© Springer International Publishing Switzerland 2016
Aamir Ahmad and Shirish Gadgeel (eds.)Lung Cancer and Personalized MedicineAdvances in Experimental Medicine and Biology89310.1007/978-3-319-24223-1_9Anaplastic Lymphoma Kinase (ALK) Signaling in Lung Cancer
(1)
Chao Family Comprehensive Cancer Center, Department of Medicine, Division of Hematology-Oncology, University of California Irvine School of Medicine, 101 City Drive, Bldg 56, RT81, Rm 241, Orange, CA 92868, USA
(2)
MUSC Hollings Cancer Center, Department of Medicine, Division of Hematology-Oncology, Medical University of South Carolina College of Medicine, Charleston, SC 29403, USA
Abstract
Chromosomal rearrangement in the anaplastic lymphoma kinase (ALK) gene was identified as an oncogenic driver in non-small cell lung cancer (NSCLC) in 2007. A multi-targeted ALK/ROS1/MET inhibitor, crizotinib, targeting this activated tyrosine kinase has led to significant clinical benefit including tumor shrinkage and prolonged survival without disease progression and has been approved by US FDA since 2011 for the treatment of advanced ALK-rearranged NSCLC (Ou et al. Oncologist 17:1351–1375, 2012). Knowledge gained from treating ALK-rearranged NSCLC patients including the presenting clinicopathologic characteristics, methods of detecting ALK-rearranged NSCLC, pattern of relapse and acquired resistance mechanisms while on crizotinib, and the clinical activities of more potent ALK inhibitors has led us to a detailed and ever expanding knowledge of the ALK signaling pathway in lung cancer but also raising many more questions that remained to be answered in the future. This book chapter will provide a concise summary of the importance of ALK signaling pathway in lung cancer. Understanding the ALK signaling pathway in lung cancer will likely provide the roadmap to the management of major epithelial malignancies driven by receptor tyrosine kinase rearrangement.
Keywords
Anaplastic lymphoma kinase rearrangement non-small cell lung cancerReceptor tyrosine kinase fusion positive tumorsCrizotinibALK breakapart FISHChromosomal rearrangementMolecular Basis of ALK Rearrangement in NSCLC
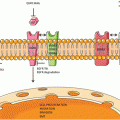
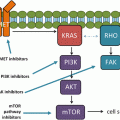
Chromosomal rearrangements have long been recognized as oncogenic drivers in hematological malignancies. Although it has been predicted in early 2000 that chromosomal rearrangements will be found in solid malignancies there was no reports of such rearrangements well into the mid 2000s [1]. This all changed in 2007 when two research groups independently discovered the chromosomal rearrangement in the anaplastic lymphoma kinase gene in non-small cell lung cancer [2, 3]. These two initial reports revealed in NSCLC there was both intra-chromosomal deletion and inversion of the echinoderm microtubule associated protein like 4 (EML4) gene to ALK on chromosomal 2 generating an EML4-ALK fusion protein containing the N-terminal portion of EML4 protein that retains the “coil-coiled” dimerization and the C-terminal portion of ALK which contains the kinase domain that is present in the cytoplasm and constitutively active [2]. Further elegant experiments demonstrated that transgenic mice expressing EML4-ALK in the lung results in multiple adenocarcinomas in the lung [4]. ALK is one of 58 human receptor tyrosine kinase receptors (RTKs) [5] and is the namesake rearrangement in anaplastic large cell lymphoma [6]. Although there are multiple pathways could be activated by the constitutively EML4-ALK [7], the common sequala of the ALK activation is the increase in survivin level and a decrease in pro-apoptotic BIM protein allowing lung cancer cells driven by ALK activation to escape apoptosis [8]. Since 2007, several different breakpoints in the EML4 have been identified that have been fused to the ALK generating several EML4-ALK fusion variants [9]. Currently, there are three main EML4-ALK fusion variants. The most common variant among EML4-ALK fusion is variant 1 (54.5 %) followed by variant 3 (34 %) and variant 2 (10 %) [9, 10]. Additionally, other fusion partners to ALK kinase domain have been identified in NSCLC including TFG (Trk-fused gene), KIF5B (kinesin family member 5), and KLC1 (kinesin light chain 1) [9]. As recently as in 2014 new fusion partner, HIP1 (Huntington interacting protein 1), to ALK in NSCLC is continuously being discovered [11, 12]. Indeed any protein that contains a “coil-coiled” domain that can serve as a dimerization domain and can localize to the cytoplasm can potentially act as a fusion partner to ALK. Hence it is likely more fusion partners to ALK in lung cancer will be identified in the future. Hence ALK-rearranged NSCLC is a molecularly heterogeneous subset of lung cancer. In vitro experiments have shown that there are potential differential responses among the various EML4-ALK fusion variants to various ALK inhibitors [13]. Therefore is reasonable to hypothesize that these various ALK fusion variants will have subtle but different clinicopathologic characteristics and likely differential response to ALK inhibitors. Additionally activation of epidermal growth factor receptor (EGFR) mutations have been identified in ALK-rearranged lung cancer; however, whether activation EGFR mutations and/or ALK rearrangement is the main oncogenic driver remained to be determined [9].
