Fig. 3.1
Classes of interspersed repeats with their examples. (termini of the branches, and their taxa of origin in square brackets). For each class their copy number and contribution (%) to the human genome summarised in the grey boxes. TSD: Target Site Duplication, P: Promoter, ORF1/2: Open Reading Frames, S: Spacer, An: poly Adenosine tract, A and B: RNA polymerase III conserved regions, LTR: Long Terminal Repeat, gag: group specific antigen, pol: polymerase, env: envelope, ITR: Inverted Terminal Repeats, Adapted from lander et al. 2001.
The question of why TEs have been so successful throughout evolution is the subject of ongoing discussion. TEs have been called “selfish genes” (Dawkins 1976) and “genomic parasites” (Yoder et al. 1997) in relation to their host genome, but evidence has accumulated over the last several decades demonstrating that, despite their disease-causing potential (reviewed in Kazazian 1998) , TEs might have some overall beneficial effect. For example, TEs can increase genomic diversity and consequently drive genome evolution within a species (Boeke and Pickeral 1999; Nekrutenko and Li 2001; Seleme et al. 2006) ; they can play a role in the stress response of the host cell (Li and Schmid 2001); and in some lineages can take over vital cellular functions, such as telomere function (Pardue et al. 1996) .
TEs can also have practical uses. For example, human specific mobile element insertions (mostly L1 and Alu) can be used for inferring human geographical origin, sex identification, DNA identification and quantification (Xing et al. 2007) . However, while the contribution of mobile elements to host genomic architecture and fluidity is undeniable, relatively little is currently known about the evolutionary dynamics of their mobilisation in humans.
3.1.1 Human Transposable Elements
In Homo sapiens, TEs are responsible for the formation of at least 45 % of the genome (Lander et al. 2001) . Figure 3.1 illustrates the different types of mobile elements that have been involved in mammalian and human genome expansion.
TEs can be classified into two groups based upon their genomic integration method (Pace and Feschotte 2007) . Class I elements transpose via an RNA intermediate, utilising a reverse transcriptase activity, and include long and short interspersed elements (LINEs and SINEs) , as well as long terminal repeat elements (LTR). The Class I transposition mechanism can be thought of as a ‘copy and paste’ method and as such is inherently replicative. Class II mobile elements integrate into the human genome, using a DNA intermediate, through a ‘cut and paste’ mechanism (Pace and Feschotte 2007; Kazazian et al. 2002) .
3.1.1.1 DNA Transposons; Class II Transposable Elements
The mechanism of DNA transposition is a ‘cut and paste’ mechanism that is not inherently replicative. DNA transposons mobilise via a DNA intermediate, which is mediated by a transposase. Only about 3 % of the human genome is derived from DNA transposons (Fig. 3.1) (Lander et al. 2001).
The evolutionary history and genomic impact of transposons have been well studied in mammals. All ~ 300,000 DNA transposons identified in the human genome reference sequence are genomic fossils that have been inactive for at least 50 Myr (Lander et al. 2001; Pace and Feschotte 2007; Smit and Riggs 1996) . Therefore any effects of transposition in contemporary human genomes must originate from a different class of transposable element. Indeed, the most active transposable elements in humans are L1 retrotransposons. Comparative genomic analysis between the human genome reference and the draft chimpanzee genome showed that 1174 human specific L1 insertions have accumulated in the 6–8 Myr since these species common ancestor (Mills et al. 2006) . Due to their ongoing mobilization in humans, it is this group of retrotransposons that are the subject of this chapter.
3.1.1.2 Retrotransposons; Class I Mobile Elements
By far, the largest portion of human mobile DNA originates from retrotransposons. In contrast to DNA transposition, DNA retrotransposition is inherently replicative and functions via a ‘copy-and-paste’ mechanism, involving transcription of the complete element, reverse transcription of the RNA into cDNA, and integration of the cDNA into a new locus in the genome. Thus, one functional progenitor retrotransposon can generate multiple copies at new genomic locations. This circumstance, and the fact that there is at least one family of retrotransposons still active in humans (the L1Hs family), may account for the excess of retroelements in the human genome. Retrotransposons can be divided into two major classes that are phylogenetically and structurally unrelated (Craig et al. 2002) . The long terminal repeat (LTR) retrotransposons account for 8 % of the human genome, and are characterised by direct LTRs flanking the element’s coding regions (Fig. 3.1). LTR and non-LTR retrotransposons do share some important structural characteristics. They each have a robust and functional promoter (Hata and Sakaki 1997) , which is responsible for transcription of full-length RNA, and they each encode a reverse transcriptase enzyme in order to produce a cDNA copy of this RNA. However, there are also important differences: in the autonomous elements (LTR retrotransposons), the cDNA integrates into new genomic loci using its own unique protein machinery (Curcio and Derbyshire 2003) and the integration process is initiated by an element-encoded integrase (IN).
3.1.1.3 Long Terminal Repeat (LTR) Retrotransposons
LTR retrotransposons are also called ‘retrovirus-like elements’ or ‘endogenous retroviruses’ because their replication pathway is similar to that of retroviruses. They are thought to originate from retroviruses that have lost a functional env-gene, confining them to strictly intracellular replication (Esnault et al. 2008) . Thus, endogenous retroviruses cannot infect other cells, and go through their replicative cycle within a single cellular lineage. With the possible exception of HERV-K, which is a putatively active human endogenous retrovirus, all known human LTR-retrotransposons are genomic fossils that have not been active for the last 40 Myr (Costas and Naveira 2000; Lander et al. 2001) . However, there is currently no evidence for mobilization events in modern day humans, despite reports of LTR promoter reactivation in two cancers (Katoh and Kurata 2013) .
3.1.1.4 Non-Long Terminal Repeat (non-LTR) Retrotransposons
Non-LTR retrotransposons are evolutionarily more ancient than LTR retrotransposons (Furano 2000) . Sequence comparisons indicate that they share a common origin with RT-bearing group II introns of bacteria and mitochondria (Yang et al. 1999) . Comprising more than one third of human DNA (32 %), non-LTR retrotransposons clearly have had a great impact.
Based on the structure of their coding regions, the autonomous non-LTR elements are further subdivided into the restriction enzyme (RE) type and the apurinic/apyrimidinic endonuclease (APE) type. The RE-type non-LTR retrotransposons are characterised by a single open reading frame (ORF) with a RE-like EN domain following the C-terminal end of the RT domain (Malik et al. 1999) . RE-type elements represent the oldest lineage of non-LTR retrotransposons (Malik et al. 1999).
Most retrotransposons discovered so far are APE-type non-LTR retrotransposons. They are recognised by having one or two ORFs and the existence of an EN domain that is distantly related in sequence to the apurinic/apyrimidinic (AP) endonucleases (Martín et al. 1995; Feng et al. 1996) . The EN domain is localised at the N-terminal end of ORF2p, upstream of the RT domain. Based on the elements’ structures, and on phylogenetic analyses of their RT domains, we can currently distinguish four groups of APE-type non-LTR retrotransposons, and these can further be subdivided into a further 11 clades (Burke et al. 1999; Eickbush and Malik 1999; Lovsin et al. 2001) . (Fig. 3.2).

Fig. 3.2
Schematic diagrams of RE-type and APE-type non-LTR retrotransposons Illustrating their differences in structural organisation and in their coding capacity. a. APE-type non-LTR retrotransposons, b. RE-type non LTR retrotransposons, UTR untranslated region, ORF open reading frame, APE Apurinic/APyrimidinic Endonuclease, RT reverse transcriptase, RE restriction enzyme-like endonuclease (Craig et al. 2002)
3.1.2 Autonomous and Non-Autonomous Non-LTR Retrotransposons
The non-LTR retrotransposons can also be categorised as either autonomous or non-autonomous retrotransposons. Autonomous retrotransposons are able to encode the proteins required for their own retrotransposition. However, non-autonomous elements are unable to retrotranspose without appropriating the retrotransposition machinery of autonomous elements (Lander et al. 2001; Dewannieux et al. 2003) .
