Causes of chronic liver disease
Prevalence in United States
Associated type of hypogonadism
Nonalcoholic fatty liver disease (NAFLD)
11%
Unclear, but likely more secondary
Alcoholic liver disease (ALD)
2%
Both primary and secondary
Hepatitis B/C
0.3% Hep B, 1.6% Hep C
Unclear, potentially more primary
Hereditary hemochromatosis
0.4%
Predominantly secondary, but likely a component of primary as well
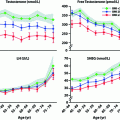
CLD or cirrhosis is associated with multiple systemic processes, and one of the underappreciated comorbidities is testosterone deficiency [3, 4]. Men with advanced liver disease present with phenotypic manifestations typical of hypogonadism. The exact prevalence of low testosterone in liver disease patients has not been carefully studied, but limited literature suggests that hypogonadism, defined by low free testosterone, occurs in 70–80% of patients with CLD, with evidence of both primary testicular failure and disruption of the hypothalamic-pituitary-gonadal (HPG) regulation (Table 11.2) [4–6]. Among men with cirrhosis, low serum testosterone has been reported in up to 90% [7–10] with worsening hypogonadism corresponding to worsening liver failure [11].
Table 11.2
Total testosterone, free testosterone, and SHBG in various chronic liver diseases
Total T | Free T | SHBG | |
|---|---|---|---|
Nonalcoholic fatty liver disease (NAFLD)a | Decreased | Decreased | Decreased |
Alcoholic liver disease (ALD)a | Decreased | Decreased | Increased |
Hepatitis B/Ca | Decreased | Decreased | Increased |
Hereditary hemochromatosisa | Decreased | Decreased | Increased |
End-stage liver disease (decompensated cirrhosis) | Decreased | Decreased | Decreased |
Post liver transplantation | Increased | Increased | Decreased |
Pathophysiology
The pathogenesis of hypogonadism in liver disease is complex, differs by diagnosis, and is hypothesized to be mediated in part by toxic metabolites affecting the hypothalamic-pituitary-gonadal (HPG) axis, hyperestrogenemia, and altered protein synthesis [12].
Cirrhosis, particularly end-stage cirrhosis, is most commonly associated with central hypogonadism [11]. Individuals with cirrhosis may lose luteinizing hormone (LH) pulsatility, suggestive of hypothalamic dysfunction [12]. The response to human chorionic gonadotropin (hCG) is blunted, consistent with an impaired gonadotropic response [12–14]. Systemic disease, in general, is associated with central hypogonadism, and is hypothesized to be due to downregulation of GnRH production by elevated inflammatory cytokines including interleukin (IL)-1, IL-6, and tumor necrosis factor (TNF)-α) resulting in functional suppression of the HPG axis [7, 15]. Hyperprolactinemia may also be seen in liver disease, particularly in alcohol-related liver disease, likely due to alterations in the dopaminergic system that contribute to GnRH suppression with suppression of the HPG axis [16–22].
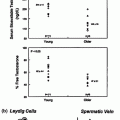
However, hypogonadism in the setting of liver disease may also be due to testicular damage. In particular, ethanol and its metabolites have been shown to exert a toxic effect on Leydig cells and are involved in onset of the fibrogenic processes [12, 23]. In vitro studies of isolated Leydig cells, isolated perfused rat testes, and testicular homogenates have consistently shown reduced testosterone synthesis [13, 24]. Despite damage to Leydig cells, testosterone levels in patients may be normal early in the disease process due to a compensatory increase in LH.
Estrogen levels can be elevated in men with cirrhosis, which is hypothesized to be due, at least in part, to reduced metabolism and clearance of estrogen, as well as portosystemic shunting further resulting in impaired/bypassed estradiol metabolism, and increased peripheral aromatization of testosterone [7, 11, 25–27]. Estrone and estradiol production may also be increased secondary to increased production of androstenedione from the adrenal glands. Increased androstenedione may be due to a combination of a shift in androgenic steroidogenesis from cortisol to androstenedione with worsening liver disease and to inflammation-mediated inhibition of conversion of DHEA to DHEA-S [12, 28]. Ethanol may also exacerbate hyperestrogenemia by increasing aromatase activity and interfering with sex hormone endocrine homeostasis before the onset of severe liver dysfunction [29, 30].
