Fig. 9.1
Macrophages foam cell formation. Under oxidative stress, macrophages generate reactive oxygen species and through activation of NADPH oxidase complex (1) increase the production and release of superoxide ions (2), thus leading to extensive LDL oxidation (3) increased oxidized LDL (OX-LDL) uptake by macrophages via their scavenges receptors SRA and CD36 (4) thus leading to foam cell formation. Conversion of macrophages into foam cells also involves decreased HDL-mediated cholesterol efflux (5) and increased cellular cholesterol biosynthesis (6)
It was shown in previous studies that LDL is oxidized in vivo under certain atherogenic conditions and, indeed, circulating OX-LDL exist in human plasma of patients with CAD and their levels correlate positively with the severity of CAD [24]. OX-LDL is present in atherosclerotic lesions from human and animal models [25]. Unlike the LDL receptor, OX-LDL receptors are not regulated by cellular cholesterol levels. The uptake of OX-LDL occurs via the scavenger receptors CD36 and scavenger receptor A (SRA), which leads to cholesterol accumulation and foam cell formation [23].
The process of LDL oxidation is unlikely to occur in plasma because of the high concentrations of antioxidants and metal cation chelation agents. LDL oxidation is more likely to occur within the artery wall, an environment depleted of antioxidants [26]. It is not clear which cells in the arterial wall are responsible for LDL oxidation, but it is postulated that endothelial cells [27] and smooth muscle cells (SMC) [28] could contribute to LDL oxidation, a process which requires the presence of transition metal ions such as copper and iron ions. LDL oxidation by macrophages also has a major role during early stage of atherosclerosis [29].
Cell mediated oxidation of LDL depends on the balance between cellular pro-oxidants such as NADPH oxidase, lipoxygenase, or myeloperoxidase and cellular antioxidants such as the glutathione system and superoxide dismutase, as well as the balance between pro-oxidants and antioxidants in the lipoprotein particles themselves [23].
LDL oxidation starts with the consumption of its antioxidants. After depleting LDL of its antioxidants, transition metal ions catalyze propagation reactions, which include the breakdown of lipid hydroperoxides and the formation of aldehydes. These reactions are responsible for the oxidative modification of Apo B 100, which alter the charge and three-dimensional configuration of this apoprotein. Affinity for the LDL receptor decreases and the LDL particles are taken up by macrophages [30].
Several biological and biochemical mechanisms were suggested to be involved in macrophage mediated oxidation of LDL including transition metal cations, superoxide anions, NADPH oxidase, lipoxygenase, myeloperoxidase, and reactive nitrogen species [23, 30].
Cellular LDL oxidation requires the presence of iron or copper ions. Several mechanisms are involved in this process but the most important is the ability of macrophages to reduce those metals which then rapidly react with lipid hydroperoxides, leading to the formation of reactive lipid radicals and to the conversion of reduced metal back to its oxidized form [31]. Superoxide ions are required for the initiation of LDL oxidation. In macrophages the predominant source of superoxide is the NADPH oxidase system [32]. We have demonstrated that LDL in the presence of copper ions activates the NADPH oxidase complex. Further evidence for the role of superoxide anions in the oxidation of LDL by macrophages was demonstrated in cells from patients with chronic granulomatous disease, who lack NADPH oxidase and do not generate superoxide anions or oxidized LDL [33]. Previous studies with an in-vitro knockout mouse model of P47 phox (one of the cytosolic subunits of NADPH oxidase) showed that both superoxide anion and LDL oxidation are inhibited [34]. Thus, superoxide and other free radicals derived from NADPH oxidase activation contribute to lipid peroxidation.
Another oxygenase that may participate in oxidation of LDL is myeloperoxidase (MPO). MPO is a heme protein released by activated neutrophils and monocytes and is present in tissue macrophages such as those in atherosclerotic plaques. MPO may play a role in monocyte-macrophage oxidation of LDL by several pathways, such as amplifying the oxidizing potential of H2O2 to reactive oxygen species (ROS), aldehydes, and nitrating agents [35, 36]. MPO-mediated oxidation reactions occur in the absence of metal ions.