3.2 Human Long Interspersed Elements (LINEs)
In this chapter we focus on human LINE1 elements, as they are the only active autonomous retrotransposons in our genome, and so are more likely to contribute to cancer. First, we will expand on what is known about the LINE1 family and their structure, the major roles of LINE1 in our genome and finally, we will discuss the role of LINE1 in different cancers and its potential to contribute to disease progression.
Long interspersed elements-1 (LINE-1s or L1s) are the only autonomous non-LTR retrotransposons in the human genome, i.e. they encode the proteins required for their own retrotransposition. LINE retrotransposons are further classified into three sub-groups in the human genome: LINE1 (L1), LINE2 (L2) and LINE3 (L3). LINE1 is the only active member of this family and it has a copy number of over 500,000, and makes up about 17 % of the genome. LINE2 and LINE3 are older lineages that together comprise less than 4 % of the genome. They have accumulated numerous mutations during the course of evolution, and so are unlikely to be still actively retrotransposing (Lander et al. 2001) . In addition ~ 99 % of LINE1s are inactive due to a 5’ truncation, internal rearrangements or deletions, but it has been estimated that in an average diploid human genome there are 80–100 full-length L1s with intact ORFs, which are likely to be competent for retrotransposition (RC-L1s) (Deininger et al. 2003; Brouha et al. 2003; Beck et al. 2010) .
During their mobilization process, LINE-1 element proteins display strong cis preference, i.e. the proteins preferentially retrotranspose their encoding RNA, largely ensuring that only functional copies are propagated (Wei et al. 2001) . This cis preference, from an evolutionary point of view, minimises the impact of the accumulation of mutated elements on active L1 retrotransposition. However, it is known that the LINE1 autonomous machinery can also act in trans to retrotranspose non-autonomous retrotransposons such as short Interspersed Elements (SINEs), SVA (SINE/VNTR/Alu) elements (Callinan et al. 2006) and other cellular RNAs (Esnault et al. 2000; Boeke 1997) . In rare cases, the cis preference of LINEs is circumvented by spliced mRNAs of cellular genes. This results in an intronless and promoterless retropseudogene copy of the original gene transcript, followed by a poly-A tail flanked by target site duplications (Vanin 1985) . Therefore processed retro-pseudogenes are also a direct result of LINE activity (Esnault et al. 2000). Indeed, recent studies have revealed that cancer genomes contain new processed pseudogenes absent from healthy tissues establishing that retrotransposition is ongoing in some cancers (Cook et al. 2014) .
3.3 L1 Retrotransposon Structure and Retrotransposition
To date, the human LINE-1 element is the most thoroughly characterised mammalian APE-type non-LTR retrotransposon (Ostertag and Kazazian 2001a; Moran and Gilbert 2002) . Human specific L1s are further divided into pre-Ta (Transcribed, subset a), Ta0, Ta1, Ta1nd, and Ta1d subfamilies based on lineage specific sequence variants.
The pre-Ta subfamily is characterised by an ACG diagnostic trinucleotide in its 3’ UTR at nucleotide positions 5954–5956 (relative to the reference element L1.3, Accession: L19088, henceforth the basis for all element coordinates). Moreover, Salem et al. (2003) demonstrated that pre-Ta elements preferentially integrate into low GC content (36 %) genomic DNA. The majority of pre-Ta family elements are, 5’ truncated but 29 full-length pre-Ta with intact ORFs have been reported. This fact and that a pre-Ta element insertion caused one case of human genetic disease (an integration into the factor VIII gene, resulting in haemophilia A) indicates the preTa family contains active members (Kazazian et al. 1988; Salem et al. 2003) .
The Ta family (or Transcribed, subset a) is the youngest and most active L1 family, and has been found to cause ~ 100 identified clinical cases of various genetic disorders (Hancks and Kazazian 2012) . Over 50 % of these elements show dimorphism (presence or absence) across human populations (Boissinot and Furano 2001) . These families of L1 emerged after the divergence of humans from chimpanzees about 6 Myrs ago, and so are specific to humans. There are two main Ta subfamilies: L1 Ta0 and L1 Ta1 (Boissinot et al. 2000). ACA nucleotides at positions 5954–5956 of the 3’ UTR are diagnostic for this family. Based on the nucleotides at positions 5557 and 5560 in ORF2 elements can be assigned to the distinct Ta1 and Ta0 classes. Ta1 elements have T and G nucleotides at these two positions and Ta0 have G and C respectively (Boissinot et al. 2000). The Ta0 subfamily is more similar in sequence to the non-Ta L1s, and therefore has been suggested to be an older family of elements. By contrast the Ta1 family are younger than the pre-Ta and Ta0 families, and so have accumulated fewer inactivating mutations. The Ta1 family still actively retrotransposes and is likely to be currently increasing in copy number in the human genome (Boissinot et al. 2000). It is estimated that the Ta1 family arose about 1.6 Myrs ago and can be further divided into two subfamilies: Ta1d and Ta1nd. The Ta1d group are recognised by a deletion at position 74 in the 5’ UTR whilst the Ta1nd group lacks this deletion. There are around 90 full-length human L1s with intact ORFs in the human genome reference sequence, which are potentially RC-L1s (Brouha et al. 2003) . However, cell culture retrotransposition assays demonstrated that only 6 of these elements account for 84 % of the total retrotransposition activity (Brouha et al. 2003). This data suggests that these very active elements dominate retrotransposition activity in the human genome. Four of the “hot” L1 elements characterised by Brouha et al. (2003) belong to the Ta1d family, with the other two elements belonging to the Ta1nd and Ta0 families (Brouha et al. 2003). Recent sequence-based studies have estimated the rate of L1 insertion into the human genome to be around 1 in 212 live births (Xing et al. 2009) and 1 in 140 (Ewing and Kazazian 2010) . These estimates are much lower than was previously estimated (1 in 33 live births) for L1 insertions, based on the activity of disease-causing elements (Brouha et al. 2003; Beck et al. 2010) . However unbiased capture of full-length elements and their retrotransposition activity suggests that presence/absence variation between individuals represents a substantial reservoir of active elements is segregating in human populations (Beck et al. 2010).
3.3.1 L1 Structure
A complete retrotransposition-competent (RC) L1 element is 6 kb in length and contains two non-overlapping open reading frames: ORF1 and ORF2 (Fig. 3.4). The 5’ untranslated region (UTR) of a RC-L1 is approximately 900 bp in length. A major polymorphism of L1 elements occurs within this region: the presence or absence of a 131-bp sequence (Minakami et al. 1992) . The L1 sense promoter is also located within the 5’ UTR region and the first 155 bp have been demonstrated to be involved in L1 expression (Minakami et al. 1992; Athanikar et al. 2004) . The structure of each L1 component and their role in L1 retrotransposition (where known) are discussed in more detail in the following sections. To illustrate these sections a schematic diagram of an intact L1 retrotransposon and its modules is presented in Fig. 3.3.