Elevation in estrogen in conjunction with decreased testosterone levels results in an increased estrogen/androgen ratio, felt to contribute to feminizing features of liver disease, including altered fat distribution, gynecomastia, spider angiomatas, palmar erythema and reduced prostate volume [13].
The liver regulates testosterone bioavailability via production of SHBG and albumin and is also responsible for the production of enzymes important in the aromatization, conversion to dihydrotestosterone (DHT) and breakdown of androgens. Plasma testosterone binds with high affinity predominantly to SHBG (~60%) and albumin (~38%) [31]. SHBG levels are frequently elevated in certain forms liver disease, resulting in increased total testosterone, but lower free or bioavailable testosterone levels. SHBG levels are low in NAFLD and NASH and are often normal in hemochromatosis. Regulation of SHBG is multifactorial with hepatic signaling molecules and increasing levels of estrogen and decreasing levels of testosterone hypothesized to be important driving factors in SHBG elevation. Thus, early in liver disease, total testosterone levels may be elevated, when free testosterone levels are actually low. SHBG levels normalize after clinical decompensation, likely due to severity of impaired synthetic capacity despite hormonal stimuli. Testosterone, in turn, is important in protein synthesis (including albumin), modulation of liver regeneration, inhibition of cytokine release, and anabolic effects related to liver metabolism [12].
Hemochromatosis
Hemochromatosis is characterized by iron overloading leading to systemic illnesses such as diabetes mellitus, hyperpigmentation, arthralgias, cardiomyopathy, cirrhosis and hepatocellular carcinoma, and hypogonadism [32]. The most notable iron overloading syndrome, hereditary hemochromatosis (HH), is one of the more common autosomal genetic disorders in Caucasians with an estimated prevalence of 1 per 220–250 individuals. HH can be divided into the primary HFE gene defect and non-HFE-related disease. The most common HFE gene defect is a missense mutation in which tyrosine is substituted for cysteine at amino acid position 282 of the protein product C282Y. The defective HFE protein signals that the body is incorrectly deficient in iron because it fails to stimulate normally the production of hepcidin, leading to increased iron absorption from the upper intestine. In addition to HH, there are multiple secondary iron overload syndromes such as thalassemia major and parental iron overload from frequent blood transfusions [32].
Liver injury and hypogonadism are two of the more common abnormalities in HH [33]. Reported prevalence rates of hypogonadism have ranged from 10 to 100% [33–35]. While liver disease is seen earlier in the disease course, hypogonadism is generally seen in its later stages. The exact timeframe for the manifestation of these symptoms is not clear. However, McDermott et al. in 2005 reported the 20 year follow-up of 141 men age 24–77 years who were diagnosed with HH with evidence of iron deposition on liver biopsy at a single center in Ireland. At the time of diagnosis of HH, those with hypogonadism were on average 56 years of age while nonhypogonadal males averaged 52 years of age [33]. They found that only 9 men (6.4%) had abnormally low testosterone levels with low gonadotropin concentrations. This is in contrast to patients with juvenile (non-HFE) HH who have more severe iron overload, and generally present with delayed pubertal development rather than cardiac or liver disease [35].
Liver injury is hypothesized to occur as a consequence of cellular degradation products from hepatocyte injury from iron deposition, activating Kupffer cells which stimulate hepatic stellate cells to synthesize collagen leading to fibrosis and cirrhosis. In HH, C282Y homozygotes with serum ferritin levels >1000 micrograms/L have a higher risk of cirrhosis with prevalence of 20–45% while fewer than 2% of patients with ferritin <1000 micrograms/L have cirrhosis [32].