Clinicopathologic Presentations of ALK-Rearranged Lung Cancer
Despite being a molecularly heterogeneous disease, several general patterns can be ascribed to ALK-rearranged NSCLC. First, the median age of diagnosis of ALK-rearranged NSCLC is in the early to mid 50s which is about 15 years younger than the typical age of diagnosis of lung cancer in the US [14–16]. However the age of diagnosis can range from the early twenties to the eighties subtly underlining this is indeed a molecularly heterogeneous disease [15, 16]. Second, the vast majority (>95 %) of the ALK-rearranged lung cancer is adenocarcinoma [15, 16] with a portion of these adenocarcinoma also had signet ring features although signet ring features is not pathognomonic for ALK rearrangement [14, 17]. However, case reports have shown that ALK rearrangement has also been found in squamous cell carcinoma [11, 12, 18]. Interestingly, a recent report has shown that EML4-ALK fusion transcript is found in small cell lung cancer [19]. Thus as we broaden our screening of ALK rearrangement in all histologies of lung cancer, we may gain further knowledge about ALK rearrangement in lung cancer. Third, similar to lung cancer driven by activating EGFR mutations, between two-thirds to three quarters of ALK-rearranged lung cancer patients were never-smokers (defined as <100 cigarettes lifetime) [20]. Therefore, smoking status should not be used as a clinical criterion to determine whether to screen for ALK rearrangement in lung cancer. Fourth, unlike EGFR mutated lung cancer there does not seem to be a propensity for ALK-rearranged lung cancer to be more common in Asian patients although whether a specific variant of EML4-ALK is more prevalent among a certain racial/ethnic group remains to be determined [9]. Fifth, in terms of tumor burden and sites of metastasis at the time of initial presentation, there seems to be some differences between ALK-rearranged lung cancer and EGFR mutated lung cancer [21]. One particular interests among clinical oncologists treating ALK-rearranged lung cancer is whether there is an increase incidence of brain metastasis. This question arises from the observation that close to half of ALK-rearranged lung cancer patients who progressed on crizotinib had brain relapses. It does not seem that patients with ALK-rearranged lung cancer inherently presents with higher incidence of brain metastasis at the time of diagnosis [21].
Biology of ALK-Rearranged Lung Cancer
Our aggregate understanding of the biology of ALK-rearranged lung cancer come from differential responses ALK-rearranged lung cancers to various chemotherapy agents, the diagnostics methods used to identify ALK-rearranged lung cancer patients, patterns of relapse on crizotinib, acquired resistances during crizotinib treatment, and the clinical activity of more potent ALK inhibitors in clinical development.
A randomized trial (PROFILE1007) comparing crizotinib to single chemotherapy agent (docetaxel or pemetrexed) as second line treatment ALK-rearranged lung cancer have clearly demonstrated that crizotinib conferred statistically significant progression-free survival (PFS) [22]. However, the response rate of ALK-rearranged lung cancer patients treated with pemetrexed was much higher than those treated with docetaxel [22]. This observation is consistent with case series that reported ALK-rearranged lung cancer patients had a higher response rate to pemetrexed [23–26] likely due to a lower level of thymidylate synthase (TS) [26], which is a target of pemetrexed, in the ALK-rearranged tumor. It has been postulated that the aberrant activation of ALK lead to repressed transcription of the TS gene.
The current FDA approved companion diagnostic test which was approved simultaneously with the approval of crizotinib in August, 2011 was the Abbott Vysis breakapart fluorescence in situ hybridization (FISH) assay [9, 27]. However FISH is expensive, labor intensive, and requires certain technical competence to interpret the FISH signals [9]. Since 2011, it is becoming clear that immunohistochemistry (IHC) is a equally effective but much cheaper way to detect ALK rearrangement in lung cancer [28]. ALK IHC test using a highly sensitive antibody D5F3 can now be automated taking the observer variability out of the testing component [29]. ALK protein is not expressed in normal tissues and thus any aberrant ALK expression represents rearrangement in ALK. IHC has been used to detect ALK rearrangement in ALCL for many years using the ALK-1 antibody from Daiko. However, the promoter of EML4 is less active than the promoter for nucleophosmin that is fused to ALK in ALCL hence ALK-1 from Daiko is not a sensitive antibody for detecting ALK rearrangement in lung cancer [30]. Reverse transcription-polymerase chain reaction (RT-PCR) [10] and target deep sequencing [31] have also been used to detect ALK rearrangement in lung cancer. One of the major advantages of the sequencing approach is that the exact fusion variant is known [31] and that any unique clinicopathologic characteristics associated with each ALK fusion variant may be identified. Finally there are conflicting data on the prognostic significance of ALK rearrangement in lung cancer ranging the wide spectrum as a poor prognostic factor [32, 33] to no significance [34] to being a favorable prognostic factor [35]. In the future when next generation sequencing is in wide use then the biology of each unique ALK fusion will be even better identified and the prognostic significance of ALK rearrangement can be answered also. It is now believed that most of the ALK-rearranged lung cancer reminds dependent on ALK signaling for its pathogenesis as a majority of ALK-rearranged lung cancer patients continued to derive clinical benefit with continual ALK inhibition with an overall survival close to 30 months [36]. Hence ALK rearrangement in lung cancer is generally a favorable prognostic factor since currently there are several potent ALK inhibitors in clinical development and the motto of precision cancer medicine of “the right drug for the right disease” did certainly prolong life [34].