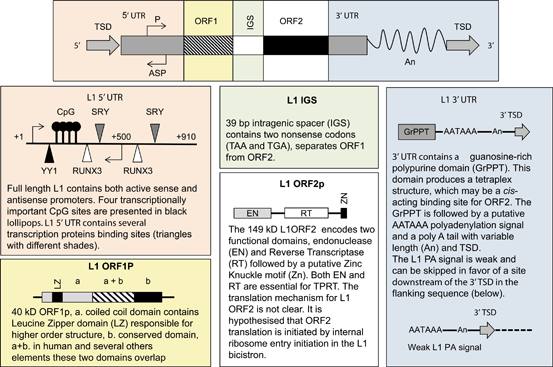
Fig. 3.3
L1 structure and the summaries of its components: L1 5’ and 3’ untranslated region (UTR), open reading frame 1 and 2 (ORF1/2) Intragenic spacer (IGS), poly A tail (An) and target site duplication (TSD). L1 5’ UTR structure is adapted from Badge, poster publication 2007, L1ORF1P adapted from Martin et al. 2000 , L1ORF2p structure adapted from Goodier et al. 2004, and L1 3’UTR adapted from Craigie et al. 2002 .
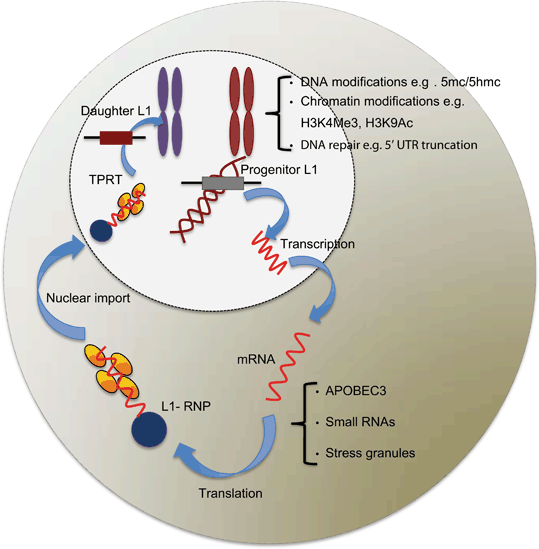
Fig 3.4
Schematic diagram of host defence mechanisms deployed against endogenous L1 retrotransposition at different stages of L1 retrotransposition. Based on the location in the cell, the defence mechanisms against L1 retrotransposition can be divided into two categories: nuclear and cytoplasmic. Few of the defence mechanisms against endogenous L1 retrotransposition, and their timing are understood in detail. Many studies have contributed to this diagram, which are cited in Sect. 1.7
3.3.1.1 The L1 Promoter and Transcription of the L1 Element
The 5’ UTR of the L1, which is about 900 bp in length, accommodates two internal promoters (+ 1 to + 670). The region between + 1 to + 100 shows the highest promoter activity, although no TATA box is present (Swergold 1990) . The L1 5’ UTR contains a sense promoter (SP), which starts at + 1 of the L1 sequence and an antisense promoter (ASP), positioned between + 399 to + 467 bp of the L1 sequence (Speek 2001) . Both sense and antisense L1 promoter sites are highly conserved in human L1PA10-L1PA1 families, covering over 40 Myr of evolution. In vitro, luciferase reporter-based experiments have demonstrated that the L1PA6 elements have an active ASP (Macia et al. 2011) , despite their antiquity. It has been suggested that over 1/3 of L1Hs contain highly active ASPs, which are capable of interfering with normal gene expression (Nigumann et al. 2002; Speek 2001) when located intragenically .
The L1 sense promoter possesses characteristics of both RNA polymerase II (Pol II) promoters, which control transcription of all protein-coding genes, and RNA polymerase III (Pol III) promoters that are responsible for synthesis of tRNA, 5S RNA and several small and non-coding RNAs (Kurose et al. 1995) . The L1 transcript is about 6 kb long and it has two protein-coding regions and a polyadenylated extension at the 3’ end of the transcript. These characteristics suggest L1’s is a Pol II dependent promoter. However inhibition studies have shown the L1 promoter is less sensitive to α-amanitin, a Pol II inhibitor, and more sensitive to tagetitoxin, a Pol III inhibitor (Kurose et al. 1995). These data suggest that the L1 promoter is Pol III dependent, but produces transcripts more characteristic of Pol II. This unusual sensitivity may be explained by the importance of YY1 transcription factor in L1 transcription initiation (Athanikar et al. 2004) , which is utilised at both Pol II and Pol III promoters.
The L1 sense promoter creates a long, protein encoding, polyadenylated transcript and the promoter acts as a Downstream Promoter Element (DPE), such that it initiates transcription at position + 1 of the L1 sequence, but lacks features characteristic of PolII promoters such as upstream TATA and CAAT boxes (Kurose et al. 1995; Swergold 1990) . The L1 5’ UTR also contains several PolII transcription factor binding-sites, such as MECP2, SOX2 and RUNX3, which have been shown to be involved in the transcriptional regulation of L1s (Rosser and An 2012) . The DNA methylation status of the L1 promoter has a great potential to impact its activity, especially during cancer progression. We discuss the methylation of the L1 promoter and its relation to cancer in later sections of this chapter.
YY1 Binding Site
The ubiquitous transcription factor YY1 (Yan Ying 1) binding site, which is a PolII and PolIII transcription, has been established as an important sequence in L1 transcription, and is located at + 13 to + 26 of the L1 5’ UTR sequence (Becker et al. 1993; Kurose et al. 1995) . Since YY1 is capable of both activating and repressing transcription, this protein may play a role in down-regulating L1 transcription in some cell types, while activating it in others (Becker et al. 1993). YY1 regulates L1 transcription by enhancing accurate transcription initiation rather than being required for initiation as even truncated L1s, which lack the YY1, site have functional promoters (Athanikar et al. 2004). It has been demonstrated that inhibition of the YY1 binding site in tissue culture assays has a minor effect on L1 transcription activation and retrotransposition (Athanikar et al. 2004). However, it has also been demonstrated that the deletion of the YY1 site in the first 20 bp significantly reduces (5 fold) L1 retrotransposition in cell culture assays (Singer et al. 2010) .
Since deletion of the YY1 binding site does not abrogate L1 transcription, L1 elements must be able to be transcribed from upstream or downstream of this site. Transcription initiation from downstream of the YY1 binding site leads to 5’ truncated progeny, which may not be retrotranspositionally competent, due truncation. It has been shown that most RC-L1s are transcribed from + 1 or very nearby, such that their progeny are potentially able to retrotranspose autonomously (Athanikar et al. 2004).
Other L1 Transcription Factor Binding Sites
Previous studies have demonstrated that the L1 5’ UTR contains four methyl-CP2-responsive elements at the following positions: + 36, + 101, + 304 and + 481 (Hata and Sakaki 1997) . These C-methyl binding proteins bind to methylated DNA (Feng and Zhang 2001) . Based on their recognition-binding site these proteins are divided into two types: the MBP group binds to methylated DNA, while the second group, MeCPs and MDBP, has no sequence specificity for methylated DNA (Feng and Zhang 2001). Among these, the MeCP2 are the most abundant methyl-Cytosine binding proteins and it has been demonstrated that MeCP2 binds to methylated DNA only in the context of chromatin contributing to long-term repression and nuclease-resistant methyl-CpGs (Meehan et al. 1992; Hata and Sakaki 1997) .
Moreover, Tchenio et al. (2000) demonstrated that the human L1 promoter contains two functional sites for SRY (sex determining region Y) transcription factors. SRY transcription factors are members of the SOX protein family, and are expressed in the urogenital ridge of the embryo and in adult, testis, hypothalamus and midbrain (Lovell-Badge 2009). Cell culture studies have shown that ectopic over-expression of one of the SRY families, Sox11, results in 10 fold trans-activation of endogenous L1Hs (Tchenio et al. 2000). The two potential binding sites for SOX transcription factors are located in the first 670 nucleotides of the L1 promoter. It is possible that L1 activity in the brain is mediated by SOX2, as a decrease in SOX2 expression during the early stages of neuronal differentiation, when recapitulated in cell culture, is associated with increases in L1 transcription and retrotransposition (Muotri et al. 2005) . The first site, SRYA, is located between nucleotides 427–477, and SRYB is located between 572–577. In addition in vivo experiments have demonstrated that SRY transcription factor binding at the L1 promoter can drive transcription in cell culture, and congruently mutations at the SOX binding site can inhibit L1 transcription (Tchenio et al. 2000). However, L1 transcription can be transiently stimulated by transcriptional binding switch from a SOX2/HDAC1 repressor complex to a wnt-mediated T-cell factor/lymphoid enhancer factor (TCF/LEF), which briefly activates L1 transcription in models of human and rodent neuronal differentiation (Muotri et al. 2010).