In HH, testosterone and LH levels are often low [33]. Iron deposition is found in the pituitary with high predilection for gonadotrophic cells. On the other hand, the secretion of ACTH and TSH is usually normal. There is decreased expression of transferrin and transferrin receptor in iron loaded gonadotrophs secondary to iron deposition [36, 37]. The depressed LH and testosterone with pituitary iron deposition is most consistent with secondary hypogonadism, but there may also be a component of primary hypogonadism since iron deposition is also found in testicular cells. Given that many of these studies did not involve a concomitant liver biopsy, it is not clear to what degree liver disease may contribute to hypogonadism in this setting.
SHBG levels are sometimes increased in HH [38]. Yeap et al. [36] showed that heterozygotes with the C282Y polymorphism without evidence of iron overload tend to have higher SHBG levels than do men who do not carry this mutation, but have normal total and free testosterone levels. These data suggest that the C282Y polymorphism may somehow influence SHBG levels, while the hypothalamic-pituitary-gonadal axis remains intact in the absence of significant iron overload [34]. It is nevertheless important to note that patients with HH may have other causes of hypogonadism such as obesity or Klinefelter syndrome [39].
Phlebotomy, with the goal of normalization of ferritin levels, remains a key treatment for hemochromatosis. Limited data suggest that phlebotomy can partially reverse hepatic fibrosis in approximately 30% of cases, and significantly lower the rate of HH-related HCC if iron removal is achieved before cirrhosis develops [40]. However, it is unclear if testicular atrophy or biochemical hypogonadism can be reversed with phlebotomy since it is hypothesized that hypogonadism may be a manifestation of irreversible iron deposition and damage to the pituitary gland [41].
In conclusion, HH is a genetic iron overloading syndrome with liver disease usually presenting before hypogonadism. Although advanced liver disease may contribute to hypogonadism, HH seems to occur predominantly due to iron deposition and damage to the pituitary gland. Primary hypogonadism due to iron deposition in the testes is less commonly seen [42]. Phlebotomy has been shown to reverse some degree of CLD before progression to cirrhosis, but further studies are needed to understand its potential impact on hypogonadism.
Alcohol-Induced Liver Injury
Alcoholic liver disease (ALD) is classified into three histologic stages: alcoholic fatty liver, alcoholic hepatitis, and cirrhosis. Fatty liver is a result of steatosis in the setting of heavy drinking, occurring in about 90% of individuals who drink more than 60 mLs per day [43]. In general, fatty liver can be arrested or significantly reversed with alcohol cessation [44]. Alcoholic hepatitis is characterized by inflammatory changes and degeneration of hepatocytes. Patients may have clinical signs of nausea, abdominal pain, hepatomegaly, fever, jaundice, and bleeding. Alcoholic hepatitis is associated with a very high short-term mortality rate and chronically, approximately 40–50% of patients are at risk for progression to cirrhosis [45]. Cirrhosis remains the final stage of ALD with about 10–15% of patients with alcoholism generally at risk [46].
While dose relationship is the most important risk factor for ALD, other important risk factors need to be considered. In general, men who ingest more than 2–3 oz (60–90 mL) of alcohol per day, or woman who ingest more than 1 oz (30 mL) per day for 10 or more years, are at higher risk of cirrhosis [43]. Two important categories include individuals with (1) alcohol dependence which includes those who drink excessively and develop physical tolerance; and (2) alcohol abuse which includes those engaged in negative social and health consequences of drinking [46]. A large study from Denmark also suggested that the type of alcohol was important, with beer or spirits more likely to be associated with liver disease compared to other drinks such as wine [47]. Binge drinking, female gender, obesity, malnutrition, and family history of alcohol dependence were also associated with a higher risk of ALD [48].
The bidirectional nature of ALD and hypogonadism remains an active area of research. ALD is known to contribute to both biochemical and clinical hypogonadism (Fig. 11.1).‘ In fact, alcoholic patients seem to have more profound phenotypic and biochemical evidence of hypogonadism than other forms of liver disease potentially since alcohol has a direct toxic effect at each level of the hypothalamic-pituitary-gonadal axis.
