(1)
Medical Sciences Division Northern Ontario School of Medicine West Campus, Lakehead University, Thunder Bay, Ontario, Canada
Key Topics
Issues with lung cancer (LnCa) screening
Molecular pathology of LnCa
Circulating cell-free nucleic acid content as LnCa biomarkers
Circulating LnCa epigenetic biomarkers
Circulating LnCa genetic biomarkers
Circulating LnCa miRNA biomarkers
Circulating LnCa protein biomarkers
Circulating LnCa cells
Key Points
The potential for early detection of LnCa can be realized given the well-charted molecular pathology and subtypes. This expansive knowledge on LnCa molecular genetics is also propelling the development of effective targeted therapies for the various molecular subtypes.
Extensive work has culminated in the discovery of a plethora of circulating LnCa biomarkers with profound clinical potential. Traditional serum biomarkers are still of important clinical utility in LnCa management. However, targeting molecular imprints in circulation, such as Resolution Bio ctDx™ Lung assay, enables the liquid biopsy paradigm to be realized.
Circulating LnCa cells are used for disease prognosis and treatment monitoring and have potential for mutation detection (e.g., EGFR mutations) for real-time therapeutic decision-making.
3.1 Introduction
For several decades, LnCa has remained a challenge in oncology by being the highest in both incidence and prevalence. The global incidence for 2012 and 5-year prevalence were 1.825 million and 1.893 million, respectively. Sadly, of the 1.825 million cases, 58 % occurred in the resource-poor parts of the world. Added to this unfavorable statistics are the dismal survival rates. An estimated 1.59 million global deaths were expected for 2012, and again many (963,000) occurred in the developing world. The prevalence of LnCa in the USA alone is at 430,090, and the estimated incidence in 2016 is 224,390, with 158,080 expected deaths. Globally, LnCa is the leading cause of cancer-related deaths. Even more worrisome is the fact that the global incident rate mirrors the mortality rate, which is partly due to the high case fatality coupled with the lack of regional variability in survival rates. This dismal statistics of LnCa is due to a number of reasons among which are:
The lack of evidence-based screening modalities (imaging or biochemical markers), even for those at elevated risk above the general population, such as smokers.
Because of the lack of recommended screening guidelines, the vast majority of patients are diagnosed with late and advanced stage disease, when the prognosis is poor. If detected early, the 5-year survival rate can be as high as 70 %. However, this rate is ~20 % for stage IIIb and 5 % for stage IV LnCa. Improved imaging and treatment has enabled some progress to be made over the past several decades in the 5-year survival rates.
Patients with late stage disease are subjected to chemotherapy and various targeted therapies. But lack of response and evolution of resistant clones lead to relapse. Additionally, field cancerization accounts for some relapse due to the emergence of second primary tumors.
Although LnCa is histopathologically dichotomized into non-small cell LnCa (NSCLC, 80 % of all cases) and small cell LnCa (SCLC, the remaining 20 %), the disease is very heterogeneous at the molecular level, thus, hampering efficient targeted therapy delivery.
Clearly, early detection of LnCa is key to improving the dismal 5-year survival outcome. The need for validated biomarkers and molecular imaging techniques that can be deployed at the point of care is urgent. Of even more importance will be biomarkers that can be assayed noninvasively in body fluids for LnCa screening, diagnosis, treatment stratification, longitudinal monitoring for treatment response, detection of early recurrences, and for the study of tumor evolution over time so as to inform clinical decision-making. Thus, various investigators are diligently pursuing these biomarkers and this chapter provides a synthesis of these findings.
3.2 Screening Recommendations for LnCa
Why has LnCa early detection and possible chemoprevention been a problem when it appears to be the easiest and straightforward cancer to pickup early when treatment is optimal? The answer may appear simple but is more complicated than can be appreciated, due probably to the issues to overcome before implementation of screening programs.
Because of the lack of evidence that screening reduces mortality, LnCa screening is not recommended even for high-risk individuals. Historically, LnCa screening had relied on chest X-ray (CXR), sputum cytology, and recently low-dose spiral CT (LDCT) scans of the lung. Chest X-ray screenings are not good for early LnCa detection because they pick up lesions that are 2 cm or larger. They also have low specificity to differentiate between mimickers of cancer such as sarcoidosis, pulmonary tuberculosis, and other lung infections from LnCa. Sputum cytology is an established diagnostic approach for LnCa, but it lacks sensitivity, especially for early detection, because frank cancer cells are rarely found in sputum from patients with early stage disease.
The first published prospective study on LnCa screening using CXR was in 1968. In this large cohort study, it was concluded that annual mortality rate was substantially similar between screened and unscreened populations, although the 5-year survival rate was much higher among the screened population. There have been five randomized control trials published between 1984 and 2011. These are the Mayo Lung Project (MLP), Johns Hopkins Lung Project (JHLP), Memorial Sloan Kettering Lung Project (MSKLP), Czechoslovakia Study, and recently the Prostate, Lung, Colorectal, and Ovarian (PLCO) screening trial. The consistent findings from all these studies are that:
LnCa detection rate is similar between the study and control groups.