Nitric oxide (NO) is formed by multiple vascular cells including monocytes and macrophages [37], and the expression of NO synthase was demonstrated in human coronary atherosclerotic plaques. NO may have both anti and pro-oxidant effects. NO exerts an antioxidant effect, attenuating the extent of cell-mediated oxidation of LDL [38], but NO could also be a pro-oxidant as evidenced by the observation that, when tissue oxidant defense mechanisms become depleted, NO can initiate lipoprotein lipid peroxidation [38].
The paraoxonase gene family includes PON1, PON2, and PON3 which are lactonase enzymes [39]. In humans, PON1 and low levels of PON3 (but not PON2) are found in serum in association with HDL. In humans, PON1 mRNA expression is limited mainly to the liver. In contrast, PON2 is more widely expressed in a variety of tissues, including human and mouse macrophages [40]. All PON proteins were shown to protect from atherosclerosis development, but their mechanism of action is currently unknown. All PONs also have the capacity to protect cells from oxidative stress [41]. PON2, unlike PON1, is not present in the circulation but rather found in cells including macrophages. PON2 like PON1 possesses antioxidant and anti-atherosclerotic properties [41]. The balance between cellular pro oxidants (such as NADPH oxidase) and antioxidants (such as PON2) in arterial wall macrophages determines the extent of LDL oxidation. Macrophage PON2 was shown to be increased under oxidative stress, probably as a compensation mechanism.
Lipoprotein Oxidation in Diabetes Mellitus
Several studies have shown that diabetes is associated with increased oxidative stress which is considered to be one of the major mechanisms for induction of atherosclerosis in diabetes [42]. The increased oxidative stress is accompanied by increased ROS generation, oxidative stress markers, and decrement of antioxidants. Diabetic patients are highly prone to oxidative stress because hyperglycemia depletes natural antioxidants and facilitates the production of oxygen and nitrogen free radicals [43].
A consequence of diabetes is hyperglycemia, which in turn contributes to the progression and maintenance of an overall oxidative environment. Macrovascular and microvascular complications are the leading cause of morbidity and mortality in diabetic patients, but the complications are tissue specific and result from similar mechanisms with many being linked to oxidative stress.
Oxidative stress is thought to be a major risk factor in the onset and progression of diabetes. Many of the common risk factors such as obesity, increased age, and unhealthy eating habits all contribute to an oxidative environment that may alter insulin sensitivity either by increasing insulin resistance or impairing glucose tolerance. The mechanisms by which this occurs are multifactorial. The mechanism by which oxidative stress may induce diabetes includes increased insulin resistance, increased β cell dysfunction, impaired glucose tolerance, and increased mitochondrial dysfunction [44].
In our studies we have examined the relationship between diabetes, increased lipid peroxidation, and enhanced atherosclerosis by using the Apo E knockout mouse model which develop severe hypercholesterolemia and extensive atherosclerosis on a chow diet [45]. We have shown previously that accelerated atherosclerosis in this animal model is associated with enhanced lipid peroxidation [46]. Diabetic induction in APO E knockout (E°) mice for three months using streptozotocin (STZ) injection led to an increase of their atherosclerosis lesion area. This phenomenon was associated with a significant increment of macrophage oxidative stress (Fig. 9.2a, b).