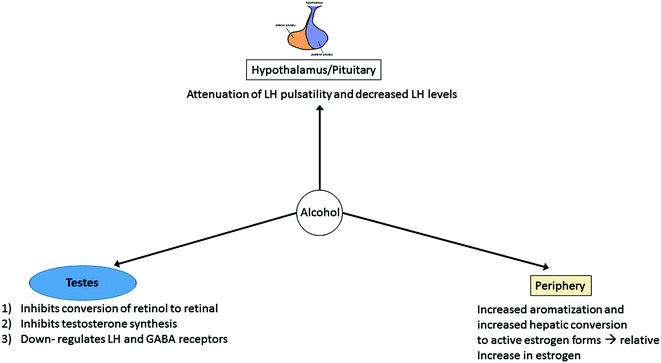
Fig. 11.1
Proposed mechanisms for hypogonadism in the setting of alcoholic liver disease. Alcohol is postulated to affect the hypothalamus/pituitary via attenuation of LH pulsatility, though the role of GnRH pulsatility remains unclear at this time. Alcohol also affects the testes with inhibition of conversion of retinol to retinal (important in spermatogenesis), inhibition of multiple enzymes necessary for testosterone biosynthesis, and downregulation of LH and GABA receptors. Alcohol additionally acts peripherally via increased aromatization of testosterone to estrogen in the setting of portosystemic shunting of androgens and increased conversion to active estrogen forms, overall resulting in higher relative estrogen levels
Most studies suggest that the hypogonadism in alcoholism far precedes, sometimes by years, the development of liver disease. In men with significant history of alcohol abuse, but without histological or biochemical evidence of liver disease, more than 50% showed primary gonadal damage with testicular atrophy and infertility, and notably seminiferous tubular atrophy, marked peritubular fibrosis, and loss of germ cells [28]. Additionally, impairment of conversion of testosterone to its active metabolite dihydrotestosterone (DHT), in the setting of reduced levels of 5-alpha reductase, has been reported [49]. Ylikahri et al. reported in 1974 that short-term alcohol administration (<4 h) to young healthy male volunteers did not alter testosterone levels, but longer administration (weeks) resulted in a reduction in testosterone concentrations [50]. Badr et al. demonstrated that mice treated with high volumes of alcohol (one-third of caloric intake) showed much higher reductions in testosterone and marked testicular changes compared to iso-calorically pair-fed controls without alcohol intake [51].
Ethanol has several direct adverse effects on the testes. Alcohol inhibits the conversion of retinol to its active metabolite retinal, which is essential for normal spermatogenesis [52]. High doses of ethanol impair testosterone synthesis from the precursor cholesterol by inhibiting multiple enzymes including 3-beta-hydroxysteroid dehydrogenase, 17-oxoreductase and 17–20 desmolase [53]. While short-term alcohol exposure leads to increased expression of LH receptors in Leydig cells, long term exposure downregulates LH receptors as well as gamma amino butyric acid (GABA) receptors, suggesting alcohol may affect LH receptor through cell membrane potential [54].
If alcohol produced solely direct testicular atrophy, a much higher level of gonadotropins, especially LH would be expected, but that is often not the case, suggesting that ethanol also affects hypothalamic-pituitary function. While some older data suggest that ethanol may reduce the pulsatility of LH secretion independent of the liver disease [52], the effect of ethanol on GnRH pulsatility has not been thoroughly studied. Bannister et al. [55] measured LH levels in blood samples drawn every 15 min for 6 or 8 hours in men with chronic alcohol liver disease without clinical signs of gonadal failure compared to those with phenotypic signs of hypogonadism such as testicular atrophy and hair loss. In men without clinical signs of hypogonadism, the pulsatility of LH was preserved, but those with overt signs of gonadal failure showed severely decreased LH hormone levels and an attenuation of pulsatility [52].