The RUNX3 family contains heterodimeric transcription factors, which can potentially bind to three regions in the L1 promoter: nucleotides + 83 to + 101 and + 526–508 of the L1 5’ UTR. These binding sites mean RUNX3 can potentially influence L1 transcription by regulating both sense and antisense promoters (Yang et al. 2003) . Mutation analysis at each of the three sites has demonstrated that mutation of the first binding site reduces L1 transcription, while mutations at the other two binding sites do not have any significant effect (Yang et al. 2003). This may be due to the second and third binding sites being located outside the first 100 nucleotides of L1 5’ UTR, which is important for transcription initiation (Yang et al. 2003) . Moreover, a recent study of L1 5’ UTR fragments driving luciferase reporter genes identified several novel transcription start sites at position + 525 and + 570. It is likely that these central sites are involved in the recruitment of transcription initiation complexes and it is possible that they can drive bi-directional L1 transcription (Alexandrova et al. 2012) .
3.3.1.2 L1 ORF1 and ORF2 and Translation of the L1 Retrotransposition Machinery
Despite host genome defence mechanisms acting against L1 retrotransposition, these potentially mutagenic insertions occur in germline and somatic tissues, as shown by disease causing insertions. Because the L1 translational machinery has a strong cis-preference, functional protein crosstalk between individual elements is greatly reduced, and lack of competition from partially functional mutants may explain the longevity of L1 activity. However this hypothesis requires both ORFs to be expressed from the same transcript, so co-expression of the ORF encoded proteins is likely a marker of active L1 retrotransposition. Co-expression of the two L1-encoded proteins, ORF1p and ORF2p, has been detected by immunohistological analyses in pre-spermatogonia of human foetal testis and in germ cells of human adult testis (Ergün et al. 2004) . Also, most disease-causing L1 insertions are apparently germline in origin (Kazazian 2004). These data and parallel observations of ORF1p expression in mouse pachytene spermatocytes (Martin and Bushman 2001) are consistent with the expectation that potentially mutagenic transposable elements confine their replication to germlines, where they can maximise their probability of transmission, without compromising host viability. As co-expression of both ORFs is required for retrotransposition their translation in quite different amounts from a bi-cistronic transcript is central to retrotransposition, but is far from clearly elucidated. However in the following section we review the current understanding of the structure, function and translation of each ORF in more detail.
Translation and Role of L1-ORF1 in L1 Retrotransposition
The first open reading frame of L1 (L1 ORF1) is 1017 bp in length and encodes a 338 amino acid cytoplasmic protein, also known as p40 (Hohjoh and Singer 1997) . The centrally located leucine zipper (LZ) domain in human L1ORF1 is involved in the formation of higher order ORF1p multimers and it has been demonstrated that the LZ domain is required for RNP assembly and retrotransposition (Craig et al. 2002 and Doucet et al. 2010) . The carboxyl domain of ORF1 is basic and contains several conserved amino acids that are likely to play a role in RNA binding. However, this carboxyl domain lacks common functional motifs, found in RNA binding proteins such as the RNP motif, and the Arginine-rich motif (Craig et al. 2002). The sequence of ORF1p is not related to any protein with known function and its role in the L1 retrotransposition cycle is incompletely understood (Basame et al. 2006) . It has been demonstrated that efficient translation initiation of L1 5’ UTR is strictly cap dependent, rather than as previously suggested via an internal ribosome entry site (IRES) mediated model (Dmitriev et al. 2007) . Results of co-immunoprecipitation experiments demonstrate that ORF1p is a high affinity RNA binding protein with no sequence binding specificity (Kolosha and Martin 2003) . It has also been demonstrated that the nucleic acid chaperone activity of ORF1p is important for successful L1 retrotransposition (Martin et al. 2005) . Also, cell culture and in vivo experiments have each demonstrated that L1ORF1p exists in many copies in the cytoplasm (Hohjoh and Singer 1996) . Moreover L1ORF1p contains non-canonical RNA recognition motifs (RRMs) that have RNA-binding properties, supporting its function as an unconventional RNA binding protein (Khazina and Weichenrieder 2009) .
Several roles have been proposed for ORF1p in the L1 retrotransposition process. One concept is that the L1 RNA is unstable: ORF1p, with its RNA binding activity, is required to coat and protect the L1 RNA intermediate in the cytoplasm before its translocation to the nucleus where TPRT occurs. It is thought that cis preference acts to ensure that the L1 proteins associate with their functional encoding RNA (Moran and Gilbert 2002) . Although ORF1p has only been definitively detected in the cytoplasm it could still be involved in the later stages of L1 retrotransposition, such as TPRT (Martin and Bushman 2001). It is hypothesised that the nucleic acid chaperone activity of ORF1p is involved in strand transfer, which allows the annealing of the DNA primer from the target site to the RNA primer during the process of reverse transcription (Martin and Bushman 2001). It is also possible that ORF1p facilitates the reverse transcription process by enabling movement of polymerase through RNA secondary structures formed during first cDNA synthesis (Martin and Bushman 2001).
Translation and Role of L1-ORF2 in L1 Retrotransposition
The L1 s open reading frame (ORF2) encodes a protein of ~ 150 kDa containing 1275 amino acids (Scott et al. 1987) . The initiator methionine of ORF2 in the human L1 element is separated from ORF1 by a 66-bp in-frame spacer region containing three stop codons. It is not clear how the separate translation of both ORFs from the bi-cistronic RNA is accomplished; this problem is made even more intriguing by the fact that the spacer region is not conserved between L1 elements of different species (McMillan and Singer 1993) . It was first suggested that translation of ORF2 must be accomplished either by reinitiating translation or by internal initiation via an internal ribosomal entry site (IRES) (McMillan and Singer 1993). However, using an engineered LINE1 retrotransposition assay, it was later demonstrated that L1-ORF2p is translated by an unconventional termination/re-initiation mechanism (Alisch et al. 2006).
The ORF2 protein has proven to be very hard to detect, largely due to the lack of robust and specific ORF2p antibodies (Wagstaff et al. 2011) . Thus, indirect methods, such as measuring its enzymatic activity have been used to study the role of this protein in the L1 retrotransposition cycle. It seems that ORF2p has two major activities, each of which can be assigned to specific domains. The N-terminal contains a conserved endonuclease activity domain. Its sequence and crystal structure is similar to AP-like endonuclease APE1, which is involved in the base excision repair pathway (Ergun et al. 2004; Feng et al. 1996; Weichenrieder et al. 2004) . Despite its conservation, it has been demonstrated that L1s lacking an EN domain are still able to retrotranspose, at a lower efficiency than wildtype, likely by using pre-existing nicked DNA sites for integration (Morrish et al. 2002) . The central domain of ORF2p is responsible for the reverse transcriptase activity, and it contains a conserved Z-motif (Mathias et al. 1991) . The L1 RT domain is related to those in other non-LTR elements (Malik et al. 1999) and also shows some sequence similarity to LTR retrotransposons and retroviruses (Xiong and Eickbush 1990) . At the C-terminal end, there is a conserved “C-domain” containing a cysteine-rich region whose function is not clear. It has been suggested that this region has evolved in response to interactions with other L1 sequences or host factors (Wagstaff et al. 2011). Also, it has been shown that mutations in this region abolish the ability of ORF2p to interact with L1 RNA and ultimately block L1 retrotransposition in cultured cells (Feng et al. 1996; Moran et al. 1996; Doucet et al. 2010) .