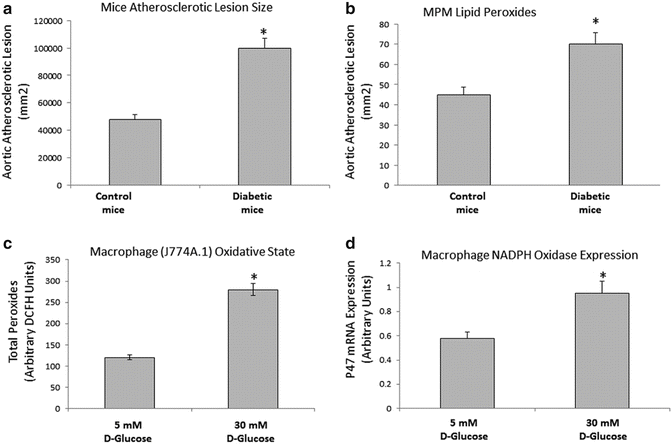
Fig. 9.2
Increased atherosclerotic lesion area and macrophage lipid peroxidation in diabetic mice and increased macrophage lipid peroxidation and NADPH oxidase expression in glucose-enriched macrophages. Aortic segments from STZ-induced diabetic mice for 3 months and age-matched untreated E0 mice were analyzed for their aortic atherosclerotic lesion area (a), n = 10,*p < 0.01. Peritoneal macrophages (MPM) from STZ-induced diabetic mice for 3 months were analyzed for their lipid peroxides content (b). J-774 A.1 macrophages were grown for 7–10 days in glucose-enriched media (5–30 mM) and analyzed for their lipid Peroxides content (c), and NADPH Oxidase expression (d), n = 4,*p < 0.01
Numerous studies have attempted to evaluate the role of high glucose conditions on cells of the artery wall, including endothelial cells, smooth muscle cells, and macrophages. It has been proposed that glucose might act directly via the generation of advanced glycation end-products (AGEs) or reactive oxygen species. Thus, hyperglycemia, one factor shared by both type 1 and type 2 diabetes, is a major contributor to oxidative stress [47, 48]. Cellular glucose could be toxic and directly linked to increased cellular lipid peroxidation, since in vitro studies showed that pre-culture of endothelial cells or smooth muscle cells in glucose-enriched media, increased the cells ability to oxidize LDL which was accompanied by enhanced secretion of superoxide anion. This could be the result of direct generation of ROS or by altering the redox balance. This is thought to occur via several well-studied mechanisms, including activation of protein kinase C [6] and overproduction of superoxide by the mitochondrial electron transport chain [49], increased intracellular formation of advanced glycation end-products (AGEs), and increased flux through the polyol pathway [50]. Hypergly-cemia specifically induces cellular oxidative stress by the following pathways:
1.
Glucose auto oxidation leads to an overproduction of NADPH, which increases the production of superoxide radicals and leads to the formation of several reactive oxygen species such as superoxide anion, which can facilitate LDL oxidation in vitro [51].
2.
Glucose induces the formation of advanced glycation end products (AGEs) which activates the receptor for AGE (RAGE) present on many vascular cells [52]. Stimulation of RAGE causes the production of reactive oxygen species (ROS) [53]. Scavenger receptors on arterial macrophages can take up modified lipoproteins, including LDL that have become oxidized as a result of glucose-mediated oxidative stress [24], or modified by AGEs [54].
3.
Activation of the polyol pathway reduces the availability of NADPH, which in turn reduces glutathione regeneration and increases oxidation [50].
4.
Activation of Protein Kinase C resulting from enhanced de novo synthesis of diacylglycerol from glucose via triose phosphate, whose availability is increased because increased ROS inhibit activity of glycolytic enzymes [55]. The enhanced activity of PKC isoforms could also result from the interaction between AGEs and their cell-surface receptors [56].
All these mechanisms are activated by a single upstream event: mitochondrial overproduction of reactive oxygen species (ROS) [57].
It is known that multiple pathways contribute to accelerated atherosclerosis in diabetes including endothelial dysfunction, increased plasma lipoprotein oxidation and our studies demonstrate another possible mechanism related to increased macrophage lipid oxidation in STZ-induced diabetic mice [45]. These data, however, do not establish a causal relationship between glucose and cellular lipid peroxidation since additional factors associated with diabetes might have been involved in increased macrophage lipid peroxidation. In order to examine whether the effect of glucose on macrophage lipid peroxidation is a direct effect, in vitro studies were performed using the J774 cell line. Glucose enrichment of macrophages after exposure to high glucose concentrations was shown to induce oxidative stress (macrophage oxidative state and macrophage NADPH oxidase expression) in a dose-dependent way (Fig. 9.2c, d). In this study, increased cellular lipid peroxidation was associated with increased ability to take up OX-LDL, increased macrophage CD36 mRNA expression and increased macrophage cholesterol content. A glucose upregulating effect on OX-LDL uptake was specific for the modified LDL since glucose enrichment did not affect the macrophage uptake of native LDL [58].