Early stage tumors are detected in screened populations (except for the MSKLP study).
No difference in overall survival or disease-free mortality is observed between the groups (except that the 5-year survival rate was better in the screened group in the MLP study).
Long-term (20 years) follow-up revealed no difference in disease-specific mortality between the screened and controlled groups.
The PLCO study of 1993 randomized 154,901 people between the ages of 55–74, who either had CXR annually for 3 years or received conventional care. This study also concluded that CXR with or without sputum analysis does not reduce disease-specific mortality.
The MLP study showed that CXR or CXR combined with sputum cytology enabled the detection of early stage cancers and increased patient survival but had no effect on reducing mortality. A confounding variable with this study was the increased detection of insignificant lesions, resulting in the problem of overdiagnosis. Thus, CXR with or without sputum cytology does not appear to reduce disease-specific mortality in high-risk populations.
Computerized tomography scans are becoming very sensitive for LnCa early detection with improved scanner resolutions. They can even detect nodules as small as 1 mm, but the problem is that not all nodules are cancerous. This increased sensitivity has led to increased detection of benign nodules (~95 % of all nodules are benign), thus making it costly, aside from exposing patients to unnecessary radiation as well as causing increased patient anxiety. While CT is associated with high false positives and costs, low-dose spiral CT (LDCT) has shown promise as a screening modality for LnCa, especially in high-risk populations. Multiple large uncontrolled studies indicate that LDCT detects early stage I, small tumors with favorable 5-year survival outcomes. Bach et al. reviewed three uncontrolled trials (Moffitt Cancer Centre, Tampa, Fl; Mayo Clinic; and Istituto Tumori in Milan studies) involving 3246 asymptomatic at-risk individuals [1]. The conclusion was that LDCT significantly detected more LnCas than expected, including many patients with early stage I/II cancers (78 % of cases). Disappointingly though, this screening did not significantly reduce mortality rates as expected, and the number of advanced stage LnCas diagnosed did not decrease in the screened population. However, guidelines by the National Comprehensive Cancer Network (NCCN) for LnCa screening recommends an annual LDCT scanning for high-risk individuals between the ages of 55–74 who quit smoking 15 years or earlier with at least 30-pack years of smoking history. Multiple randomized control trials are in progress with initial published data from the National Lung Screening Trial (NLST) study indicating a 20 % relative reduction in LnCa-specific deaths among high-risk individuals being attributable to LDCT screening compared to CXR [2]. While recommended by the NCCN for screening high-risk individuals 55–74 years of age, LDCT is costly and not universally available or accessible.
Bronchoscopy allows direct visualization and even biopsy of lung lesions. However, this method is invasive and expensive and only scans the major airways. It will therefore miss up to 40 % of all LnCas located in the periphery. In the face of all these evidences, screening decisions are varied and rest on the physician and patient. Tobacco avoidance or cessation still remains the first-line preventive measure of reducing the societal burden of LnCa. However, recent efforts on clinical utility of biomarkers could change the dismal outlook on screening. LnCa is a disease of epigenetics and genetics, and hence several alterations in biomolecules are potential clinical useful biomarkers. These epigenetic and genetic changes impinge on cellular regulatory pathways involved in apoptosis, proliferation, and senescence. Aberrant effects of oncogenes and tumor suppressor genes primarily mediate the development of both SCLC and NSCLC. Validated assays targeting these biomarkers noninvasively should permit implementation of acceptable screening programs for at least the high-risk population.
3.3 Molecular Pathology of LnCa
From a clinical and molecular pathologic perspective, primary LnCa is classified into SCLC and NSCLC (Table 3.1). NSCLC, which accounts for the majority (80 %), is a broad category of LnCas that are nonresponsive to SCLC treatment regimes. This group is further subdivided into adenocarcinoma (accounts for >50 % of LnCas), squamous cell carcinoma (SCC), and large cell carcinoma. SCLC tends to be aggressive and metastatic, and hence is often treated by chemotherapy. NSCLC, however, is less aggressive and is amenable to surgery. Neuroectodermal tumors include SCLC, large cell neuroectodermal carcinoma (LCNEC), and carcinoid tumors. These are very aggressive tumors with neuroendocrine morphology. They therefore express at least one of the following neuroendocrine markers: synaptophysin, chromogranin, or CD56. The three subgroups differ primarily in their level of mitosis and the presence or absence of necrosis.