3.3.1.3 L1 3’ UTR and Poly A Tail
The L1 3’ UTR covers the terminal 205 bp of full-length elements, includes a polyadenylation (PA) signal, and terminates in a poly (A) tail. One of the characteristics of the L1 PA signal is the ability to transduce genomic DNA (up to 1.6 kb in vitro) downstream of its 3’ UTR (Holmes et al. 1994) . In the process of polyadenylation the poly-A tail is added to the putative AAUAAA polyadenylation specificity factor (CPSF1) binding site. However, the L1 PA signal lacks the conserved elements that normally reside downstream of the poly-A site in canonical RNA polymerase II transcripts. Hence it has been suggested that the L1 PA site is weak and can be bypassed by the transcription machinery in favour of a stronger PA site in the 3’ flanking genomic sequence (Moran et al. 1999) . L1’s weak PA signal is suggested to be an evolutionary adaptation that allows L1 to reside within introns with minimum effect on gene expression through the induction of premature polyadenylation (Moran et al. 1999) . Around a third of L1 elements carry a 3’ transduction and they are estimated to have contributed ~ 33 Mb of DNA to the human genome (Moran et al. 1999; Pickeral et al. 2000; Goodier et al. 2000; Szak et al. 2003) .
The L1 3’ UTR also contains the sequence motif (CACAN5GGGA) at position 5796–5884 nt, which has a high binding affinity for nuclear export factor 1 (NXF1) (Lindtner et al. 2002) . The role of NXF1 is similar to that of constitutive transport elements (CTE) , which facilitate the nuclear transport of viral intronless mRNA, such as simian type D retroviruses (Lindtner et al. 2002).
The 3’ UTR of the L1 element is poorly conserved within and between species (Scott et al. 1987) . Interruption of this region by additional nucleotides does not seem to have severe effects on retrotransposition, as illustrated by reporter assays, where L1 tolerates marker genes of up to 3500 bp in length in its 3’ untranslated region (Moran et al. 1996; Ostertag et al. 2000; Gilbert et al. 2002; Symer et al. 2002) .
All the classifications above apply to full-length copies of L1. However, only 5 % of endogenous human L1 elements are full length (6 kb). The remaining 95 % are 5’ truncated, internally rearranged or deleted (Szak et al. 2002). Some of this damage to L1 structure may be the result of mutations and genomic rearrangements after integration of the retrotransposon. Indeed, Coufal et al. (2011) demonstrated that in ataxia telangiectasia mutated (ATM) deficient cells , there were either more or longer L1 retrotransposition events, compared to ATM wild type cells. This suggests that cellular proteins involved in the DNA damage response may modulate L1 retrotransposition. 5’ truncation and inversion most probably occur during the retrotransposition process itself (Ostertag and Kazazian 2001a) . In inverted L1 elements, the 5’ truncated region is commonly orientated in an antisense direction to its 3’ end. This structure is thought to be the consequence of a model of retrotransposition called ‘twin priming’, whereby second strand cDNA synthesis initiates before first strand cDNA is completed (Ostertag and Kazazian 2001b). Inversions can be detected in about 25 % of insertions in members of the Ta family (Ostertag and Kazazian 2001a; Skowronski et al. 1988) .
L1 integrants are usually flanked by variable TSDs with lengths of up to 60 bp (Szak et al. 2002) . These TSDs are generated during the process of L1 replication. Some TSDs are difficult to identify due to statistical uncertainties about the occurrence of short duplications; the presence of multiple mutations in TSDs of ancient integrants; the presence of blunt end nicking sites (Van Arsdell and Weiner 1984) ; or the presence of a staggered double strand break with a 5’ overhang instead of a 3’ overhang. The latter process causes a deletion of the target site instead of duplication (Gilbert et al. 2002; Symer et al. 2002). However the vast majority of L1 insertions have identifiable TSDs, suggesting they originate from an endonuclease dependent process.
3.3.2 Mechanism of L1 Retrotransposition
The mechanism of retrotransposition of non-LTR retrotransposons is not entirely understood. However, the first steps of integration of these classes of elements have been elucidated by biochemical experiments using the site-specific RE-type retrotransposon R2BM from the silkworm Bombyx mori (Luan et al. 1993) . These studies led to the model of L1 retrotransposition called ‘target primed reverse transcription’ (TPRT) (Cost et al. 2002) .
Although RE-type and APE-type elements belong to different families of non-LTR retrotransposons that share very few structural similarities, the basic mechanism of transposition initiation by TPRT is relatively conserved. This has been demonstrated by reconstitution of the initial steps of L1 element transposition in vitro, by providing only the complete L1 ORF2 protein, L1 RNA, and a target DNA (Cost et al. 2002). Also, further experiments have shown that the EN domains of the two types of retrotransposons (RE and APE) initiate the integration process by nicking the target DNA (Cost et al. 2002; Eickbush and Malik 2002) . The resulting 3’-hydroxyl group serves as a primer for reverse transcription of the element’s RNA. On the other hand, it has been demonstrated that L1 integration can also occur at pre-formed nicks and double strand breaks in the target DNA, known as endonuclease independent-TPRT (Morrish et al. 2002) . However, this mode of insertion is prevalent only in cell lines with defects in DNA repair machinery. Therefore, endonuclease-independent insertion provides an alternative pathway for L1 retrotransposition in the human genome (Sen et al. 2007) . As a result it is likely that nicking and reverse transcription are two independent steps in TPRT (Cost et al. 2002; Eickbush and Malik 2002). The EN domain, can also cleave the second strand of target DNA at a slower rate compared to the nicking of the first strand (Cost et al. 2002). Depending on the position of the second nicking site relative to the initial one, TPRT can generate a target site deletion, a simple ‘blunt’ integration, or a target site duplication (TSD) which flanks the inserted element (Cost et al 2002; Eickbush and Malik 2002).
A major unresolved issue regarding the mechanism of LINE retrotransposition is what occurs after second-strand cleavage. Despite extensive efforts, in vitro experiments with the R2 protein did not lead to the detection of intermediates expected for second-strand synthesis (Luan et al. 1993) . In contrast, in vitro TPRT of L1 yielded 5’ junctions between the L1 sequence and the target DNA. This result indicates that the RT is able to accept cDNA as a template for second-strand synthesis, probably by a second round of TPRT (Cost et al. 2002; Eickbush and Malik 2002) .
However, this in vitro process is very inefficient and it does not necessarily reflect the natural mode of retrotransposon integration and still leaves open the major question of how the damaged genomic DNA is repaired. It is generally assumed that cellular DNA repair pathways are involved in these final steps of integration and that these activities generate the observed TSDs (Gilbert et al. 2005) .