The above results for the direct effect of glucose on macrophages in vitro was demonstrated only by using D-Glucose which is taken up by cells and not L-glucose which cannot penetrate the surface membrane of cells. Thus glucose uptake by macrophages is needed in order to increase macrophage oxidation and this effect could be linked to previously reported data that diabetes induction led to an increased activity of NADPH oxidase [59]. These results are in accordance with previous data showing that human monocyte derived macrophages (HMDM) incubated with high glucose concentrations increased the expression of CD36. Some evidence demonstrates also a role for AGE and RAGE in the induction of CD36 mRNA expression in macrophages [60].
Antioxidants in DM
Cells and tissues contain antioxidant defense mechanisms, which help prevent ROS production and maintain the redox balance of the cell or tissue [61]. Epidemiological studies suggest that low levels of antioxidants are associated with increased risk for cardiovascular disease (CVD), and that increased intakes appear to be protective. Several antioxidants such as vitamin E and flavonoids such as glabridin, tannins, and red wine were all shown to substantially inhibit macrophage-mediated oxidation of LDL by increasing cellular GSH [62]. During early atherogenesis, the extent of LDL oxidation by macrophages is determined by the balance between antioxidants in cells [23]. We have previously found that GSH content and glutathione peroxidase activity are both inversely related to macrophage-mediated oxidation of LDL. We have shown that MPM from E° mice contain decreased GSH levels and a profound increase of lipid peroxide content compared to MPM from controls. The E° MPM also demonstrated increased ability to reduce superoxide anions and to oxidize LDL [63].
Flavonoids are powerful antioxidants that act against LDL oxidation, and their antioxidant capacity is related to their localization in the LDL particle, as well as to their chemical structures [64]. Flavonoids, which are mostly hydrophilic and thus not LDL-bound, can act as potent inhibitors of LDL oxidation via several mechanisms, which include: (1) scavenging of free radicals by acting as reducing agents, as hydrogen atom donating molecules, and as singlet oxygen quenchers; (2) chelation of transition metal ions, thereby reducing the metal’s capacity to generate free radicals; (3) sparing of vitamin E and of carotenoids ([beta]-carotene, lycopene) in the LDL particle, thus protecting LDL from oxidation; and (4) preserving or increasing serum paraoxonase activity, thus promoting hydrolysis of LDL-associated lipid peroxides.
Diabetes is associated with reduced levels of antioxidants such as GSH, vitamin C, and vitamin E [65, 66]. Glycation of antioxidant enzymes during hyperglycemia can impair cellular defense mechanism, leading to the development of oxidative stress and the progression of diabetes with its complications [67]. Glycation of superoxide dismutase and esterases can inhibit their enzymatic activity. Glycation of thioredoxin inhibits its antioxidant activity. Thus, a reduction in antioxidative enzymes and inhibition of enzymatic activity due to glycation in diabetes significantly contributes to the oxidative environment in diabetes.
Diabetic patients are highly prone to oxidative stress. Consequently, antioxidant treatment in diabetes could be beneficial. Indeed it was shown that vitamin E or red wine supplementation to diabetic patients significantly reduced their serum oxidative stress [68, 69]. In addition to vitamin E and red wine, other types of antioxidants were shown to be effective in diabetes such as vitamin C, N-acetyl cysteine, garlic, and aged garlic [70].
In spite of these reports, recent trials of supplementation of antioxidants to the general population and also diabetic patients were all negative regarding vascular disease endpoints [71]. These studies have reduced the interest for antioxidant supplementation in general including in diabetics. The current American Diabetes Association recommendations do not encourage vitamin supplementation to diabetic patients unless a deficiency state is evident [72].
The potent antioxidant pomegranate juice (PJ) possesses impressive antioxidative properties due to its polyphenols, and consumption of PJ in humans for a period of one year significantly reduced LDL oxidation [73]. We have shown that PJ consumption in patients (non-diabetics) with carotid artery stenosis for three years caused a significant reduction of oxidative stress in their blood and significant reduction of carotid atherosclerotic lesions as measured by B-mode ultrasonography [74].