Table 3.1
Pathologic classification of LnCa
Tumor (frequency) | Pathologic features |
|---|---|
NSCLC (~80 %) | |
Adenocarcinoma | Glandular differentiation with or without mucin |
Squamous cell carcinoma | Squamous differentiation with keratinization |
Large cell carcinoma | Diagnosis of exclusion. Classified as NSCLC-NOS; can have squamous or adenomatous differentiation; diagnosed on surgically resected samples |
Adenosquamous carcinoma | At least 10 % of both squamous and glandular differentiation. Definite diagnosis on surgically resected samples |
Sarcomatoid carcinoma | Poorly differentiated NSCLC with features of sarcoma or sarcoma-like; spindle and/or giant-cell carcinoma |
Carcinoma of salivary gland type | Mucoepidermoid, adenoid cystic, or epithelial-myoepithelial forms |
Neuroendocrine tumors (20–25 %) | |
SCLC | Small tumor cells, scant cytoplasm, fine granular chromatin, no evident nucleoli; necrotic with high mitotic rate (≥11 mitotic figures/10 high-power field (HPF) |
Large cell neuroendocrine carcinoma | Cytologically resemble NSCLC but with neuroendocrine morphology and positive for at least one NE marker; necrotic with high mitotic rate (≥11 mitotic figures/10 HPF |
Carcinoid | Common in children; neuroendocrine morphology; typical carcinoid tumors have no evidence of necrosis and low mitotic rate (<2 mitoses/10 HPF); atypical forms have focal necrosis and intermediate mitotic rate (2–10 mitoses/10 HPF) |
LnCa is a conglomerate of heterogeneous diseases, and this complex or diverse histology reflects on the molecular pathology as well. Important signaling pathways with multiple gene alterations on LnCa include the PI3K, RAS-MAPK, JAK/STAT, among others. Multiple oncogenes (KRAS, EGFR, BRAF, MEK1, HER2, MET, ALK, RET) and tumor suppressor genes (TP53, PTEN, and LKB-1) are activated or silenced by diverse mechanisms and at various frequencies and patterns in LnCa.
3.3.1 Genetic Alterations in LnCa
Chromosomal structural and numeric instabilities are common in LnCa. These include nonreciprocal translocations and deletions involving tumor suppressor genes, amplifications of oncogenes, and aneuploidy. Allelic losses at 3p, 4p, 4q, 5q, 8p, 10q, 13q, 17p, and 22q are common in SCLC, while losses at 3p, 6q, 8p, 9p, 13p, 17p, and 19q are associated with NSCLC. Microsatellite instability (MSI) is observed in ~35 % of SCLC and ~22 % of NSCLC. Efforts to use MSI in early detection of LnCa using body fluids and sputum have been encouraging. Here are synopses of some genes altered in LnCa.
3.3.1.1 TP53 Alterations in LnCa
TP53 is inactivated in a significant proportion of LnCas and is one of the major genetic alterations in this disease. For example, LOH at 17p13, the TP53 locus is observed in ~90 % of SCLC and ~65 % of NSCLC. Inactivating mutations, mostly involving the DNA-binding domain occurs in 80–100 % of SCLC and in ~47 % of NSCLC. The Cancer Genome Atlas estimates 81 % mutation frequency in SCLC. Additionally, ~45 % of adenocarcinomas harbor these TP53 mutations, which are tobacco smoke carcinogen induced. Consistently, the mutations are higher in smoking-associated G to T than G to C transversions. Never smokers often harbor G to A transitions in these cancers. These mutations are associated with treatment resistance and poor prognosis.
3.3.1.2 KRAS Alterations in LnCa
The KRAS oncogene is frequently mutated in LnCa, primarily NSCLC of the adenocarcinoma histology. The mutations are virtually absent in SCC and SCLC. KRAS driver mutation frequencies vary geographically and with sex and lifestyle exposures. Thus, it appears to be more common in Western populations than Asians and more frequent in males and smokers of both sexes. As a member of the RAS-MAPK pathway, at least one component of this pathway is mutated in ~70 % of tumors, with most mutations clustered in the KRAS. Recorded mutation frequency varies from 25 to 40 % in adenocarcinomas. The identified hotspot mutation is a single amino acid substitution in codon 12, but also rarely in codons 13 and 61. In smokers, G to T transversions occur at a much higher frequency of ~84 %. Tumors from never smokers rather tend to harbor G to A transitions. KRAS mutations are mutually exclusive in tumors with EGFR mutations, and tumors with these mutations confer resistance to EGFR-TKI therapy because of constitutive pathway activation downstream of EGFR. KRAS mutations have prognostic value, and this appears to vary with the types of mutation as well. Data from the Biomarker-Integrated Approaches of Targeted Therapy for LnCa Elimination (BATTLE) indicate that G12C or G12V mutant KRAS is associated with shorter progression-free survival than other mutations or wildtype genotype.