Treatment of ALK-Rearranged Lung Cancer
The discovery of ALK rearrangement in lung cancer would have been a historical academic footnote for awhile had it not been the existence of a phase I trial of crizotinib, a multi-targeted MET/ALK/ROS1 inhibitor [15, 16]. Crizotinib was initially being developed as a MET inhibitor [37] but its anti-ALK activity was known [38]. The discovery of a RTK rearrangement in a common epithelial malignancy is unexpected but the crizotinib phase I investigators and Pfizer were able to modify the protocol to accommodate this newly discovery cohort of molecularly defined patients. Within 2 years crizotinib was able to show impressive anti-tumor activity with overall response rate of approximately 60 % and progression-free survival of approximately 9 months essentially independent of the line of therapy [15, 16]. Based on the expanded phase I and a global phase 2 study of crizotinib received US FDA approval on August 26, 2011 for the treatment of advanced ALK-rearranged lung cancer [9]. Subsequently crizotinib has been demonstrated to be superior to chemotherapy in first-line (Pfizer Press release, March 25, 2014) or second-line treatment of ALK-rearranged lung cancer [22] in terms of statistically significant improved progress-free survival.
Invariably, these patients develop disease progression while on crizotinib. Many of these patients are likely to be still dependent on ALK signaling as they can benefit from continuation of crizotinib beyond treatment especially if the new and/or progressing metastatic site can be controlled by loco-ablative therapy [36, 39]. Recent retrospective analysis of data of crizotinib trials suggested progression free survival from the date of progression among patients who continued crizotinib beyond progressive disease (CBPD) was 16.4 months comparing to 3.9 months who discontinued crizotinib at progression (Hazard ratio [HR] 0.27, 95 % confidence interval [CI]: 0.17−0.42; p < 0.0001) [36] and an overall survival of 29.6 months among CBPD patients comparing to an overall survival of 10.8 months among patients who discontinued crizotinib on progression (HR 0.30, 95 % CI: 0.19−0.46; p < 0.0001) [36]. Furthermore the continual dependence of ALK signaling was the phenomenon of “disease flare” where there is rapid tumor progression when crizotinib is discontinued after the patient has progressed on crizotinib [40]. We can generally view there are two major mechanisms that result in disease progression on crizotinib. First close to 50 % of patients on crizotinib will develop progression in the brain (new or existing) likely indicating a pharmacodynamics failure of crizotinib to reach adequate therapeutic level in the central nervous system (CNS) [41]. Second the ALK gene acquired secondary resistance mutations to crizotinib including gatekeeper mutation (L1196M) and solvent front mutations (G1202R) [31, 42–46]. There are other resistance mechanisms reported such as activating of other by-pass signaling pathway such as EGFR [46] or the loss of ALK dependence altogether [43]. There are now several more potent ALK inhibitors in clinical development (ceritinib, alectinib, AP26113, ASP3026, X396, TSR-011, PF0663922, RXDX101) [47]. Among these ALK inhibitors, alectinib [48, 49] and ceritinib [50] are the farthest along in clinic development where phase I results of both compounds have been published or presented and global phase 2 trial of both compounds in crizotinib-resistant patients have been completed. In general most of the ALK inhibitors can overcome most of the acquired resistance mutations except the solvent-front mutation G1202R [31, 51]. Thus an ideal ALK inhibitor for the treatment of ALK-rearranged lung cancer should have two major properties: the ability to penetrate CNS and have clinical activity in existing brain metastasis and the ability to overcome secondary acquired ALK mutations especially the solvent front mutation G1202R. However there are evidence both in vitro and from patient series that these ALK inhibitors have differential sensitivity to the various secondarily acquired mutation [13, 31, 52]. Thus rebiopysing crizotinib resistant tumor to try to understand exactly the resistance mechanism(s) will be necessary to tailor the treatment and serve as a paradigm for precision cancer medicine. Crizotinib and other ALK inhibitors in clinical development have put ALK-rearranged lung cancer in the parlance of precision oncology medicine. We also gained insight into the biology of ALK-rearranged lung cancer when we understand the mechanism of resistance to these inhibitors.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