3.4 Genomic Distribution of Human L1s
Human LINEs are distributed across the genome, but not distributed evenly. There are some parts of the genome, which have very low repeat density. This could be because these regions cannot tolerate insertion of repeats due to essential cis regulatory architecture. An example of repeat poor regions is the homeobox (HOX) gene clusters, which contain the lowest reported density of interspersed repeats (Lander et al. 2001; Simons et al. 2006) . In contrast to this, some parts of the genome are very rich in repeats, such as chromosome Xp11, which contains a 525 kb region comprised of 89 % repeats. Overall it is suggested that LINEs are more abundant in gene poor, and AT rich regions, which usually show low recombination rates (Lander et al. 2001) . In comparison to Alu, LINEs have been reported to insert at a four fold higher density in GC poor regions, while Alus have a lower tendency (five fold lower) to insert in AT rich regions (Lander et al. 2001). One reason for this insertional bias of LINEs towards AT rich regions could be due to the consensus L1 endonuclease target site TT/AAAA, which is intrinsically more common in AT rich regions (Lander et al. 2001; Jurka 1997; Cost and Boeke 1998) . However, Alu elements also use the L1 machinery in trans to integrate into the genome, but Alus have a high density in GC rich regions. Therefore, the biasing of L1 insertion in AT rich regions may not be only due to endonuclease site selection but also post-insertion selection. It has been suggested that L1 insertion occurs in AT and GC rich regions, but that insertions in GC-rich regions are lost through selection. It is clear that L1s inserted within genes can have a variety of negative effects on their host gene such as altered splicing, interference with gene regulation and level of expression, and premature polyadenylation (Cost and Boeke 1998; Lander et al. 2001) .
3.5 Impact of L1 Integration on Human Genome Plasticity
Recently, efforts have been directed towards unveiling the molecular mechanisms by which L1 impacts gene expression and mammalian cell development, differentiation, and cancer. New L1 integrations have a great impact on host genome diversification and evolution. The ways that L1 retrotransposition can alter the host genome are discussed in detail below.
3.5.1 Increasing the Size of the Human Genome
An orthologous sequence comparison of the human and chimpanzee genomes suggested that the human genome continues to expand, either because of inherently more active insertional mutation processes or through being less efficient at deleting such events (Liu et al. 2003) . Therefore, one of the greatest impacts of L1 on the human genome is their contribution to expanding genome size (Liu et al. 2003). Considering that L1 is also responsible for Alu retrotransposition in the genome, it has contributed about 750 Mb to the human genome (Lander et al. 2001). Moreover, the ongoing expansion of L1 has also created significant inter- and intra-individual variation by introducing L1 insertional polymorphisms (presence/absence) at orthologous loci.
3.5.2 Disease Causing L1 Retrotransposition
There are ~ 100 cases of human genetic diseases caused by L1 integration into genes (Hancks and Kazazian 2012) . Based on L1 retrotransposition assays it has been suggested that about 10 % of de novo L1 retrotransposition events occur in the introns of actively transcribed genes (Moran et al. 1999) . In fact, it is likely that evolutionarily successful L1s (active L1s) preferentially insert into genes, which are transcriptionally active and therefore have an open chromatin configuration (Macia et al. 2011) .
The first L1 disease-causing insertion was reported in two patients with haemophilia, where an L1 was integrated into exon 14 of the human factor-eight gene (Kazazian et al. 1988). Subsequently cases of L1 disruption of the dystrophin gene have been reported to cause muscular dystrophy and cardiomyopathy in four unrelated individuals (Holmes et al. 1994; Matsuo et al. 1991 and Yoshida et al. 1998) . It has also been shown that a heritable full length L1 insertion into intron two of the ß-globin gene (L1ß-thal) is responsible for some cases of ß-thalassemia (Divoký et al. 1996; Kimberland et al. 1999) . Additionally insertion of a full length L1 into an intron of the X-linked RP2 gene is responsible for progressive retinal degeneration and ultimately retinitis pigmentosa (XLRP) (Schwahn et al. 1998) . Moreover, a case of colon cancer has reported to be caused by somatic insertion of a truncated L1 into the APC gene (Miki et al. 1992) . More recently it has been reported that somatic de novo L1 retrotransposition events are detectable in lung cancer cells (Iskow et al. 2010) . Also, up regulation of L1 RNA and ORF1p has been reported in several tumours including breast sarcomas and in 10 % of tumours of germline origin, such as ovarian and testicular tumours (Asch et al. 1996; Bratthauer and Fanning 1993) . The role of L1 in cancer will be covered in more detail in the following sections.
3.5.3 Genome Instability Caused by L1 Retrotransposition
In addition to mutagenic insertions, L1 retrotransposition can generate local genomic instability through several other mechanisms, which are explored in this section. All of these mechanisms are compatible with tumorigenic potential for these elements. DNA double strand breaks (DSBs) can be caused by the endonuclease activity endogenous L1ORF2p (Gasior et al. 2006) . It is been shown that the number of DNA DSBs generated by L1ORF2p is much higher than the number of actual L1 insertions (Gasior et al. 2006). However, the extent of genome instability induced by endogenous L1 retrotransposition is not clear due to a lack of sensitive antibodies to target ORF2p and also because the repair of L1-mediated DSBs may not leave any sign of L1ORF2p involvement. As a result, the attribution of L1ORF2p to genomic DSBs, which are highly mutagenic and prone to induce recombination, is likely underestimated (Cordaux and Batzer 2009) . In addition to generating local genome instability, L1 can also cause genomic rearrangements through insertion-mediated deletions. Studies of L1 retrotransposition in cell culture have demonstrated that about 20 % of L1 insertions are associated with structural rearrangements, including flanking genomic deletions at the insertion site (Gilbert et al. 2002; Gilbert et al. 2005; Symer et al. 2002) . Another study reported a lower frequency of deletion (2 %) than in cell culture assays, with endogenous L1 retrotransposition causing deletions with an average size of 800 bp in the human genome (Han et al. 2005) . Since L1-mediated insertion deletions are generally grouped into two sizes classes (< 100 bp and > 1 kb), it has been suggested that each group is caused by a different mechanism. In general, small deletions may arise due to template switching with subsequent 5’ to 3’ exonuclease activity on both the exposed 5’ ends. Larger deletions can be mediated by non-homologous end joining when the nascent cDNA invades a double strand break with a 3’ overhang located upstream of the integration site. Subsequent gap repair will remove the cDNA and the adjacent segment to cause a large deletion (Han et al. 2005) . A study by Chen et al., (2007) demonstrated a 46 kb full length L1 insertion-mediated deletion event that possibly occurred through the template jumping process. This deletion resulted in removal of seven exons of the pyruvate dehydrogenase complex, component X (PDHX) gene, which caused a case of pyruvate dehydrogenase complex deficiency (Chen et al. 2007).
3.5.4 Ectopic Recombination upon L1 Retrotransposition
Due to the high copy number of L1s in the human genome, they can also create structural variation at the post-integration stage, through non-allelic homologous recombination or ectopic recombination. Ectopic recombination events seem relatively rare and are usually mediated by truncated elements (Boissinot et al. 2000) . Indeed there is no evidence of polymorphic L1 associated ectopic recombination in humans. This can be explained by the low activity of retrotransposition competent L1s in the modern human genome (Boissinot et al. 2000), or perhaps by the frequency with such mutations are deleterious (Wang et al. 2006) . Ectopic recombination potentially causes various types of genomic rearrangements, including duplications, deletions, and inversions.
Segmentally duplicated regions can contain paralogous copies of genes, promoters and other regulatory components (Samonte and Eichler 2002). It is likely that segmentally duplicated regions are associated with the creation of novel genes and the formation of pseudogenes (Lynch and Conery, 2000) . Alternatively, ectopic recombination can cause recombination-associated deletion events (RADs). Genome-wide comparisons of the human and chimpanzee genomes have identified 73 human specific L1RAD events that occurred following the divergence of humans from chimpanzees (Han et al. 2008) . Although L1RAD events are not very common, it has been suggested that they are responsible for the deletion of about 450 kb of the human genome (Han et al. 2008). This event is most frequent in heterochromatic regions, which suggests that there may be negative selection against L1RADs in euchromatin (Graham et al. 2006) .