In the periphery, alcohol may increase aromatase activity, converting androgens to estrogens even beyond what can be explained by obesity. In alcoholic liver disease, estrogen sulfate may be converted to active estrogens in the liver through NF-kb [55]. Relative estrogen levels may further be increased as a result of portosystemic shunting leading to increased androgen substrate for bioconversion by extra-hepatic peripheral aromatase. It is unclear to what degree the relative excess estrogen may contribute to the inhibition of LH secretion [49]. Estrone and estradiol levels may also be increased secondary to increased production of androstenedione from the adrenal cortex [16].
Many of the studies that examined alcoholism and hypogonadism were performed in the 1970s and 1980s, highlighting the need for new research. In summary, existing data suggest that acute alcohol consumption results in transient primary hypogonadism while longer term alcohol use, even in the absence of liver damage, is associated with both primary and secondary hypogonadism. In those who develop cirrhosis, hypogonadism seems to be more profound than in other causes of liver disease. The reversibility of hypogonadism in those with chronic liver disease who abstain from alcoholism has not been well studied.
Hepatitis B/C
Hepatitis B (HBV) and Hepatitis C (HCV) are the leading cause of hepatocellular carcinoma, cirrhosis, and transplantation. Within the United States, HBV and HCV are responsible for 15 and 26% of cirrhosis cases, respectively. This section will focus on predominantly HCV given HBV rates have been declining with increasing vaccination use [2].
It is estimated that between 2.5 and 4.7 million people in the United States had chronic HCV between 2003 and 2010, with major risk factors including current or prior intravenous drug use, and treatment with blood products before 1987 [56]. About Approximately 20% of patients clear HCV without additional treatment. Of the 75–80% who develop chronic infection, up to 25% will develop cirrhosis over a period of 20–30 years. It also important to note that more than 50% of HIV-infected intravenous drug users have HCV [56].
While HCV is associated with multiple immune-mediated conditions such as mixed cryoglobulinemia, glomerulonephritis, thyroiditis, arthritis, and peripheral neuropathy [57], it remains to be seen if there is a direct relationship between HCV with hypogonadism, especially in the setting of compensated liver disease. Multiple studies have suggested that hypogonadism is not seen with hepatitis C infection until liver architectural damage is present, as evidenced by elevated SHBG and total testosterone levels [58].
Ferri et al. (2002) compared 207 men with HCV (all with compensated liver disease) to age-matched men without liver disease, and demonstrated that plasma levels of total and free testosterone were generally lower in the former group [59]. However, testosterone levels were not significantly correlated with the severity of the liver disease based on ultrasound and laboratory testing.
It is postulated that one mechanism by which HCV induces hepatocyte injury is through the production of reactive oxygen species (ROS) via expression of core protein with direct mitochondrial injury [60]. HCV may also induce ROS-mediated damage to sperm given that somewhere between 30–80% of those with hypospermatogenesis have evidence of ROS damage, including damage to sperm membranes and DNA [61]. Lorusso et al. showed that patients with chronic HCV have impaired sperm quality, particularly the percentage of spermatozoa with normal progressive motility and morphology [62].
The exact prevalence of low testosterone in HCV has not been thoroughly studied, likely due to multiple co-morbidities in HCV patient such as alcohol use, HIV, and HBV infection. Also, many studies looking at the relationship between hypogonadism and HCV were conducted in the setting of interferon treatment. More recently, antiviral drugs such as ledipasvir (inhibitor of viral phosphoprotein) and sofosbuvir (RNA chain terminator) have shown sustained virologic response up to 94–99% in those with genotype 1 HCV infections [63]. Additional studies are needed to examine the impact of these new treatments on testosterone levels.
