgery (TCS) has been used in the past and is occasionally used today.
Patients unresponsive to surgical treatment, or those in whom surgery is deemed inappropriate (large invasive tumors, or high surgical risk), are referred for pituitary irradiation with adjunctive medical therapy [80]. There is some evidence that stereotactic radiation may lead to biochemical remission faster than with conventional radiation therapy, while estimates of new hypopituitarism at 4–5 years range from 5 to 35%.
The development of novel medical therapies may limit the use of radiotherapy to patients who are unresponsive to or intolerant of medical treatment, or it may result in the deferral of radiotherapy until after a period of control of cortisol secretion by medical therapy.
Current medical therapies include drugs to decrease ACTH secretion, inhibit corticosteroid synthesis or block their peripheral actions [78]. The first class of drugs (serotonin antagonists, dopamine agonists, GABA agonists and somatostatin analogues) inhibit ACTH release. However, few cases so treated have achieved permanent remission. The second group of drugs includes inhibitors of steroidogenesis (ketoconazole, metyrapone, aminoglutethimide and mitotane). These drugs have the advantage of being rapidly effective in most cases. However, the mechanism(s) of action has little selectivity, and extra-adrenal effects are likely to occur. Ketoconazole inhibits various cytochrome P450 enzymes, including C17-20 lyase involved in the synthesis of sex steroids. Thus, the drug not only lowers cortisol levels, but also decreases testosterone levels such that gynecomastia, impotence, loss of libido and oligozoospermia could occur [81]. The third class of drugs is represented by the glucocorticoid antagonist (mifepristone). Although a potentially important therapy that has been approved by the FDA to control hyperglycemia in adults with Cushing’s syndrome, few patients have been treated with mifepristone, and further clinical studies are needed to assess long-term effectiveness and safety, and the impact on gonadal function. Few studies have reported the efficacy of cabergoline treatment in patients with Cushing’s disease and Nelson’s syndrome [82]. With each of these drugs, improvement in hypercortisolemia and insulin sensitivity may result in a rise in SHBG, LH and testosterone levels.
Finally, bilateral adrenalectomy is reserved for cases in which other treatment modalities have failed. However, the risk of Nelson’s syndrome is high with this approach. Adrenal surgery is followed by a rapid and definitive control of cortisol excess in nearly all patients, but it induces adrenal insufficiency.
Non-functioning/Gonadotroph Adenomas
Physiopathology of Hypogonadism
Clinically non-functioning pituitary adenomas (NFPA) represent about 30% of pituitary tumors and are the most commonly encountered macroadenomas [83]. The prevalence of NFPA is 1.5 per 100.000 people, and among them, gonadotroph adenomas account for 43–64% of silent adenomas. Hypogonadism is present in about half of the patients with NFPA, of whom about 50% have mild hyperprolactinemia [83], reflecting stalk compression that might contribute to impaired gonadal function.
The majority of “non-functional” tumors are inefficiently secreting rather than non-secreting. They usually synthesize and immunostain for FSH and/or LH, and/or free α- and β-subunits [84]. Gonadal dysfunction in men with NFPA is primarily related to intra-sellar expansion of the tumor with compression of residual glandular tissue and may cause hypopituitarism both by direct compression of the portal vessels that limits delivery of hypothalamic releasing factors and by focal ischemic necrosis of the normal pituitary tissue [85]. LH secretion is impaired in about 53% of NFPA [103–105]. However, disruption of ACTH, TSH and GH secretion may further inhibit gonadal function.
Diagnosis
The majority of NFPA are diagnosed incidentally or as a result of headache, visual disturbances or neurological symptoms. Less often, deficient pituitary hormone secretion, including gonadotropin deficiency with hypogonadism, brings the patient to medical attention. These tumors can attain considerable size before manifesting clinically. A mild or moderate increase in the prolactin level is common. A complete endocrine evaluation, including measurement of FSH, LH, α-subunit, testosterone, TSH, FT4, ACTH, cortisol, GH and IGF-1, as well as prolactin should be performed in order to recognize functional lesions and diagnose hypopituitarism. Magnetic resonance imaging with gadolinium enhancement represents the best way to detect a pituitary tumor.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