As mentioned earlier, L1-mediated ectopic recombination is also involved in gene inversion events. It is suggested that L1 contributes to genomic inversion possibly through the formation of secondary structures or by providing a target site for double strand breaks (Lee et al. 2008) . Among the characterised inversions mediated by L1 insertions, some loci include the exonic regions of known genes, which suggests that L1-mediated inversions can generate alterations in gene function (Lee et al. 2008; Cordaux and Batzer 2009). Therefore, although this type of recombination does not affect the size of the genome it can produce genomic variation.
3.5.5 L1-Mediated Sequence Transduction
In addition to duplicating themselves, L1s sometimes carry with them upstream or downstream flanking genomic sequences (termed 5’ and 3’ transduction, respectively), providing a novel mechanism for genome evolution. L1-mediated sequence transduction occurs when L1 transcripts extend upstream or downstream of the genomic flank and then transduce these sequences into new genomic locations through the L1 retrotransposition process. L1 5’ sequence transduction is usually very short, ranging between 5–8 nt sequences and it is not a common process. Additionally, due to 5’ truncation during L1 retrotransposition, there is a severe ascertainment bias to determine how often L1 mRNAs may contain 5’ transduced sequences. This process occurs when L1 sequences are transcribed by a host promoter upstream of the L1 5’ terminus, and subsequently mobilised during the L1 retrotransposition cycle (Pavlicek et al. 2002a; Pickeral et al. 2000; Szak et al. 2003) . The 3’ sequence transduction process is more common, and occurs when transcription of the L1 bypasses the weak polyadenylation (PA) signal in favour of a stronger canonical PA signal in the 3’ genomic flank followed by mobilisation of the genomic flanking DNA to a new location. The sequence transduction process seems to be more common in active or recently active elements: it has been demonstrated, in cell culture assays that between 10–20 % of recent active human insertions contains sequence transductions (Goodier et al. 2000) . During the process of sequence transduction, exons, promoters and other regulatory sequences upstream and downstream of the L1 can be transduced into the new genomic location, causing exon shuffling and potentially altering the expression and or structure of the recipient gene (Moran et al. 1999) . This process maintains genome plasticity and genome evolution (Goodier et al. 2000). Indeed if 5’ truncation occurs during retrotransposition, removing the L1 sequences, exon shuffling events are expected to be difficult to identify, and so their frequency may be greatly underestimated.
3.5.6 Regulation of Gene Expression
As mentioned above, L1s can affect the genome at the DNA level. In this section the effect of L1 at the RNA level are considered in more detail. It has been demonstrated that L1 can affect transcription in several distinct ways. They can generate alternative splice sites resulting in the exonization of L1 sequences, at least in rodents (Zemojtel et al. 2007; Huang et al. 2009) . Also intronic L1s may sometimes interfere with transcriptional elongation and so produce different lengths of mRNA from a gene (Han et al. 2004) . If the L1 inserts in the antisense orientation relative to the host gene, it can potentially produce truncated cellular transcripts by premature polyadenylation (Han et al. 2004). Moreover, L1 can produce novel transcripts through the activity of its antisense promoter (ASP). Nearly 1/3 of the L1Hs studied contain active ASPs (Speek et al. 2001) . Therefore it is possible that some of the transcripts initiated from the L1 ASPs are competent for translation. On the other hand, it has been recently demonstrated that a large proportion of regulatory RNAs, termed long-non coding RNAs (lncRNAs), are derived mostly from TE sequences, and are frequently generated from TE-derived promoters (Kapusta et al. 2013) . In addition, insertion of full length L1 sequences into intronic regions of a gene can potentially “break” a gene. “Gene breaking” occurs where an L1 inserted in the opposite orientation to a host gene can generate two novel partial transcripts: one from the endogenous promoter including exons upstream of the L1 insertion, and a second internal transcript driven by the L1 ASP. Indeed, bioinformatic analysis on the human genome has highlighted 15 genes and transcription units that have potentially been affected by L1 insertions in this way (Wheelan et al. 2005) . Additionally a recent study of intragenic L1s in lung cancer cells has shown that L1 pre-mRNA binds to the Ago2 complex to suppress the transcription of cancer genes (Aporntewan et al. 2011) . Therefore, with transcriptional interference from the endogenous L1 sense and antisense promoters, its polyadenylation signal, and alternative L1 transcripts, L1 exhibits a great potential to impact human transcriptome composition.
3.5.7 Epigenetic Regulatory Role of Human L1s
Because L1 elements are frequently found in or near genes, it is possible that heterochromatin formed at retrotransposons could spread and repress the transcription of nearby genes. It has been suggested that L1’s principle epigenetic regulatory role is in X chromosome inactivation (XCi). XCi is a well-established mechanism of gene regulation that acts to achieve gene dosage compensation between male and female embryos (Heard and Disteche 2006) . XCi initiates at the X inactivation centre (XIC) (Rastan 1983) , which contains several genes that produce non-coding RNAs (Chureau et al. 2002) . Little is known about how inactivation spreads across the chromosome, although it has been proposed that L1s play a role in the cis spreading of X chromosome inactivation (Lyon 1998) . L1s are enriched on the X chromosome compared to autosomes, and significantly so at Xq13 where the XIC is located. To support this idea, it has been demonstrated that genes on the X chromosome which escape X inactivation are generally located in L1 poor regions (Ross et al. 2005) . For young L1s, the proposed involvement in X inactivation is also linked to methylation. Indeed, It has been shown that demethylation and activation of the L1 ASP can drive the transcription of neighboring genes: Weber et al. (2010) have shown that demethylation of the L1 ASP in colon cancer cell lines induces the expression of L1 and proto-oncogene cMet (L1-cMet) transcripts. This result demonstrated the involvement of L1 in gene regulation and a clear link to methylation. However, the formal demonstration of direct retrotransposon-mediated epigenetic control of neighboring genes in humans and the evaluation of the extent of this phenomenon at a genome-wide scale are active topics of investigation, and will be discussed more in following sections.
3.6 Host Defence Mechanisms Against L1 Retrotransposition
As well as the direct mutational effects of L1 insertion, various forms of genetic instability caused by L1 integration include the generation of L1 chimeras, intrachromosomal deletions (chromosomal deletions of > 11 kb), intrachromosomal duplications, and chromosomal inversions (approximately 120 kb in length) (Gilbert et al. 2002; Han et al. 2005; Symer et al. 2002) . It was demonstrated by Gilbert et al. (2005) that the L1 reverse transcriptase can faithfully replicate its own transcript and has a base mis-incorporation rate of ~ 1 in 7000 bases. All these observations indicate that L1 retrotransposition can lead to a variety of genomic rearrangements suggesting that hosts should be under selection to restrict L1 activity, as integration of L1 and other retrotransposons poses a potential threat. As a result organisms have apparently evolved diverse mechanisms to combat retrotransposon activity. Indeed, the initial step in L1 retrotransposition was described as a host/parasite “battleground” that serves to limit the number of active L1s in the genome (Gilbert et al. 2005). Since L1 has been actively mutating mammalian genomes for millions of years, it is likely that the host has evolved multiple mechanisms to combat L1 mobility at discrete steps of the retrotransposition cycle. In the following sections the mechanistic strategies used by the host to restrict L1 retrotransposition are discussed in more detail.
3.7 Epigenetic Modifications Regulate L1 Retrotransposition
Different types of epigenetic regulation are suggested to keep L1 retrotransposition activity in check. Some of the well-studied epigenetic regulatory modes are outlined in the following subsections.
3.7.1 Cytosine Methylation in Host Defence and Genome Instability
A possible mechanism, by which the activity of many potentially active human L1s could be suppressed, is methylation of cytosine bases in their promoters, some of which are known to be critical for promoter activity (Hata and Sakaki 1997) .