3.3.1.3 EGFR Alterations in LnCa
Similar to KRAS, almost all EGFR mutations occur in adenocarcinomas and rarely in adenosquamous carcinomas. They are almost exclusive in SCCs, but EGFRvIII mutations that alter the extracellular domain of the receptor, as well as copy number gains and increased receptor expressions, are more common in SCC than adenocarcinomas. EGFR mutations are more common in tumors from younger people, females, and nonsmokers. Mutation prevalence also varies geographically, being 30–40 % in Asians compared to 10–15 % in people from the Western world. Activating mutations are clustered in the first four exons that encode the intracellular tyrosine kinase domain. Most are inframe deletions in exon 19 (45 %). Of the over 20 reported deletions, del E746-A750 is the commonest in NSCLC. Missense L858R mutation in exon 21 is the next frequently observed mutation (40 %). In early stage cancer, exon 18 mutations can account for 14 % of EGFR mutations, while L858R comprises about 29 %. Inframe duplications and insertions in exon 20 that often confer resistance to EGFR-TKIs account for 5–10 % of EGFR mutations in LnCa. However, the commonest EGFR-TKI resistant mutation is the activating point mutation c.2369C>T (T790M) in exon 20, which accounts for 50 % of all resistant mutations. The T790M substitution interferes with the binding efficiency of TKIs. Usually these mutations develop during treatment and are clonally selected for. However, exon 20 mutations including T790M have been found in patients who had no previous treatments.
3.3.1.4 BRAF Alterations in LnCa
Activating mutations in BRAF that increase kinase activity occur in ~3 % of NSCLCs, mostly adenocarcinomas. Other mutations affect the G-loop of the activation domain. Kinase domain mutations include V600E, D594G, and L596R, while activating domain mutations include G465V and G468A. About half of all mutations in adenocarcinomas are accounted for by the common V600E, followed by G469A and D594G. V600E mutation is common in female nonsmokers, while the other mutations are more frequent in smokers than never smokers. Mutations are also mutually exclusive of other pathway gene (e.g., KRAS, EGFR) mutations.
3.3.1.5 MET Alterations in LnCa
MET oncogene is amplified in at least 1–7 % of NSCLC. Copy number increase is more common in SCC than adenocarcinoma where they occur in 3–5 % of cases. MET amplification leads to overexpression, constitutive receptor phosphorylation, and activation of downstream pathways including PI3K and RAS-MAPK. MET amplification accounts for ~20 % of resistance to EGFR-TKIs through signaling via the PI3K pathway, thus obviating the need for EGFR signaling.
3.3.1.6 HER2 Alterations in LnCa
Human epidermal growth factor receptor 2 (HER2) encodes membrane bound receptor tyrosine kinase, a member of the ERBB/EGFR receptor family. Although HER2 does not bind to ligands, it can heterodimerize with other ligand-bound family members to signal via the PI3K, MAPK, and JAK/STAT pathways. Activating mutations occur in up to 4 % of NSCLC. Gene amplifications occur in 2 % of NSCLC, but increased expression is demonstrated in as many as 20 % of NSCLC. Exon 20 inframe insertions of 3–12 bases are the common activating mutations in HER2. These alterations are common in adenocarcinomas and are mutually exclusive of EGFR and KRAS mutations. HER2 alterations are also associated with nonsmokers, people of Asian descent, and female gender.
3.3.1.7 PI3K Pathway Alterations in LnCa
PI3K is an important signaling pathway in LnCa, being altered in 50–70 % of NSCLC, especially SCCs that demonstrate a mutation frequency of 47 % (according to the Cancer Genome Atlas Project). Activating mutations in EGFR, KRAS, AKT, PI3K, and PIK3CA, as well as loss of PTEN, are altered pathway components. Mutations and amplifications in PIK3CA cause constitutive pathway activation. Mutations, mainly in the catalytic domain, are observed in 1–3 % of NSCLC, and increased copy number is observed in ~5 % of SCLC. PIK3CA mutations may occur in association with KRAS and EGFR mutations. AKT mutations occur in 0.5–2 % of NSCLC, especially SCC. PTEN mutations occur in ~5 % of NSCLC (more in SCCs than adenocarcinomas; in ~10 % SCC compared with ~2 % of adenocarcinomas). Transcriptional repression of PTEN with decreased protein levels is more common in NSCLC (~75 %).
3.3.1.8 Alterations of Cell Cycle Regulators in LnCa
RB1 was the first described tumor suppressor gene in LnCa, and this gene is activated in a vast majority of LnCas (~90 % of SCLC and 10–15 % of NSCLC). Inactivation of CDKN2A, a negative regulator of the cell cycle, occurs in ~80 % of NSCLC, especially SCCs in which alterations occur in ~72 % of cases. CCND1 overexpression is observed in ~40 % of NSCLC.
3.3.1.9 FGFR1 and DDR2 Alterations in LnCa
Membrane-associated receptor tyrosine kinase FGFR1 signals via MAPK and PI3K pathways. FGFR1 amplifications are demonstrated in ~20 % of SCCs (uncommon in adenocarcinomas). Membrane-associated receptor tyrosine kinase DDR2 is mutated in ~4 % of SCCs.
3.3.1.10 LKB-1/STK11 Alterations in LnCa
LKB encodes serine-threonine kinase that inhibits mTOR and mTOR pathway components (excluding KRAS). LKB-1 inactivation via mutations and deletions alone occur in 11–30 % of adenocarcinomas. Alterations are associated with smoking and male gender and correlate with KRAS mutations.