There have been ongoing concerns that elevated testosterone levels may be a risk factor for the development of HCV-related HCC given that males have a higher risk of HCC than females. One prospective Japanese study of 46 males with HCV-related cirrhosis found that increased total testosterone was associated with HCV–HCC risk, but a case-control study in Australia with 35 HCV patients did not find any significant association [64]. White et al. (2012) performed one of the largest cross-sectional studies in the U.S. of 308 HCV compensated liver patients with mild to advanced fibrosis. They reported a significant 27% increase in advanced fibrosis risk and 16% inflammatory activity risk for each 1 ng/ml increase in total testosterone [65]. The study examined risk of fibrosis and inflammatory activity based on three groups of total testosterone: low (≤4.11 ng/mL), middle (4.12–5.91 ng/mL), and highest (≥5.91 ng/ml). HBV was reported to augment androgen receptor (AR) signaling using HepG2 carcinoma cells, which may lead to elevated testosterone and subsequent proliferation, apoptosis, and inflammatory response of the hepatocyte, increasing risk of HCC [66]. Increases in total testosterone may be explained by high SHBG levels, and SHBG has been proposed as a marker for HCC [67]. Given somewhat similar mechanism of invasion between HCV and HBV, it may be that HCV could have a similar response.
In summary, whether HCV causes hypogonadism independent of liver disease remains unclear, and should be a focus of further studies. There are data to suggest that HCV may induce ROS-mediated damage in the testes. Additionally, limited data suggest that patients with higher levels of endogenous testosterone and SHBG may be at increased risk for HCC, fibrosis, and inflammatory activity, but the mechanism is not well understood at this time.
Nonalcoholic Fatty Liver Disease
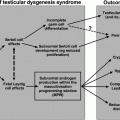
According to the American Association for the Study of Liver Diseases (AASLD), the diagnosis of nonalcoholic fatty liver disease (NAFLD) requires (1) evidence of hepatic steatosis by imaging or histology, and (2) no other causes for secondary hepatic fat accumulation such as alcohol consumption or steatogenic medications. In general, NAFLD is associated with metabolic syndrome including obesity, diabetes mellitus, and dyslipidemia. NAFLD should be differentiated into nonalcoholic fatty liver (NAFL) and nonalcoholic steatohepatitis (NASH). While NAFL is defined by the presence of hepatic steatosis without hepatocellular injury, NASH includes hepatocyte injury. NAFLD is a spectrum of liver injury progressing from lipid accumulation within hepatocytes, to inflammation and ballooning of hepatocytes, and to eventual scarring and cirrhosis (Fig. 11.2). Estimates of the worldwide prevalence of NAFL range from 6.3 to 33%, with a median of 20%, while the prevalence of NASH is estimated to be 3–5% [68].
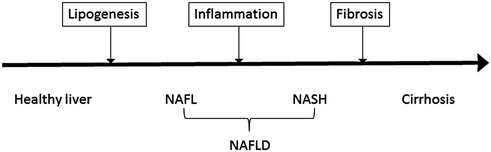
Fig. 11.2
Schematic of progression from fatty liver to cirrhosis. Nonalcoholic fatty liver disease (NAFLD) is subdivided into nonalcoholic fatty liver (NAFL) and nonalcoholic steatohepatitis (NASH). The former has a prevalence of 6.3–33% and is characterized by hepatic steatosis without hepatocellular injury. The latter has a prevalence of 3–5% and is characterized by hepatocellular injury. Importantly, NASH can progress to cirrhosis, which could potentially lead to decompensated liver disease and/or hepatocellular carcinoma
It is well documented that men with metabolic syndrome have an increased prevalence of hypogonadism [68, 69]. However, the bidirectional nature of the relationship is complex and multifactorial (Fig. 11.3; see Chaps. 15 and 17). It is important to understand these associations in order to determine if these risk factors are independently associated with the development of hepatic steatosis to inform the development of relevant treatment strategies [70]. The actual prevalence of hypogonadism in NAFLD is not established at this time.