In a short term study we have shown that consumption of PJ by diabetic patients for three months did not change glycemic indices (glucose and HgbA1C) nor the levels of cholesterol and triglycerides in these patients, but it resulted in a significant reduction of serum lipid peroxidation and TBARS (that were high before PJ treatment in diabetics vs. healthy volunteers), whereas PON1 activity was significantly increased [75]. In diabetic patients the amount of macrophage cellular lipid peroxidation was significantly increased compared to controls. PJ consumption for three months significantly reduced cellular peroxides to levels lower than those observed in healthy volunteers. These effects of PJ on both serum and macrophages in diabetics could contribute to the accelerated atherogenesis in these patients. The PJ antioxidative effect could be related to its potent tannins which scavenge a wide spectrum of free radicals [76], as well as PJ-induced increment of PON1 in the serum of diabetic patients that can also contribute to the antioxidative effect of PJ in diabetics [77]. The increased uptake of OX-LDL by macrophages in diabetic subjects could be related to the increased expression of the scavenger receptor CD36 which is induced by glucose and/or the high oxidative state. In vitro incubation of PJ with the macrophages of diabetic patients resulted in a significant reduction of OX-LDL uptake [75]. Similar observations were seen when PJ was administrated to STZ-diabetic mice and also when J774 macrophages were incubated with PJ [78].
We have also shown by using different models with low vs. high oxidative states that in STZ-injected mice, the development of hyperglycemia and the severity of oxidative stress are both significantly reduced [79]. Vitamin E supplementation of APO E knockout (E0) mice with enhanced atherosclerosis and increased oxidative stress reduced serum oxidative stress and significantly reduced diabetes development, 12 days after STZ injection (Fig. 9.3a, b).
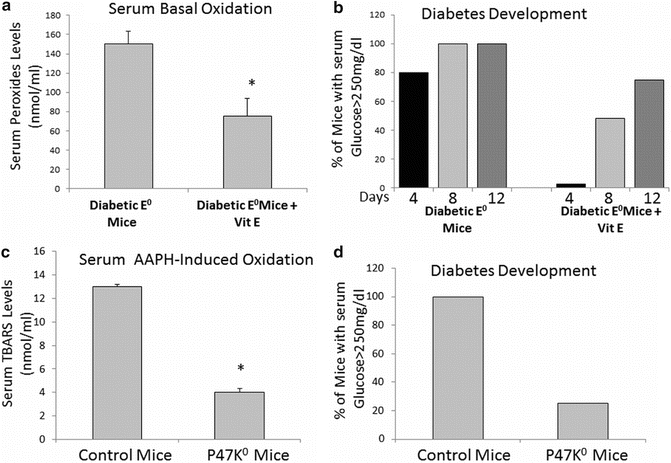
Fig. 9.3
Decreased serum oxidative state and diabetes development in atherosclerotic (E0) mice treated with vitamin E and in NADPH oxidase knockout mice. Blood drawn from fasting Vit E-treated diabetic mice were analyzed for their serum basal oxidation (a) as well as for their glucose levels in order to assess diabetes development (b) in comparison to untreated diabetic E0 mice. Blood drawn from fasting NADPH oxidase knockout mice diabetic mice were analyzed for their serum basal oxidation (c) as well as for their glucose levels in order to assess diabetes development (d) in comparison to diabetic control mice
Similar findings were shown by using P47 knockout mice lacking NADPH oxidase activity and characterized by a very low oxidative state compared to control mice. In this model, glucose levels were much lower after STZ injection compared to controls and only 25 % of the mice developed diabetes (Fig. 9.3c, d). Thus, it seems that enhanced oxidative stress could be responsible for the development of processes that lead to hyperglycemia and consequent diabetic development. On the other hand decreased oxidative stress in mice could be responsible for the reduction in new-onset diabetes mellitus.
Paraoxonase 1 (PON1) in Diabetes Mellitus
PON1 is an HDL-associated esterase/lactonase, found mainly in serum and it protects against lipid peroxidation in lipoproteins, macrophages, and atherosclerotic lesions. The activity of PON1 is inversely correlated to the risk of CAD [41]. Diabetes is associated with increased oxidative stress and low serum PON1 activity [80].
PON1 activity has been shown to be reduced in patients with DM. These patients also appear to be predisposed to the development of severe multivessel CAD [81]. PON1 activity was found to decrease in parallel to the DM duration, and this phenomenon may be related to acceleration of CHD in DM patients. We have shown that in diabetes, a significant amount of serum PON1 is dissociated from HDL to the lipoprotein deficient serum (LPDS) fraction (as a free PON1). PON1 in LPDS, unlike PON1 in HDL, is less able to protect against lipid peroxidation [82]. PON1 in HDL from type 2 diabetic patients is glycated and, as a result, has decreased ability to metabolize membrane lipid hydroperoxides [83, 84]. Similarly, in vitro studies demonstrated that glucose inactivates PON1, and glycated PON1 has reduced ability to hydrolyze membrane hydroperoxides [83].