3.3.1.11 Fusion Genes in LnCa
While gene fusions are often more of a feature of hematologic malignancies, recent advancements indicate their presence in some solid tumors as well. Thus, subsets of LnCas harbor RET, ALK, and ROSI fusion rearrangements.
RET encodes receptor tyrosine kinase that is normally involved in neural crest cell development. Alterations are common in papillary and medullary thyroid cancers. RET activation through chromosomal rearrangement is however observed in some LnCas. The functional kinase domain (exons 12–20) are fused to kinesin family 5B (KIF5B) that is 10 Mb from the RET locus (10q11.2). KIF5B-RET fusion occurs in 1–6 % of LnCas, primarily adenocarcinomas from never smokers. This activated fusion gene is mutually exclusive of other LnCa driver mutations including KRAS, HER2, EGFR, ALK, BRAF, and ROSI.
Activation of ALK receptor tyrosine kinase signals through the PI3K, RAS-MAPK, and JAK/STAT pathways. ALK is activated by chromosomal rearrangements when fused to echinoderm microtubule-associated protein-like 4 (EML4). Various variants with different lengths of EML4 have been identified. Fusions involve inversion in chromosome 2p; the commonest is intron 13 of EML4 fusion to intron 19 of ALK. Other reported fusion partners are KIF5B, TRK-fused gene (TFG), and Kinesin light chain 1 (KLC-1). Rearrangements occur in ~4 % of NSCLC, especially in adenocarcinomas from young nonsmokers.
ROSI activation signals via the PI3K, RAS-MAPK, and STAT3 pathways. It encodes membrane-bound tyrosine kinase receptor that is homologous to ALK kinase domain. Multiple 5′ fusion partners have been identified in 1–3 % of LnCas, mainly adenocarcinomas from younger people, nonsmokers, and people of Asian descent (similar to ALK). Fusion partners include TPM3, CD74, E2R, SLC34AZ/NaPi2b, FIG, KDELR2, SDC4, and LRIG3. However, oncogenic functions of these partners are yet to be determined.
3.3.2 Molecular Classification of LnCa and Personalized Medicine
From the standpoint of unbiased and accurate classification that informs treatment selection, molecular phenotyping of LnCa is desirable. Several gene expression profiling, mutation detection, and other genetic approaches provide strong evidence for the molecular heterogeneous nature of LnCa. Thus, from the perspective of actionable oncologic practice, LnCa is viewed from the molecular profile.
Targeted therapies are mainly focused on the larger group of NSCLC, because the molecular pathology of this histologic subgroup has been fairly characterized. In regard to pharmaceutical development, biotherapies are targeted mostly at altered cancer “driver genes,” mainly oncogenes and tumor suppressor genes. Given experiences with evolving resistance to monotherapies, it is likely that combinatorial therapies may be a more useful approach. For example, the T760M mutation in EGFR causes resistance to erlotinib and gefitinib, while ALK C1156Y and L1196M mutations render cancer cells resistant to crizotinib. Furthermore, as sequencing costs become more affordable, it will be possible to sequence tumors at a depth and resolution that will enable tumor heterogeneity to be identified more accurately. This should inform the use of therapies targeting all clones (even the very occult ones) in tumors. In NSCLC, a few targeted genes have been identified. It should be noted that the molecular underpinnings of many LnCas (e.g., over 40 % of adenocarcinomas) are still elusive. Well-established alterations in lung adenocarcinoma are KRAS, EGFR, NRAS, BRAF, ALK, ROS1, RET, HER2, AKR1, PIK3CA, and MAP2K and in SCC are EGFR, EGFRvIII, DDR2, PIK3CA, and amplifications in FGFR1 [3]. Targeted therapies are either available or being developed for several members of these pathways (Table 3.2). For example, EGFR-TKIs, gefitinib and erlotinib target tumors with exons 18–21 mutations. Similarly, ALK-TKI, crizotinib targets ALK translocations. With the advent of personalized medicine, targeted molecular therapy is imperative for all cancers. Hence, for LnCa, the IASLC/CAP/AMP recommends that all lung adenocarcinomas be tested for EGFR mutations and ALK translocations.
Table 3.2
Molecular targeted therapies for lung adenocarcinoma
Targets | Agents |
|---|---|
ALK | Crizotinib |
BRAF | Vemurafenib, GSK2118436 |
EGFR | Afatinib, erlotinib, gefitinib |
FGFR1 | Brivanib, ponatinib, AZD4547, S49076 |
HER2 | Afatinib, dacomitinib, neratinib |
MEK | AZD6244 |
MET | Onartuzumab, rilotumumab, cabozantinib, crizotinib, tivantinib |
PD-1/PD-L1 | Nivolumab, MPDL3280A |
PIK3CA | GDC-0941, XL-147, BKM120 |
PTEN | Vandetanib |
RET | Cabozantinib, sunitinib, sorafenib, vandetanib |
ROS1 | Crizotinib |
NSLC34AZ/NaPi2b | DNIB0600A |
3.3.3 Multistep Carcinogenesis and Field Cancerization in LnCa
LnCa develops in two stepwise fashions, depending on whether it is SCC or adenocarcinoma. The sequence of events for bronchial tumors is hyperplasia–dysplasia–carcinoma in situ–invasive squamous cell carcinomas, and for adenocarcinoma of peripheral bronchioles and alveolar, the progression pathway is atypical alveolar hyperplasia–adenomatous and alveolar hyperplasia–adenocarcinoma.