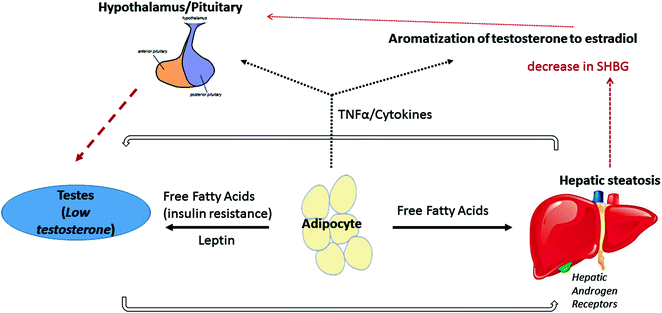
Fig. 11.3
Proposed mechanism of the bidirectional relationship between hypogonadism and nonalcoholic fatty liver disease. Adipose tissue may play a central role in the bidirectional relationship between hypogonadism and nonalcoholic fatty liver disease. Increased free fatty acid secretion results in insulin resistance and subsequently decreased testosterone production. Leptin may also be directly be toxic to the testes. Downregulation of testosterone production may lead to reduced activation of androgen receptors on hepatocytes, further increasing hepatic steatosis. Free fatty acid production from adipocytes can also have a direct steatogenic effect on the liver. Adipose tissues can also increase cytokine production, leading to central downregulation of the HPG axis and promoting peripheral conversion of testosterone to estrogen [69–72]
One leading hypothesis is that obesity is the predominant driver of hypogonadism in patients with NAFLD/metabolic syndrome. Excess of leptin, an adipokine, in obese men directly inhibits Leydig cell androgenesis. Both subcutaneous and visceral adipose tissue may directly affect gonadotropin release by the pituitary by either increased secretion of cytokines such as TNF-α or IL-2, or by increased conversion of circulating androgens into estrogens, which further contribute to negative feedback. Aromatase is expressed in adipose tissues, and obese patients may have increased expression of the enzyme. Additionally, SHBG, a major testosterone transport protein, is decreased in obese patients, contributing to low total testosterone levels [71] (see Chap. 15).
Another idea is that increased visceral adipose tissue which is partly due to testosterone deficiency increases hepatic exposure to free fatty acids, which can lead to worsening hepatic insulin resistance and systemic insulin resistance [71]. Leydig cell function may also be directly impaired in men with insulin resistance [72]. There is some evidence that testosterone treatment can decrease visceral adiposity and improve insulin sensitivity [73].
There have been a number of studies that suggest that androgen deficiency and low SHBG levels in metabolic syndrome patients may be independently associated with hepatic steatosis. In a large retrospective cohort study of 1912 Korean men, hepatic steatosis was associated with lower serum testosterone levels (14.2 vs. 17.2 nmol/l). When these results were adjusted for confounders such as age, BMI, lipid panel, diabetes, exercise, and high-sensitivity reactive protein (hs-CRP), low serum testosterone still remained associated with hepatic steatosis [70]. Additionally, there have been other studies showing that testosterone replacement in hypogonadal men is followed by reduction in visceral adiposity and circulating TNFα levels [74], and in obese men with obstructive sleep apnea, by reduced liver fat measured by CT scan [75]. Furthermore, low SHBG has been associated with NAFLD, which is likely one of the major reasons for low testosterone. SHBG levels are lower with increasing obesity [76], type 2 diabetes [77], and metabolic syndrome. The TNFα and IL1β increase seen in metabolic syndrome has been shown to reduce SHBG expression in HepG2 cells by decreasing HNF4α (hepatocyte nuclear factor 4-α), which is responsible for activating promoters of multiple genes expressed in liver that function in lipid metabolism [78].
Finally, there is growing research in the relationship between testosterone and microRNAs (miRNAs) in the liver. Some studies suggest that downregulation of miRNAs in liver may ultimately lead to liver diseases, and that testosterone is linked to miRNA regulation in the mouse liver [79]. Specifically, testosterone seems to affect miRNA upregulation of 6 miRNAs: miR-22, miR-690, miR-122, let-7A, miR-30D, and let-7D.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