The majority of cytosine methylation in plants and mammals resides in repetitive elements and a large proportion of this lies in retrotransposons, which constitute more than 42 % of the human genome (Goll and Bestor 2005) . Transposons can only proliferate in genomes where the fitness of transposons is greater than that of the host. Therefore, host defence mechanisms are under selective pressure to suppress these elements (Bestor 2003) ; as judged by its distribution, DNA methylation is primarily a mechanism of transposon suppression. In somatic cells L1 promoters are generally hypermethylated, but in malignancy-derived cells, the global hypomethylation of CpG dinucleotides is correlated with L1 activity (Kitkumthorn and Mutirangura 2011) . This correlation was supported by the recent identification of several de novo L1 insertions in a cohort of lung tumours (Iskow et al. 2010) with more frequent insertions being observed in tumours showing significant genomic hypomethylation.
As previously mentioned, a variety of studies have suggested that de novo L1 retrotransposition is more likely to occur in germ cells and/or during early embryonic development (Garcia-Perez et al. 2007b; Van den Hurk et al. 2007) , where a pair of global de-methylation events occur at the genome reprogramming stages. Although it has been frequently suggested that methylation of CpG dinucleotides has a regulatory role, especially in suppressing repetitive elements, there is evidence against this hypothesis (Walsh and Bestor 1999) , such as the somatic inheritance of genomic methylation patterns in mammals (Riggs 2002) . Therefore, chromatin modifications such as DNA methylation could be a consequence of active transcription rather than a cause, and the causal relationship of these phenomena remains to be fully elucidated.
Studies on 5-methylcytosine residues in the L1 promoter, especially at the four transcriptionally important CpG sites, show that DNA methylation can repress L1 activity both in vivo and in vitro (Hata and Sakaki 1997) . In contrast to the suppressive effect of DNA methylation on L1 promoters, it has been demonstrated that 5-hydroxylation of the methylcytosine moiety (hm5c) can be an activating factor. However, a study of hm5c protein interactions showed that it does not interact with the same proteins as the 5mc pathway, which suggests that hm5c must regulate the L1 promoter through other mechanisms (Williams et al. 2011) . Indeed, Ficz et al. (2011) demonstrated that hm5c methylation modifications are enriched in euchromatic regions and show a positive correlation with L1 expression. Also, a recent study has demonstrated that the Tet protein can generate other cytosine modifications downstream of hm5c (Ficz et al. 2011). These modifications are 5-formylcytosine (5fc) and 5-carboxylcytosine (5ca5) (Ito et al. 2011) . Whether these newly discovered DNA cytosine modifications have any direct and controlling effect on L1 promoters and L1 expression remains to be investigated, but their existence suggests that epigenetic DNA modification is more complex than suspected.
Many studies have shown that a variety of epigenetic modifications can regulate L1 activity, and these are not limited to DNA modifications. Chromatin modifications are also likely to have an important role in controlling L1 activity. For example, Teneng et al. (2011) have recently demonstrated the direct association of H3K4 and H3K9 modifications with L1 activity. In fact they have demonstrated that the exposure of HeLa cells to Benzo (a) pyrene (Bap) causes L1 reactivation in HeLa cells through induction of early enrichment of the transcriptionally active chromatin markers histone H3 trimethylation at lysine 4 (H3K4Me3) and histone H3 acetylation at lysine 9 (H3K9Ac), and also reduces the association of DNMT1 with the L1 promoter. These processes cause depletion in cellular DNMT1 expression, which subsequently reduces cytosine methylation within the L1 promoter CpG island (Teneng et al. 2011) .
Other evidence for chromatin modifications regulating L1 activity was uncovered in hippocampus neural stem (HCN) cells . Muotri et al. (2005) showed that histone deacetylase 1 (HDAC1) and methylation of H3 at Lys9 (K9), which both associate with transcriptional silencing in undifferentiated HCN cells, was directly correlated with L1 reporter construct activity in transgenic mice. In contrast acetylation of H3K9 and methylation of H3K4 (associated with transcriptional activation) was associated with high levels of L1 transcripts in HCN differentiated cells. This data supports the idea that chromatin remodelling during the early stages of neuronal cell differentiation allows transient stimulation of L1 retrotransposition (Muotri et al. 2005). Additionally, recent studies of L1 expression in undifferentiated human embryonic stem cells have demonstrated that retrotransposition processes in pluripotent cells are subjected to strong epigenetic control (Macia et al. 2011; Munoz-Lopez et al. 2011) .
3.7.2 Role of Small RNAs in Regulation of L1 Retrotransposition
Small RNAs inhibit retrotransposon proliferation in the host genome via two mechanisms, which are independently mediated by either small interfering RNAs (siRNAs) or PIWI-interacting RNAs (piRNAs) (Meister et al. 2004; Soifer and Rossi 2006) . The mechanisms by which these small RNAs are generated and how they inhibit retrotransposon mRNAs are still not fully understood, but there is strong evidence for a connection. It has been reported that host siRNA can repress retrotransposition through the post-transcriptional disruption of L1mRNA (Yang and Kazazian 2006) . It is suggested that L1 bidirectional transcripts can be processed into small interfering RNAs (siRNAs) that supress L1 retrotransposition by an RNA interference mechanism (Yang and Kazazian 2006) . Multiple RNA silencing pathways might act as a defence mechanism against L1 retrotransposition. Consistently, very recently it has been demonstrated that Dicer and Ago2-dependant RNAi restricts L1 retrotransposition in undifferentiated mouse embryonic stem cells (Ciaudo et al. 2013) .
Another independent mechanism that has been suggested to suppress retrotransposon mRNA are piRNAs , which are generated from genomic loci that encode long precursor RNAs containing the remnants of different families of TE elements (Malone et al. 2009) . It is likely that small-RNA-based mechanisms may also play role in silencing the mammalian L1 elements. Indeed it has been demonstrated that an antisense promoter located within the human L1 5’ UTR allows the production of an antisense RNA transcript (Speek et al. 2001) that, in principle, could base pair with sense-strand L1 mRNA to establish a dsRNA substrate for the Dicer protein (Levin et al. 2011) . Furthermore, mouse mutants lacking the murine PIWI family proteins (MILI or MIWI2) exhibit a loss of methylation of L1 and IAP elements. This loss correlates with the elements transcriptional activation in male germ cells and suggests that MILI and MIWI2 play essential roles in establishing de novo DNA methylation of L1 retrotransposons in the fetal male germline (Kuramochi-Miyagawa et al. 2008) . Recently it has been demonstrated that Drosha-DGCR8, components of the microprocessor machinery responsible for the generation of miRNAs, recognize and binds L1 RNA derived sequences; additionally, cultured cells lacking these proteins support elevated levels of L1 and Alu retrotransposition. Overall, these observations suggest that the microprocessor complex is involved in post-transcriptionally suppressing L1 and Alu retrotransposition (Heras et al. 2013) .
Related posts:
 Retinoic Acid Receptor-β, From Gene to Clinic
Epigenetics and Three Main Clinical Aspects of Breast Cancer Management
Predictive Role of O6-Methylguanine DNA Methyltransferase Status for the Treatment of Brain Tumors
Molecular and Biological Aspects of Microcephalin Gene: Directions in Brain Tumor and Methylation
Epigenetics of Thyroid Cancer
Malignant Rhabdoid Tumor: Epigenetic Mechanism of Tumorogenesis
Retinoic Acid Receptor-β, From Gene to Clinic
Epigenetics and Three Main Clinical Aspects of Breast Cancer Management
Predictive Role of O6-Methylguanine DNA Methyltransferase Status for the Treatment of Brain Tumors
Molecular and Biological Aspects of Microcephalin Gene: Directions in Brain Tumor and Methylation
Epigenetics of Thyroid Cancer
Malignant Rhabdoid Tumor: Epigenetic Mechanism of Tumorogenesis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