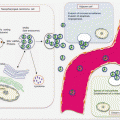
Auerbach and colleagues elegantly demonstrated the tobacco-induced lung field defect in 1961 [4], and several molecular events preceding LnCa have been demonstrated thereafter. Their findings are consistent with tobacco-related histological changes in lung epithelia, with the development of widespread multifocal premalignant lesions. Because smoking is a known risk factor for development of LnCa, studies have established a molecular field defect in smokers with or without cancer. In addition to other carcinogens, toxins from tobacco smoke are established agents that create the LnCa field, either directly or through induction of epithelial inflammation. About 85 % of smokers are at risk for developing LnCa (that is having established LnCa fields evidenced by molecular alterations), but only ~15 % will actually progress to develop the disease. Smoking causes aberrant promoter methylation of multiple genes in the epithelium of cancer-free individuals. These genes include CDKN2A, DAPK, GSTP1, RAR-β2, RASSF1A, CDH13, and APC. Thus, smokers have multiple foci of “damaged” epithelium with epigenetic alterations. Many lungs from smokers without cancer demonstrate numerous genetic changes as well, including loss of heterozygosity (LOH) and microsatellite alterations. Even in bronchial washes from both ipsilateral and contralateral lungs from smokers, LOH and TP53 mutations are present. Additionally, in histologically normal lung tissue adjacent to LnCa, mutations in EGFR and KRAS, as well as LOH at chromosome 3p (DDUT and FHIT loci) and chromosome 9p (CDKN2A loci), have been characterized. Figure 3.1 is a summary of the molecular alterations in tobacco smoke-associated pulmonary field cancerization. While this is the tip of the iceberg for LnCa, molecular field cancerization occurs in all tumors.
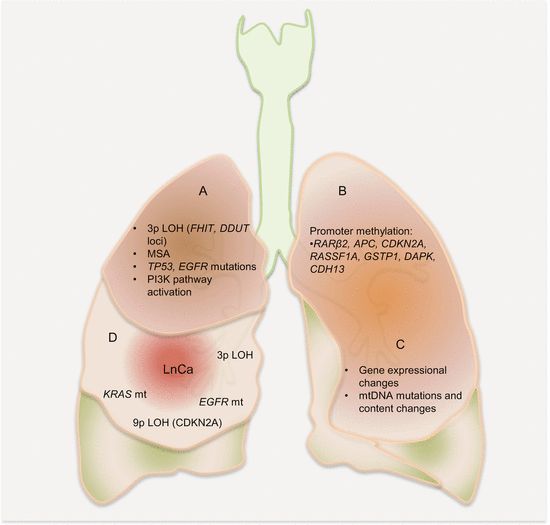
Fig. 3.1
Molecular pathology of lung field cancerization. In lung exposed to tobacco smoke without cancer (A, B, C), LOH, MSA, promoter methylation of tumor suppressor genes, mutations, gene expressional changes, mtDNA mutations, and activation of the PI3K pathway characterize the field. In normal appearing lung tissue close to LnCa (D), similar alterations are present
3.4 Circulating LnCa Biomarkers
Given the inherent problems with LnCa screening, there is a need for noninvasive biomarkers for this disease. Thus, circulating biomarkers have been extensively explored for LnCa management.
3.4.1 Circulating Cell-Free Nucleic Acid Content as LnCa Biomarkers
The clinical relevance of circulating cell-free DNA (ccfDNA) and RNA (ccfRNA) has been extensively evaluated in LnCa patients. Both quantitative analyses as diagnostic and prognostic biomarkers and more specific qualitative studies of epigenetic and genetic alterations in key genes involved in LnCa have been pursued. Various targets including hTERT, ACTB, HBB, and ALU sequences have been used to measure the amounts of circulating free DNA. While the findings are compelling and have potential clinical applications, the nonspecific nature of circulating cell-free nucleic acid (ccfNA) levels discourages the clinical translation of these findings. While gene mutations have been targeted qualitatively, it is also possible with advanced technologies to quantify the levels of mutations and methylations.
