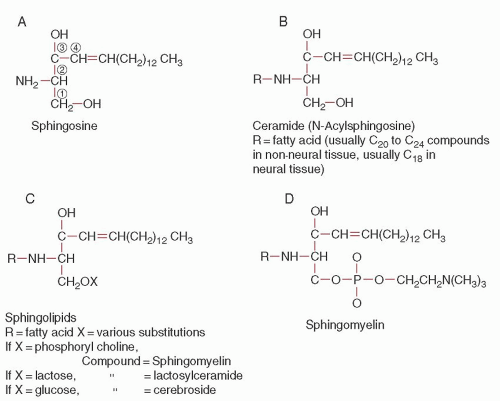
 FIGURE 59.1. Formulas of some of the sphingolipids. |
storage disease and one of the most prevalent genetic disorders among Ashkenazi Jewish individuals, with an incidence of about 1 in 1,000 and a carrier frequency of about 1 in 15.3
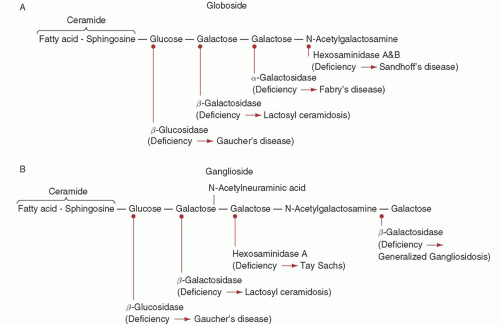 FIGURE 59.2. Schematic structure of globoside (A) and ganglioside (B) to show site of action of the several catabolic enzymes, which result in one of the storage diseases when defective. |
onset and a milder course than patients with one N370S allele and another mutant allele. However, the wide variability in clinical presentation among Gaucher disease patients cannot be fully explained by the underlying acid β-glucosidase mutations, and presumably other “modifier” genes can influence disease severity.
TABLE 59.1 BIOCHEMICAL AND PHENOTYPIC CHARACTERISTICS OF GAUCHER AND NIEMANN-PICK DISEASES | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
TABLE 59.2 MOLECULAR BASIS OF GAUCHER AND NIEMANN-PICK DISEASES | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Related posts:
 Clinical Flow Cytometry
Lymphocytes and Lymphatic Organs
Endothelium: Angiogenesis and the Regulation of Hemostasis
Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane
Thalassemias and Related Disorders: Quantitative Disorders of Hemoglobin Synthesis
Anemias Unique to the Fetus and Neonate
Clinical Flow Cytometry
Lymphocytes and Lymphatic Organs
Endothelium: Angiogenesis and the Regulation of Hemostasis
Hereditary Spherocytosis, Hereditary Elliptocytosis, and Other Disorders Associated with Abnormalities of the Erythrocyte Membrane
Thalassemias and Related Disorders: Quantitative Disorders of Hemoglobin Synthesis
Anemias Unique to the Fetus and Neonate
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