In order to examine the effect of PON1 on the development of diabetes we used mice with different PON1 expression levels:
Mice lacking expression of PON1: PON1 knockout (K0) mice.
Mice expressing mouse PON1: C57Bl control mice.
Mice expressing human PON1 in addition to mouse PON1: PON1 Transgenic (Tg) mice.
In order to study the effect of PON1 on the development of diabetes half of the three groups of mice were injected with STZ, whereas the second half served as controls. In the non-diabetic mice macrophage superoxide ion release was increased in PON1 K° mice and decreased in PON1 Tg mice vs. controls. These effects were similar after STZ injection but more pronounced, showing a direct inhibitory effect of PON1 on cellular oxidation (Fig. 9.4a). Regarding the effect of PON1 on diabetes development, we have shown that following induction of diabetes, levels of glucose significantly increased in PON1 K° mice, whereas they remained significantly lower in PON1 Tg mice in comparison to control diabetic mice (Fig. 9.4b). After 45 days of STZ injections diabetes developed in all control and PON1 K° mice but only in 67 % of PON1 Tg mice. Moreover, the rate of mortality in controls was significantly lower than in PON1 K° mice after 45 days whereas in the PON1 Tg mice there was no mortality. Interestingly the insulin levels in PON1 K° mice were decreased significantly after STZ injection compared to controls. These studies in PON1 K° mice and PON1 Tg mice suggest indirectly that PON1 has a protective role against diabetes development secondary to its unique antioxidant properties [79].
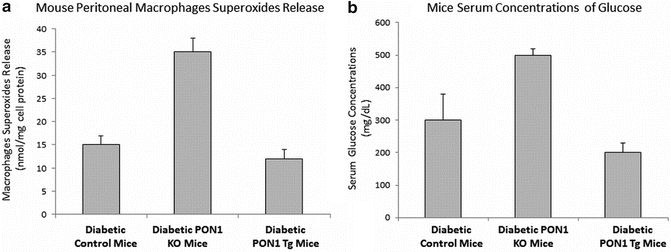
Fig. 9.4
 Lipid: Extracellular Matrix Interactions as Therapeutic Targets in the Atherosclerosis of Diabetes
Lipid: Extracellular Matrix Interactions as Therapeutic Targets in the Atherosclerosis of Diabetes
 Insulin Resistance and Atherosclerosis
Insulin Resistance and Atherosclerosis
 Lipoprotein(a): Structure, Metabolism, and Pathophysiology
Lipoprotein(a): Structure, Metabolism, and Pathophysiology
 Lipoproteins and Diabetic Nephropathy
Lipoproteins and Diabetic Nephropathy
 Fibrate Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
Fibrate Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
 Apoproteins and Cell Surface Receptors Regulating Lipoprotein Metabolism in the Setting of Type 2 Diabetes
Apoproteins and Cell Surface Receptors Regulating Lipoprotein Metabolism in the Setting of Type 2 Diabetes




Modulation of PON1 expression in mice affects macrophages superoxide release and serum glucose levels of diabetic mice: Macrophages from PON1 Knockout (KO) diabetic mice and PON1 Transgenic (Tg) diabetic mice were analyzed for their superoxide anion release in comparison to diabetic control mice (a). Serum from PON1 KO diabetic mice and PON1 Tg diabetic mice were analyzed for their glucose levels in comparison to diabetic control mice (b)
Related posts:
Lipid: Extracellular Matrix Interactions as Therapeutic Targets in the Atherosclerosis of Diabetes
Insulin Resistance and Atherosclerosis
Lipoprotein(a): Structure, Metabolism, and Pathophysiology
Lipoproteins and Diabetic Nephropathy
Fibrate Therapy: Impact on Dyslipidemia and Cardiovascular Events in Diabetic Patients
Apoproteins and Cell Surface Receptors Regulating Lipoprotein Metabolism in the Setting of Type 2 Diabetes
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