3.4.1.1 Cell-Free Nucleic Acid Content as Diagnostic Biomarkers of LnCa
The clinical applications of ccfDNA have diagnostic and prognostic relevance for LnCa. Sozzi et al. demonstrated the diagnostic and prognostic potential of quantitative plasma DNA in NSCLC patients. Mean plasma DNA levels were higher in patients, including those with early stage disease. The diagnostic accuracy achieved an AUROCC of 0.844. Microsatellite alterations and DNA levels correlated with clinical status and early detection of recurrence [5]. In a follow-up study, Sozzi et al. could discriminate NSCLC patients from matched controls for age, sex, and smoking status based on the levels of ccfDNA [6]. They amplified hTERT gene as a measure of ccfDNA in LnCa patients. In this well-designed study, median DNA concentration in patients was eight times that of controls, achieving a diagnostic accuracy with AUROCC of 0.94. Plasma DNA concentration was a strong risk factor for NSCLC. These findings have been confirmed by numerous other studies. The amount of ccfDNA from LnCa patients (~13 ng/ml of plasma) is significantly different from age and sex matched control individuals without cancer (~3 ng/ml of plasma). The diagnostic performance of ccfDNA measurement by Paci et al. achieved an AUROCC of 0.79 for NSCLC detection. Additionally, high levels of ccfDNA had prognostic value on follow-up [7]. While another study by Sozzi and colleagues found that baseline plasma ccfDNA levels did not improve the accuracy of LnCa detection by spiral CT in smokers, it was evident that higher levels of plasma ccfDNA at surgery still conferred elevated risk for the presence of an aggressive tumor [8]. Benlloch et al. found that median ccfDNA concentration was higher in both pleural effusions and sera from LnCa patients compared to controls. Additionally, higher ccfDNA in either fluid compartment correlated with poor survival [9].
Circulating cell surface-bound DNA (csb-DNA) as useful diagnostic biomarkers of LnCa has also been explored. While the amounts of ccfDNA were nondiscriminatory between cancer and controls, there was a significantly lower level of csb-DNA in cancer patients compared to controls. Consistent with other findings, these low csb-DNA levels correlated with poor survival [10].
The diagnostic potential of ccfDNA may not be of much utility as a stand-alone test for detection of LnCa; however, it could improve the diagnostic accuracy when combined with other tests, imaging assessments, and probably other clinical parameters in a nomogram.
3.4.1.2 Cell-Free Nucleic Acid Content as Prognostic Biomarkers of LnCa
It has been fairly established that the levels of ccfDNA have prognostic utility in LnCa management. High levels are indicative of aggressive disease and predictive of poor overall survival. Reduced levels of csb-DNA are also associated with poor prognosis. In multivariate analysis, plasma and serum ccfDNA have been found to be independent prognostic biomarkers in NSCLC patients [8, 9, 11, 12]. A high serum level of ccfDNA measured by targeting hTERT was an independent predictor of time to progression and OS of advanced stage NSCLC patients in multivariate analysis [13]. Circulating nucleosomal DNA is also much higher in cancer than healthy people.
While they may have limited use in cancer detection because of increased release in nonmalignant conditions such as trauma, stroke, inflammation, autoimmune diseases, and infection, ccfDNA and nucleosomal DNA levels have shown promise as prognostic biomarkers in LnCa. For example, Holdenrieder et al. found that nucleosomal DNA levels before first and second cycle chemotherapy were independent prognostic factors in multivariate analysis [14]. Circulating nucleosomal DNA is also useful for monitoring treatment response in LnCa. Patients who responded to treatment demonstrated decreasing circulating nucleosomal DNA levels, while nonresponders with progressive disease showed persistently high or increasing levels [15]. In several studies of advanced stage NSCLC patients on either first- or second-line chemotherapy, and also in SCLC patients on first-line chemotherapy, reducing levels or the absolute serum levels of nucleosomal DNA at staging investigation significantly predicted responders from nonresponders [16, 17]. Thus, plasma DNA dynamics appear useful for disease prognosis and monitoring for treatment efficacy or resistance.
3.4.2 Circulating LnCa Epigenetic Biomarkers
Epigenetic biomarkers are detected in tissue, sputum, and blood samples from LnCa patients. Commonly methylated is CDKN2A, which is detectable in almost all LnCa tissue, 63 % of sputum, and 33 % of serum samples. Also, MGMT and APC methylation are found in 66 % and 50 % of serum samples, respectively. Low levels of MGMT and APC methylation are associated with better survival, and similarly, loss of DAPK and RASSF1A through methylation has prognostic implications. Hypomethylation and loss of imprinting of IGF2 and H19 are also features of LnCa.
Several a priori LnCa biomarkers are assayed in plasma and serum samples from patients, and various methylation frequencies have been demonstrated. The list includes methylation of CDKN2A, GSTP1, DAPK, MGMT, RASSF1A, RAR-β, CDH1, CDH13, APC, and TMS1, which are assayed singly or in combination as panels. In a proof of concept study, methylation status of four genes (CDKN2A, GSTP1, DAPK, and MGMT) was analyzed in serum samples from patients with NSCLC. Methylation of at least one gene was detected in 68 % of tumor samples and 73 % of paired serum samples. Methylation was specific, as it was not detected in normal lung tissue and tumor-negative patient serum samples. All stages of cancer harbored methylation in these genes [18]. CDKN2A promoter methylation was assessed in 111 tumor tissue samples, 136 plasma, and 95 sputum samples using semi-nested methylation-specific PCR (MSP). Methylation was positive in a majority of the tumor (80.2 %), plasma (75.7 %), and sputum (74.7 %) samples. When CDKN2A methylation in sputum and plasma was combined with sputum cytology, most (92 %) of the LnCas could be detected [19]. CDKN2A methylation in paired NSCLC tissue and plasma were assayed using a modified semi-nested MSP, which increased detection in both tissue and plasma samples. Methylation was in 79.3 % of tumor samples, of which 64 of the 73 positive cases (87.7 %) were demonstrated in paired plasma samples [20].
The prevalence of methylation in multiple genes was examined in plasma and sputum from women who were at various risks (cancer survivors, cancer-free smokers, and never smokers) for LnCa to determine their possible use as screening biomarkers. Methylation of three genes in plasma and seven genes in sputum was positive in LnCa survivors. LnCa survivors had the highest prevalence of CDKN2A promoter hypermethylation in plasma, as well as a significant increase in the odds (OR, 3.6) of having one or more methylated genes in plasma than never smokers [21]. NSCLC tissue and paired plasma samples were investigated for CDKN2A promoter hypermethylation, MSA at 3p, KRAS mutations, and ccfDNA concentration. Individually, these biomarkers demonstrated diagnostic potential. Methylation was detected in 63 % of tissue and 55 % of plasma samples. MSA was present in 57 % of tissue and 50 % of plasma samples, and KRAS mutations were detected in 31 % of tissue but undetectable in plasma samples. The combination of MSA and methylation status increased the sensitivity of this plasma diagnostic assay to 62 %, which was further increased to 80 % with the incorporation of ccfDNA levels, demonstrating the obvious advantage of multi-panel assays [22]. Methylation of CDKN2A, MGMT, RASSF1A, and DAPK in sera from 200 patients undergoing bronchoscopic evaluation due to abnormal chest radiographs was investigated. Methylation was more frequent in LnCa patients than patients with nonmalignant lung diseases. The diagnostic sensitivity and specificity of this serum assay were 49.5 % and 85 %, respectively. The odds of having LnCa when one gene was methylated in serum were 5.28, and this increased to 5.89 when two or more genes were involved. Noteworthy, 50.9 % of stage I disease patients had genes methylated compared to other serum protein markers that were positive in only 11.3 %, underscoring the importance of methylation biomarkers in early detection [23]. CDKN2A and CDH13 methylation status in tissue and matched serum samples from 61 NSCLC patients was assayed with fluorescent MSP. Methylation frequencies in tissue were 79 % for CDKN2A, 66 % for CDH13, and 52 % for tumors harboring both gene methylations. Methylation in at least one gene was detectable in 92 % of tumor samples. For serum samples, the detection rate was 26 % for CDKN2A, 23 % for CDH13, and 39 % for at least one methylated gene. This study also demonstrated the diagnostic specificity because samples from healthy people were all negative [24]. Ninety-six percent of primary LnCa samples harbored APC promoter hypermethylation, of which 47 % of available serum/plasma samples were positive. No prognostic use was found for serum targets, but high levels of methylation in tumor tissue were independent predictors of poor survival, and detection of methylation in serum, however, was of diagnostic potential [25].
Gene promoter hypermethylation in circulation has also been associated with prognostic features of LnCa patients. The promoter methylation of the G2-M checkpoint control gene, SFN (14-3-3σ), was assayed in pretreatment sera as possible prognostic factor in advanced stage NSCLC patients on platinum-based chemotherapy. Methylation was positive in 34 % of serum samples from patients, and this was associated with longer median survival time. Multivariate analysis identified methylation of SFN as a novel (apart from Eastern Cooperative Oncology Group performance status) independent prognostic factor for patients with advanced stage NSCLC [26]. Methylation of multiple genes (MGMT, CDKN2A, RARβ2, RASSF1A, FHT, CDKN2A/ARF, APC1A, APC1B, CDH1, and DAPK) in sera from 46 LnCa patients was investigated using a nested MSP. All except APC1B showed various frequencies of methylation. The most frequently methylated genes were APC1A (78.1 %), CDKN2A (41.3 %), and RASSF1A (40.9 %). Methylation in at least one of these genes was present in 78.3 % of the patients and was associated with advanced tumor stage, size, and undifferentiated status [27]. Methylation of RASSF1A, DAPK, and target of methylation-induced silencing (TMS) as well as KRAS mutations was assayed in sera from 50 resected NSCLC patients. Tumor methylation status correlated significantly (p = 0.001) with serum frequencies (both ranged from 34 to 45 % for the assayed genes). Twelve KRAS mutations were found in serum samples, and these correlated significantly with survival (compared to methylation that had no prognostic utility in this study) [28]. CDKN2A methylation was detected in 42 % of NSCLC tissue samples and in only 14 % of plasma samples from patients with advanced TNM stage tumor. However, the presence of CDKN2A methylation in tissue and circulation was associated with poor survival and shorter disease-free survival. Pre- and post-resection pleural lavage fluids were positive for CDKN2A methylation that was equally associated with poor survival [29].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


